

Abstract
(−)-Lomaiviticin A (1) and the monomeric lomaiviticin aglycon [aka: (−)-MK7-206, (3)] are cytotoxic agents that induce double-strand breaks (DSBs) in DNA. Here we elucidate the cellular responses to these agents and identify synthetic lethal interactions with specific DNA repair factors. Toward this end, we first characterized the kinetics of DNA damage by 1 and 3 in human chronic myelogenous leukemia (K562) cells. DSBs are rapidly induced by 3, reaching a maximum at 15 min post addition and are resolved within 4 h. By comparison, DSB production by 1 requires 2–4 h to achieve maximal values and >8 h to achieve resolution. As evidenced by an alkaline comet unwinding assay, 3 induces extensive DNA damage, suggesting that the observed DSBs arise from closely spaced single-strand breaks (SSBs). Both 1 and 3 induce ataxia telangiectasia mutated- (ATM-) and DNA-dependent protein kinase- (DNA-PK-) dependent production of phospho-SER139-histone H2AX (γH2AX) and generation of p53 binding protein 1 (53BP1) foci in K562 cells within 1 h of exposure, which is indicative of activation of nonhomologous end joining (NHEJ) and homologous recombination (HR) repair. Both compounds also lead to ataxia telangiectasia and Rad3-related- (ATR-) dependent production of γH2AX at later time points (6 h post addition), which is indicative of replication stress. 3 is also shown to induce apoptosis. In accord with these data, 1 and 3 were found to be synthetic lethal with certain mutations in DNA DSB repair. 1 potently inhibits the growth of breast cancer type 2, early onset- (BRCA2-) deficient V79 Chinese hamster lung fibroblast cell line derivative (VC8), and phosphatase and tensin homologue deleted on chromosome ten- (PTEN-) deficient human glioblastoma (U251) cell lines, with LC50 values of 1.5 ± 0.5 and 2.0 ± 0.6 pM, respectively, and selectivities of >11.6 versus the isogenic cell lines transfected with and expressing functional BRCA2 and PTEN genes. 3 inhibits the growth of the same cell lines with LC50 values of 6.0 ± 0.5 and 11 ± 4 nM and selectivities of 84 and 5.1, for the BRCA2 and PTEN mutants, respectively. These data argue for the evaluation of these agents as treatments for tumors that are deficient in BRCA2 and PTEN, among other DSB repair factors.
Graphical Abstract
INTRODUCTION
A large portion of chemotherapies target DNA, but many of these agents possess dose-limiting side effects arising from excessive damage to normal tissues. DNA damage is ameliorated by a complex network collectively known as the DNA damage response (DDR). The DDR comprises no less than one dozen distinct pathways that repair specific types of DNA lesions, such as alkylations, base mismatches, single-strand breaks (SSBs), and double-strand breaks (DSBs).1 These repair pathways are frequently mutated in cancer and drive genomic instability and tumor progression.2 However, these mutations also provide a therapeutic opportunity because DDR-deficient cell lines ameliorate the damage induced by exogenous agents less efficiently than normal tissues.3 This may translate into increased sensitivity, lower effective doses, and fewer off-target effects. The heightened sensitivity of cell lines to genotoxic agents conferred by specific mutations in the DDR has been referred to as a synthetic lethal interaction,4 an expression originally used to describe pairwise genetic mutations that render a cell nonviable.5
The complex dimeric bacterial metabolite (−)-lomaiviticin A (1) is a powerful inhibitor of cultured human cancer cell growth, with half-maximal inhibitory potencies in the low nM–pM range (Figure 1).6 It was recently shown7 that the remarkable cytotoxicity of 1 arises from the induction of highly toxic8 DSBs in DNA. Both of the diazotetrahydrobenzo[b]-fluorene (diazofluorene)9 functional groups (see structure 4) within 1 contribute to DNA cleavage. The natural monomeric diazofluorene (−)-kinamycin C (2)10 does not display DNA-cleavage activity and is much less potent (〈GI50〉 of 2 = 340 nM against the NCI 60). However, the structural complexity of 1, its high reactivity toward reducing agents and nucleophiles, and its limited availability6b significantly constrain structure–function studies and translational development. To address this, efforts have been directed toward the identification of simpler analogs that induce DNA DSB formation and are amenable to synthesis scale-up and optimization. An evaluation11 of a panel of synthetic diazofluorenes led to the identification of the monomeric lomaiviticin aglycon [aka: (−)-MK7-206, (3)] as an inducer of DNA DSBs in tissue culture, although much higher concentrations of 3 were required (1 μM 3 vs <1 nM 1).7
Figure 1.

