

Abstract
The inflammatory response at infliximab (IFX) treatment failure due to tumor necrosis factor (TNF)-α-independent Crohn disease activity is unknown.
This is an exploratory, hypothesis-generating study based on samples collected in a clinical trial among patients failing conventional IFX dosages and treated with an intensified IFX regimen for 12 weeks. Patients with clinical response at week 12, as defined by a reduction of Crohn disease activity index by ≥70, were considered to suffer from nonimmune pharmacokinetic (PK) treatment failure (n = 18), and nonresponders had a presumed pharmacodynamic (PD) failure due to non-TNF-driven disease (n = 8). Patients failing IFX due to functional anti-IFX antibodies (n = 2) were excluded. The study population also comprised a group of 12 patients in long-term remission on IFX. A functional cell-based reporter gene assay was applied to measure IFX and anti-IFX antibodies. Circulating cytokines and cytokine receptors were assessed by enzyme-linked immunosorbent assay: granulocyte-macrophage colony-stimulating factor, interferon-γ, interleukin (IL)-1α, IL-1β, IL-1Ra, IL-6, IL-10, IL-12p70, soluble TNF receptor (sTNF-R) 1, sTNF-R2, IL-17A, and monocyte chemotactic protein 1.
The IFX levels were similar between patients with IFX failure caused by nonimmune PK or PD at treatment failure (median 1.4 vs 2.4 μg/mL; P = 0.52), during treatment intensification (8.1 vs 5.6; P = 0.85), and after 12 weeks (8.8 vs 7.7; P = 0.93), congruent with nonresponders failing IFX due to predominantly TNF-α-independent signaling pathways in their disease. Cytokine and cytokine receptor levels were comparable between patients with nonimmune PK failure and PD failure at time of manifestation of IFX failure, but with higher IL-6 and sTNF-R2 levels among IFX treatment failures as compared with patients in remission (IL-6 median 3.6 vs <3.1 pg/mL; P = 0.03; sTNF-R2 3207 vs 2547 pg/mL; P = 0.01). IL-6 and sTNF-R2 were lower after 12 weeks in nonimmune PK failures than in PD failures (<3.1 vs 4.0; P = 0.02; 3209 vs 4740; P = 0.04, respectively), and were measured at levels comparable with patients in remission. Further, trends of decreased IL-6 and sTNF-R2 levels among nonimmune PK failures during IFX intensification (P < 0.05 and P = 0.12) were observed.
These observations indicate that IL-6 and sTNF-R2 are of potential relevance in driving the inflammatory response in IFX refractory Crohn disease caused by TNF-α-independent disease activity.
INTRODUCTION
Antitumor necrosis factor (TNF)-α therapy with infliximab (IFX) is effective for management of patients with Crohn disease refractory to conventional immunomodulating therapies.1 Regrettably, approximately half of the patients lose effect of IFX therapy over time.2 The underlying mechanisms for IFX treatment failure and their corresponding interventions can be identified by measurements of circulating levels of trough IFX and anti-IFX antibodies (Abs) according to the algorithm outlined in Figure 1.3 Treatment strategy according to this algorithm has become an integrated part of clinical practice because it results in comparable or superior clinical outcomes at reduced costs as compared with intensification of the IFX treatment regimen.4–11
FIGURE 1.
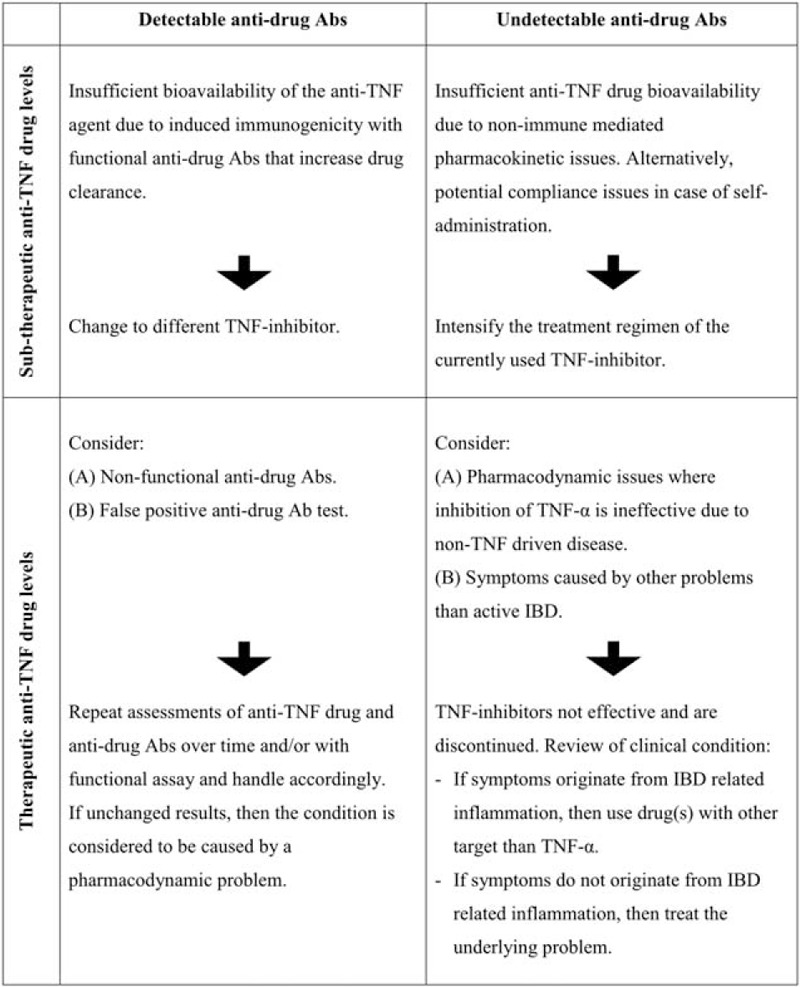
Personalized treatment strategy at anti-TNF treatment failure. Algorithm based on measurements of anti-TNF drug levels and antidrug antibodies (Abs) for identification of underlying mechanisms for anti-TNF treatment failure with, for example, infliximab (IFX) and corresponding interventions. Initially devised by our group,3 and later tested in clinical trials and observational studies.4,5,7–10 Adapted from the studies by Bendtzen et al3 and Steenholdt et al.7 Reproduced and modified with permission from BMJ Publishing Group Ltd from the study by Steenholdt et al. TNF = tumor necrosis factor.