Structures of (−)-lomaiviticin A (1), (−)-kinamycin C (2), (−)-MK7-206 (3), and the diazofluorene 4.
An understanding of the DDR induced by 1 and 3 and the identification of synthetic lethal interactions with specific DDR mutations will guide the selection of tumor types for in vivo evaluation. In this manuscript we first characterize the kinetics of DNA damage by 1 and 3 in human chronic myelogenous leukemia (K562) cells. These data allowed us to develop a time-resolved immunofluorescence assay to probe the DDR induced by 1 and 3. We then show that 1 and 3 activate nonhomologous end joining (NHEJ) and homologous recombination (HR) repair8 and are synthetic lethal with mutations in breast cancer type 2, early onset- (BRCA2) and phosphatase and tensin homologue deleted on chromosome ten (PTEN), among other factors. BRCA2 is essential to HR repair; individuals with germline mutations in BRCA2 are at a high risk for developing breast and ovarian cancers (50–80% and 30–50%, respectively), among others.12 Although the role of PTEN in the DDR is not fully understood, mutant PTEN undergoes translocation from the nucleus via an interaction with the key NHEJ kinase ataxia telangiectasia mutated (ATM),13 and over 2700 PTEN mutations have been documented across 28 tumor types.14 Our results provide a foundation for the in vivo evaluation of 1 and 3 against tumor types that are deficient in BRCA2 and/or PTEN.
RESULTS
Determination of the Kinetics of DNA Damage by 1 and 3
In order to elucidate the DDR pathways activated by 1 and 3, we first needed to characterize the kinetics of DNA damage by each agent. We used an alkaline comet unwinding assay15 which allows for the detection of both SSBs and DSBs (Figure 2). In these assays, K562 cells were treated with 1 or 3, fixed in agarose, lysed, unwound under alkaline conditions, and subjected to electrophoresis at high pH. The DNA was then visualized by staining with the fluorophore SYBR green. DNA damage decreases supercoiling, relaxing the DNA and allowing for migration toward the anode. This relaxation creates the diagnostic comet “tails” that are observed upon visualization. The amount of undamaged DNA (in the head of the comet) and the amount of damaged DNA (in the tail of the comet) were then quantified using the CometScore software (Figure 2A). This assay revealed that both 1 and 3 rapidly induced DNA damage within 15 min of exposure in a dose-dependent fashion. Extensive DNA damage was observed in the presence of only 5–50 nM 1 (Figure 2B). By comparison, approximately 1–5 μM 3 was required to attain comparable levels of DNA damage (Figure 2C). At 10 μM 3, extensive fragmentation was observed with little DNA remaining in the head of the comet.
Figure 2.

1 and 3 induce extensive DNA damage in K562 cells. A. Overview of the alkaline comet unwinding assay. Undamaged, supercoiled DNA does not migrate to an appreciable extent when subjected to electrophoresis (left). Nicking and cleavage of DNA results in relaxation and migration toward the anode, generating the characteristic “tail” of the comet (center and right). The level of DNA damage is presented as %DNA in the comet tail, which is equal to the total comet tail intensity divided by the total comet intensity, multiplied by 100. K562 cells were exposed to 0.5, 5, or 50 nM 1 (B) or 0.1, 1, 5, or 10 μM 3 (C). Exposure was 15 min. Cross denotes mean %DNA in tail (91–151 cells), box denotes 2nd and 3rd quartiles, and lines in the middle of the box denote the median. Error bars represent 10th and 90th percentile, and values individually plotted are outliers.
The neutral comet unwinding assay follows the same protocols as the alkaline comet assay with the exception that unwinding and electrophoresis are conducted at near-neutral pH, conditions that allow for the selective detection of DSBs.15,16 We implemented a time-resolved form of this assay to gain insights into the rates of production and resolution of DSBs induced by 1 and 3. K562 cells were treated with 0.5 nM 1 or 1 μM 3 for up to 24 h. To obtain each data point, the cells were collected, placed on ice, washed with cold PBS, and resuspended for analysis. These experiments showed that both 1 and 3 induced the formation of DSBs; however the kinetics of DSB production and resolution were remarkably distinct (Figure 3). The accumulation of DSBs induced by 1 reached a maximum at 2 h, and the breaks were resolved within 8–24 h. By comparison, DSB production and resolution in cells treated with 3 were much more rapid, reaching a maximum at approximately 15 min post addition and approaching resolution within 2–4 h. As in the alkaline comet assay (Figure 2), the DNA-damaging activity of 1 in this assay was much higher than that of 3. Additional experiments showed that DSB production by 3 was not measurably increased at concentrations >1 μM (Figure S1).
Figure 3.
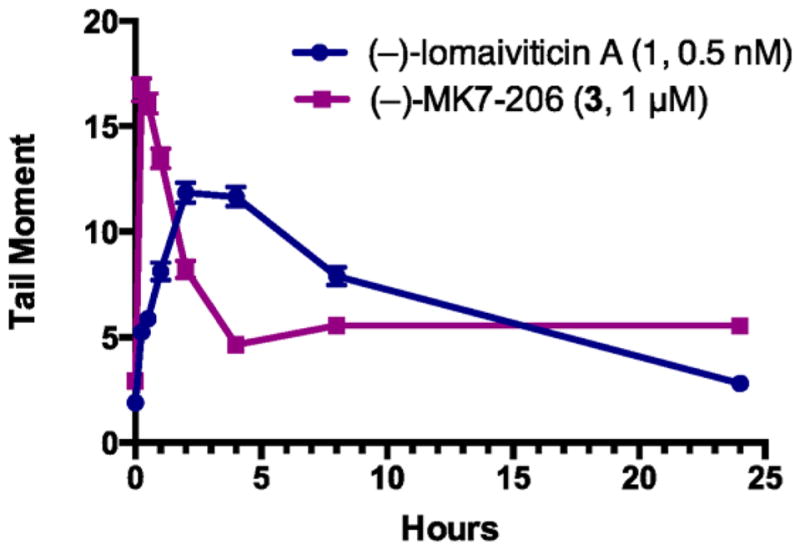
1 and 3 induce DNA DSBs in K562 cells, as determined by a neutral comet unwinding assay. This assay is similar to the alkaline comet unwinding assay (Figure 2) with the exception that unwinding and electrophoresis are conducted at near-neutral pH, allowing for the selective detection of DSBs. The amount of DNA in the tail is expressed as the tail moment, which is defined as the product of the tail length and the fraction of DNA in the tail. K562 cells were treated with 0.5 nM 1 or 1 μM 3 and incubated at 37 °C for 15 min to 24 h. Each point represents the mean tail moment (of 92–152 cells) at each time point with standard error of the mean shown.
1 and 3 Induce Time-Dependent Production of γH2AX and 53BP1 Foci
Using the kinetic data outlined above, we then sought to probe for the activation of NHEJ and HR repair in cells treated with 1 or 3. An overview of the cellular sensing mechanisms that lead to activation of these repair pathways is shown in Figure 4 (for detailed discussions, see refs 8). Activation of NHEJ is initiated by recognition of the DNA DSB by the lupus Ku autoantigen protein p70–lupus Ku autoantigen protein p80 (KU70–KU80) heterodimer, which recruits the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and p53-binding protein 1 (53BP1)17 to the site of damage. ATM kinase18 is then recruited to form phospho-SER139-histone H2AX (γH2AX).19 53BP1 and γH2AX stabilize the DSB and prevent resection. Additional factors, such as Artemis, DNA ligase 4, X-ray repair cross-complementing protein 4, and XRCC4-like factor (not shown) are then recruited to ligate the broken ends. In HR repair, the DSB is detected by the MRE11–Rad50–NBS1 (MRN) complex, which leads to activation of ATM and ataxia telangiectasia and Rad3-related (ATR) kinases. In addition to H2AX, ATM phosphorylates CtBP-interacting protein (CtIP) and breast cancer type 1 susceptibility (BRCA1), among other factors. End resection by MRE11 is followed by binding of replication protein A (RPA) to the single-stranded ends and replacement by Rad51 in the presence of BRCA2. Rad51 mediates invasion of the sister chromatid and homology searching. The single-stranded ends are extended by DNA polymerase, and the strands are annealed by recombinases or resolvases (not shown). In both NHEJ and HR, γH2AX is generated for a megabase region surrounding the site of damage and is amplified by the DNA-PK complex20 in NHEJ repair. Finally, replication stress is sensed by RPA which recruits ATR to phosphorylate H2AX21 and initiate DNA repair. The factors that determine repair pathway choice are complex and include the DSB microenvironment, availability of the repair factors in each pathway, and the stage of the cell cycle.8a
Figure 4.

Overview of the cellular sensing pathways leading to the activation of NHEJ and HR repair, the two canonical pathways that ameliorate DNA DSBs. Only factors relevant to the present study are shown; for detailed discussions, see ref 8.
γH2AX19 and 53BP117 foci are useful indicators of DNA DSB repair activity. We applied the data from our comet assays above to develop a time-resolved immunofluorescence assay for the detection of γH2AX and 53BP1 foci in cells treated with 1 or 3. 53BP1 and γH2AX foci were evident at 30 min after exposure to 0.5 nM 1 (Figure 5), and residual foci were observed after 24 h. The kinetics of 53BP1 and γH2AX foci formation from 0–4 h correlate with the induction of DNA DSBs observed by the neutral comet assay (Figure 3). However, while the neutral comet assay appeared to show high levels of DSB resolution after 8 h, the frequency of 53BP1 and γH2AX foci did not markedly decrease between 8 and 24 h. The presence of foci at later time points may be due to residual DNA DSBs, which are likely to be substrates for the slow component of DNA DSB repair.22
Figure 5.

Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with 0.5 nM 1. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 0.5, 2, 4, 8, 16, and 24 h after addition.
We then studied the time-dependent formation, colocalization, and resolution of 53BP1 and γH2AX foci in K562 cells treated with 1 μM 3. Because our comet assay data indicated that DSB production by 3 peaked at 15 min (Figure 3), we examined 53BP1 and γH2AX foci production over the time ranges 5–60 min (Figure 6A) and 1–24 h (Figure 6B). Relatively small 53BP1 foci were evident as early as 5 min post addition and appeared to plateau after 15 min, in accord with the comet assay presented above. However, as with 1, 53BP1 and γH2AX foci persisted and were evident at later time points (Figure 6B). Moreover, the levels of γH2AX appeared to increase without a corresponding increase in 53BP1, suggesting the formation of apoptotic DNA fragments.23
Figure 6.

Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with 1 μM 3. (A) 5–60 min post addition 3. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 5, 10, 15, 30, and 60 min after addition. (B) 1–24 h post addition 3. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 0.5, 1, 2, 4, 8, 16, and 24 h after addition.
3 Induces Apoptosis in K562 Cells
One of the earliest steps of apoptosis involves the translocation of the membrane phospholipid phosphatidylserine (PS) from the inner to the outer cell membrane. Once exposed to the extracellular environment, PS can bind annexin V. Accordingly, annexin V–fluorophore conjugates, such as Alexa Fluor 488-conjugated annexin V (Annexin V–A488) are used to identify apoptotic cells. The translocation of PS precedes other apoptotic processes such as loss of membrane integrity. Propidium iodide (PI) binds nucleic acids by penetrating compromised membranes and is frequently used as a secondary dye to identify late-apoptotic cells. Annexin V–A488/PI staining was conducted to determine if the increase in γH2AX induced by 1 μM 3 over the period 4–24 h was related to induction of apoptosis. Parallel experiments were conducted using 0.5 nM 1. Upon treatment of K562 cells with 3 for 4 h, approximately 70% of the cells revealed late-apoptotic PS exposure (Annexin V positive) and a compromised cell membrane (PI positive). Induction of apoptosis was not observed in K562 cells treated with 1 at a 4 h time point (Figure S2).
1 and 3 Activate NHEJ and HR Repair
To more conclusively connect the immunofluorescence experiments above with activation of NHEJ and HR repair, we evaluated the production of γH2AX and 53BP1 foci by 1 and 3 in the presence of DSB repair inhibitors. As discussed in Figure 4, ATM kinase is a major physiological mediator of the phosphorylation of H2AX in response to DSB production.18 2-(4-Morpholinyl)-6-(1-thianthrenyl)-4H-pyran-4-one (KU55933) is an ATP-competitive inhibitor of ATM that was identified in a high-throughput screen (IC50 = 13 nM).24 Cellular inhibition of ATM by KU55933 diminishes ionizing radiation (IR)-initiated phosphorylation of a range of ATM targets including H2AX.24
Co-incubation of K562 cells with KU55933 and 1 or 3 demonstrated that production of γH2AX is ATM dependent (Figure 7). K562 cells were pretreated with KU55933 (10 μM) for 1 h and then assayed for γH2AX after treatment with 0.5 nM 1 or 1 μM 3 for an additional hour. Pretreatment with KU55933 diminished the formation of γH2AX foci induced by 1 and 3. 53BP1 foci formation was also noticeably decreased upon pretreatment with KU55933.
Figure 7.

Induction of γH2AX by 3 and 1 is ATM dependent. Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with 1 μM 3 or 0.5 nM 1 in the presence or absence of 10 μM KU55933. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 0.5 nM 1, 1 μM 3, 10 μM KU55933, 0.5 nM 1 + 10 μM KU55933, 1 μM 3 + 10 μM KU55933. Cells were exposed to KU55933 for 1 h prior to addition of 1 or 3; exposure was 1 h.
ATR kinase phosphorylates H2AX in response to replication stress (Figure 4).21 Caffeine is an inhibitor of both ATM (IC50 = 0.2 mM) and ATR (IC50 = 1.1 mM) kinases.25 Pretreating cells with 5 mM caffeine for 1 h before the addition of 1 or 3 (1 h additional incubation) resulted in a marked reduction in γH2AX foci (Figure 8), and the decrease in γH2AX foci was more pronounced in cells treated with 3 than 1. As with the ATM inhibitor KU55933, the 53BP1 response was diminished in cells treated with caffeine and 1.
Figure 8.

γH2AX foci induced by 3 and 1 are ATM and ATR dependent. Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with 1 μM 3 or 0.5 nM 1 in the presence or absence of 5 mM caffeine, which inhibits ATM and ATR. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 0.5 nM 1, 1 μM 3, 5 mM caffeine, 0.5 nM 1 + 5 mM caffeine, 1 μM 3 + 5 mM caffeine. Cells were exposed to caffeine for 1 h prior to addition of 1 or 3; exposure was 1 h.
DNA-PK is a member of the PI3KK family of kinases that phosphorylates histone H2AX in response to DNA DSBs20 and apoptosis (Figure 4).23 8-(4-Dibenzothienyl)-2-(4-morpholin-yl)-4H-1-benzopyran-4-one (NU7441) is a selective inhibitor of DNA-PK (IC50 = 14 nM).26 Addition of NU7441 increases the persistence of γH2AX after DNA damage.27 Pretreating cells with 10 μM NU7441 for 1 h before addition of 1 or 3 (1 h additional incubation) demonstrated that production of γH2AX is DNA-PK dependent (Figure 9). In the presence of NU7441 and 1, γH2AX and 53BP1 foci formation was reduced, and induction of γH2AX and 53BP1 foci was almost completely inhibited by NU7441 in K562 cells treated with 3.
Figure 9.

γH2AX foci induced by 3 and 1 are DNA-PK dependent. Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with 1 μM 3 or 0.5 nM 1 in the presence or absence of 10 μM NU7441. Rows (top to bottom): γH2AX (green), 53BP1 (red), DNA (blue), and merge. Columns (left to right): control, 0.5 nM 1, 1 μM 3, 10 μM NU7441, 0.5 nM 1 + 10 μM NU7441, 1 μM 3 + 10 μM NU7441. Cells were exposed to NU7441 for 1 h prior to addition of 1 or 3; exposure was 1 h.
The experiments outlined above were repeated with the exception that immunofluorescence was conducted after incubating with 1 or 3 for 6 h. These experiments suggested that production of γH2AX foci is ATR dependent at 6 h but is ATM- and DNA-PK-independent, at this later time point (Figure S3).
1 and 3 Are Potent and Selective Inhibitors of BRCA2-and PTEN-Deficient Cell Lines
The data outlined above indicate that 1 and 3 activate NHEJ and HR repair. Accordingly, we hypothesized that 1 and 3 would be synthetic lethal4 with cell types that are deficient in these pathways. Toward this end, we evaluated the potency of 1 and 3 against cell line pairs that are proficient or deficient in specific DNA DSB repair factors but are otherwise isogenic, using a clonogenic survival assay.
These studies revealed that BRCA2-deficient V79 Chinese hamster lung fibroblast cell line derivative (VC8) and PTEN-deficient human glioblastoma (U251) cell lines are highly sensitized toward 1, with LC50 values of 1.5 ± 0.5 and 2.0 ± 0.6 pM, respectively (Figure 10). At 4 pM, 1 displayed 11.6-fold selectivity for the BRCA2 mutant, while at this same concentration, 1 completely inhibited the growth of the PTEN mutant colonies, with only 23% reduction in the growth of the PTEN-proficient colonies. In addition, 1 displayed selectivities toward other BRCA2-deficient cell lines (Peo1, DLD1) as well as KU80-, DNA-PK-, and ATM-deficient cell lines. MutL homologue 1 (MLH1)-, breast cancer type 1 susceptibility protein (BRCA1)-, fanconi anemia group D2 protein (FANCD2)-, and DNA repair protein complementing XP-A cells (XPA)-deficient cell lines were not sensitized toward 1. The activity of 3 against the same panel of tumors was also determined (Figure 11). As with 1, we observed that BRCA2-deficient VC8 cells and PTEN-deficient U251 cells are potently sensitized toward 3, with LC50 values of 6.0 ± 0.5 and 11 ± 4 nM and selectivities of 84 and 5.1, for the BRCA2 and PTEN mutants, respectively. KU80- and ATM-deficient cell lines, and other BRCA2-deficient (Peo1, DLD1) cell lines, were also sensitized toward 3. FANCD2-, MLH1-, and BRCA1-deficient HCC1937 cell lines were not sensitized toward 3, as was found with 1. Several differences in cell line selectivities were observed. For example, while the BRCA1-deficient UWB1.289 and XPA-deficient XP20S cell lines were not sensitized toward 1, these cell lines were moderately sensitized toward 3 (selectivity = 1.5, 1.5, respectively). In addition, while DNA-PK-deficient 50D cell lines were sensitized toward 1 (selectivity =1.2), these cell lines were not sensitized toward 3. Finally, the BRCA2-deficient EUFA423 cell line was more highly sensitized toward 3 than 1 (selectivities = 3.6, 1.3 respectively).
Figure 10.

Activity of 1 against pairs of cell lines that are proficient or deficient in specific DNA DSB repair factors but are otherwise isogenic. Cell viability was determined by a clonogenic survival assay. Sensitive cell lines are defined as those in which the (% survival repair-deficient cell line) < (% survival repair-proficient cell line) at 4 pM 1. Selectivity is defined as % survival repair-proficient cell line/% survival repair-deficient cell line at 4 pM 1. Complete data are presented in Table S1. The superscript “a” indicates not defined.
Figure 11.

Activity of 3 against pairs of cell lines that are proficient or deficient in specific DNA DSB repair factors but are otherwise isogenic. Cell viability was determined by a clonogenic survival assay. Sensitive cell lines are defined as those in which the (% survival repair-deficient cell line) < (% survival repair-proficient cell line) at 20 nM 3. Selectivity is defined as % survival repair-proficient cell line/% survival repair-deficient cell line at 20 nM 3. Complete data are presented in Table S2.
DISCUSSION
A recurring limitation of all DNA-damaging agents involves off-target effects arising from excessive damage to normal tissues. However, many cancers are driven by mutations in DNA repair factors,2 and these mutations can sensitize tumors to DNA-damaging agents.3 The goal of this study was to identify synthetic lethal interactions between the DNA cleavage agents 1 and 3 and mutations in specific DNA DSB repair factors. These data will guide the selection of tumor types for in vivo evaluation and translational development, which is ongoing in our laboratories. Toward this end we initially probed the kinetics of DNA DSB production by 1 and 3. While both compounds induce DSBs, the kinetics of DNA damage production are distinct. The levels of DSBs induced by 1 increase for approximately 4 h post addition (Figure 3), and this slow increase in DSB levels may be associated with the formation of secondary DSBs arising from cell cycle progression (e.g., S phase) or interference with other proteins associated with transcription and replication, such as topoisomerases. By comparison, DSBs are induced more rapidly by 3 and peak at approximately 15–30 min post addition (Figure 3). A >2000-fold higher concentration of 3 is required to induce a level of DSBs comparable to 0.5 nM 1. We hypothesize that DSBs induced by 3 arise from closely spaced SSBs, which allow separation of the helical structure. This is supported by our alkaline comet assay, which revealed extensive DNA damage in cells treated with 3 (Figure 2C), and prior mechanistic data, which indicated that both diazofluorenes of 1 are involved in DNA cleavage.7 The accumulation of SSBs may explain the higher rate of resolution of the DSBs in cells treated with 3, as each contributing SSB can be repaired by many different pathways.1,2,28
Using the kinetic data above, we employed two markers of DNA DSB repair, γH2AX19 and 53BP1,17 to probe the cellular response to 1 and 3. Our studies indicate that γH2AX production induced by 1 is ATM and DNA-PK dependent at early time points (Figures 7–9), which is consistent with activation of NHEJ and HR repair. Phosphorylation of H2AX is an early and essential step in NHEJ repair19 and is carried out redundantly by ATM18 and DNA-PK.20 Accumulation of γH2AX initiates the recruitment of DNA damage response proteins, such as 53BP1, to the site of DSBs and triggers checkpoint activation leading to cell cycle arrest (Figure 4).19e,29 At early time points, production of γH2AX in cells treated with 3 is also ATM- and DNA-PK dependent (Figures 7–9), suggesting activation of NHEJ and HR in cells treated with 3 immediately after exposure.
Interestingly, we observed that H2AX phosphorylation is ATR dependent and ATM independent at later time points (6 h) in cells treated with 1 or 3 (Figure S3). Thus, selective inhibition of ATM in cells treated with 1 or 3 with the ATM-inhibitor KU5593324 did not block H2AX phosphorylation at 6 h, while treatment with caffeine, which inhibits ATR (as well as ATM),25 decreased H2AX phosphorylation at 6 h. This is consistent with the finding that γH2AX foci formed at early times postexposure to IR is ATM- and DNA-PK dependent but ATR independent, while at later times post IR (6 h), γH2AX foci formation is ATR dependent but independent of ATM and DNA-PK.30 It has been reported that H2AX is phosphorylated in an ATR dependent, but ATM-independent, response to replication arrest, and that this γH2AX colocalizes with other repair proteins such as 53BP1 and BRCA1.21 We postulate that 1 and 3 form replication blocks that lead to ATR-dependent phosphorylation of H2AX at later time points. In addition, 3, but not 1, induces high levels of γH2AX as time progresses. This increase in γH2AX does not correlate with 53BP1 foci formation and appears to be related to induction of apoptosis, which is supported by Annexin V–A488/PI staining (Figure S2).
The data presented above conclusively point to the involvement of NHEJ and HR repair as important pathways to remediate DNA damage induced by 1 and 3. This suggested that 1 and 3 would be synthetic lethal with specific mutations in these pathways. To evaluate this hypothesis, we determined the activity of 1 and 3 against a panel of paired cell lines that are proficient or deficient in specific DNA DSB repair factors but are otherwise isogenic. We observed remarkably potent activity toward BRCA2-deficient V79 Chinese hamster lung fibroblast cell line derivative (VC8) and PTEN-deficient human glioblastoma (U251) cell lines, with LC50 values of 1.5 ± 0.5 and 2.0 ± 0.6 pM for 1 and LC50 values of 6.0 ± 0.5 and 11 ± 4 nM for 3, respectively. PTEN has recently been implicated in DNA repair,13 and BRCA2 is an important mediator of HR,12 and both BRCA212 and PTEN14 display high incidences of mutation across a wide range of cancers. The selectivities toward these mutant cell lines versus the isogenic cell lines transfected with and expressing functional BRCA2 and PTEN genes were also high, suggesting an acceptable therapeutic index might be attainable in vivo. We also discovered that ATM- and KU80-deficient cell lines are sensitized toward 1 and 3, albeit to a lesser degree, which is consistent with our mechanistic model and provides additional tumor types for preclinical evaluation.
CONCLUSION
This work establishes the kinetics and relative eficiencies of DNA damage induced by 1 and 3 and shows that 1 is a much more potent DNA cleavage agent than 3. This is consistent with an earlier model for DNA cleavage by 1, which invoked participation of both diazofluorene functional groups in its DNA-damaging effects and showed that the observed DSBs arise from a single binding event.7 Nonetheless, 3 cleaves DNA in tissue culture at higher concentrations, likely through the accumulation of unrelated SSBs, and this lower activity is mitigated to some extent by the ease of synthesis and optimization of 3. In addition, we have elucidated the cellular response to 1 and 3 and shown that the compounds activate NHEJ and HR repair. These latter data lay the foundation for evaluation of 1 and 3 as monotherapies against tumor types that are deficient in either pathway. Toward this end, we have identified synthetic lethal interactions between 1 and 3 mutations in PTEN or BRCA2. Collectively, these data reveal the cellular responses to 1 and 3 and will help to advance the translational development of this new family of chemotherapeutic agents.
Supplementary Material
Acknowledgments
Financial support from the National Institute of General Medical Sciences (R01GM090000, S.B.H.), the National Institute of Environmental Health Sciences (R01ES005775, P.M.G.), and the National Cancer Institute (R01CA168733, P.M.G.) is gratefully acknowledged.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Detailed experimental procedures, Figures S1–S3 and Tables S1–S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reed E. Clin Cancer Res. 2010;16:4511. doi: 10.1158/1078-0432.CCR-10-0528. [DOI] [PubMed] [Google Scholar]
- 2.Curtin NJ. Nat Rev Cancer. 2012;12:801. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 3.Hosoya N, Miyagawa K. Cancer Sci. 2014;105:370. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaelin WG., Jr Nat Rev Cancer. 2005;5:689. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 5.Hartman JL, Garvik B, Hartwell L. Science. 2001;291:1001. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- 6.Isolation of 1: He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362. doi: 10.1021/ja010129o.Elucidation of the relative and absolute stereochemistry of 1: Woo CM, Beizer NE, Janso JE, Herzon SB. J Am Chem Soc. 2012;134:15285. doi: 10.1021/ja3074984.
- 7.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. Nat Chem. 2014;6:504. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews of DNA DSBs and the mechanisms of repair, see: Brandsma I, van Gent D. Genome Integr. 2012;3:9. doi: 10.1186/2041-9414-3-9.Chowdhury D, Choi YE, Brault ME. Nat Rev Mol Cell Biol. 2013;14:181. doi: 10.1038/nrm3523.Aparicio T, Baer R, Gautier J. DNA Repair. 2014;19:169. doi: 10.1016/j.dnarep.2014.03.014.
- 9.For reviews of diazofluorene-containing natural products, see: Gould SJ. Chem Rev. 1997;97:2499. doi: 10.1021/cr9600215.Marco-Contelles J, Molina MT. Curr Org Chem. 2003;7:1433.Arya DP. Top Heterocycl Chem. 2006;2:129.Nawrat CC, Moody CJ. Nat Prod Rep. 2011;28:1426. doi: 10.1039/c1np00031d.Herzon SB. The Kinamycins. In: Li JJ, Corey EJ, editors. Total Synthesis of Natural Products. At the Frontiers of Organic Chemistry. Springer-Verlag; Berlin: 2012. Herzon SB, Woo CM. Nat Prod Rep. 2012;29:87. doi: 10.1039/c1np00052g.
- 10.Isolation of the kinamycins: Ito S, Matsuya T, Ōmura S, Otani M, Nakagawa A. J Antibiot. 1970;23:315. doi: 10.7164/antibiotics.23.315.Hata T, Ōmura S, Iwai Y, Nakagawa A, Otani M. J Antibiot. 1971;24:353. doi: 10.7164/antibiotics.24.353.Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Chem Pharm Bull. 1971;19:2428. doi: 10.1248/cpb.21.931.Furusaki A, Matsui M, Watanabe T, Ōmura S, Nakagawa A, Hata T. Isr J Chem. 1972;10:173.Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Chem Pharm Bull. 1973;21:931. doi: 10.1248/cpb.21.931.
- 11.Woo CM, Ranjan N, Arya DP, Herzon SB. Angew Chem, Int Ed. 2014;53:9325. doi: 10.1002/anie.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a review of BRCA2 in DNA repair, see: Roy R, Chun J, Powell SN. Nat Rev Cancer. 2012;12:68. doi: 10.1038/nrc3181.
- 13.Bassi C, Ho J, Srikumar T, Dowling RJO, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Science. 2013;341:395. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For a review of PTEN, see: Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. Trends Biochem Sci. 2014;39:183. doi: 10.1016/j.tibs.2014.02.006.
- 15.For a review of the comet assay, see: Collins A. Mol Biotechnol. 2004;26:249. doi: 10.1385/MB:26:3:249.
- 16.Olive PL, Banath JP. Nat Protoc. 2006;1:23. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 17.(a) Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. J Cell Biol. 2000;151:1381. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Anderson L, Henderson C, Adachi Y. Mol Cell Biol. 2001;21:1719. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rappold I, Iwabuchi K, Date T, Chen J. J Cell Biol. 2001;153:613. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Kastan MB, Lim DS. Nat Rev Mol Cell Biol. 2000;1:179. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]; (b) Bakkenist CJ, Kastan MB. Nature. 2003;421:499. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 19.(a) Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. J Biol Chem. 1998;273:5858. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]; (b) Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Biochem Cell Biol. 2003;81:123. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]; (c) Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. Nat Rev Cancer. 2008;8:957. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Muslimovic A, Ismail IH, Gao Y, Hammarsten O. Nat Protoc. 2008;3:1187. doi: 10.1038/nprot.2008.93. [DOI] [PubMed] [Google Scholar]; (e) Podhorecka M, Skladanowski A, Bozko PJ. Nucleic Acids. 2010;1 doi: 10.4061/2010/920161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. Cancer Res. 2004;64:2390. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 21.Ward IM, Chen J. J Biol Chem. 2001;276:47759. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 22.(a) Nunez MI, Villalobos M, Olea N, Valenzuela MT, Pedraza V, McMillan TJ, Ruiz de Almodovar JM. Br J Cancer. 1995;71:311. doi: 10.1038/bjc.1995.62. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DiBiase SJ, Zeng ZC, Chen R, Hyslop T, Curran WJ, Jr, Iliakis G. Cancer Res. 2000;60:1245. [PubMed] [Google Scholar]
- 23.Mukherjee B, Kessinger C, Kobayashi J, Chen BP, Chen DJ, Chatterjee A, Burma S. DNA Repair. 2006;5:575. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GCM. Cancer Res. 2004;64:9152. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 25.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Cancer Res. 1999;59:4375. [PubMed] [Google Scholar]
- 26.Leahy JJJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GCM. Bioorg Med Chem Lett. 2004;14:6083. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. Cancer Res. 2006;66:5354. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 28.Aziz K, Nowsheen S, Pantelias G, Iliakis G, Gorgoulis VG, Georgakilas AG. Pharmacol Ther. 2012;133:334. doi: 10.1016/j.pharmthera.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Mahaney BL, Meek K, Lees-Miller SP. Biochem J. 2009;417:639. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. Cell Cycle. 2010;9:662. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.