Treatment failure caused by a pharmacokinetic (PK) problem is characterized by insufficient IFX bioavailability to adequately suppress TNF-α-mediated inflammatory disease activity.3,12 This can originate as a consequence of immunogenicity with formation of functional anti-IFX Abs that neutralize IFX and/or increase drug clearance (Figure 1: upper left row)13; alternatively, because of nonimmune-mediated mechanisms caused for example by a heavy inflammatory load leading to high IFX turnover (Figure 1: upper right row).14–16 Treatment failure caused by a PK problem should preferably restore sufficient inhibition of TNF-α. This is characteristically done by intensification of the IFX regimen if anti-IFX Abs are absent, or by switching to a different TNF-inhibitor if anti-IFX Abs are present.3–10,12
During IFX therapy, a notable proportion of patients experiences relapse of their Crohn disease because of a pharmacodynamic (PD) problem (Figure 1: lower rows).7,10 This condition originates from predominantly or exclusively non-TNF-driven inflammatory disease pathways—either primarily or as a result of redundancy with a dynamic shift during ongoing IFX therapy.3,12,17,18 It is characterized by relatively high circulating levels of IFX at time of manifestation of failure, and lack of effect of IFX dose intensification.4,7,9,10,19,20 Continued anti-TNF therapy is inefficient in such cases.3,4,7–10,12,20 Identification of mediators maintaining active TNF-α-independent disease may uncover rational treatment targets for future biologic agents, but the inflammatory response in these patients is yet unaccounted for.14,21–27
METHODS
Objectives
The aim of this study was to explore selected characteristics of the systemic inflammatory response in patients with Crohn disease failing IFX due to a PD or a nonimmune PK problem as compared with patients in remission on IFX. Furthermore, to compare characteristics in patients’ refractory to intensified IFX due to PD treatment failure with those of patients failing IFX due to nonimmune PK.
Study Design and Patients
This was a hypothesis-generating and explorative study which was based on data and samples obtained as part of a randomized controlled trial in which Crohn's disease patients with loss of response of IFX maintenance therapy (Crohn Disease Activity Index [CDAI] ≥220 or minimum one draining perianal fistula) had been randomized to an intensified IFX regimen or to personalized therapy based on IFX and anti-IFX Abs levels at time of treatment failure as outlined in Figure 1 and detailed in references.7,8 The current study population comprised patients who had received an intensified IFX regimen throughout the entire 12-week study period (5 mg/kg every [q] 4 weeks, n = 22; q >4, n = 4), and with blood samples available both at baseline corresponding time of treatment failure (week 0) and also at end of trial (week 12) (Figure 2). Patients with treatment failure caused by fistulizing Crohn disease only were excluded along with patients who had been dose-intensified on IFX in the presence of functionally active anti-IFX Abs (Figure 1: upper left row). Clinical response after 12 weeks of intensified IFX therapy was defined by ≥70 point reduction of CDAI from baseline. Patients without response were considered to suffer from IFX failure characterized by non-TNF-α-driven disease pathways, that is, PD failure (Figure 1: lower rows). Responders were considered to suffer from IFX failure due to a nonimmune-mediated PK problem characterized by a TNF-α-driven disease activity (Figure 1: upper right row).
FIGURE 2.
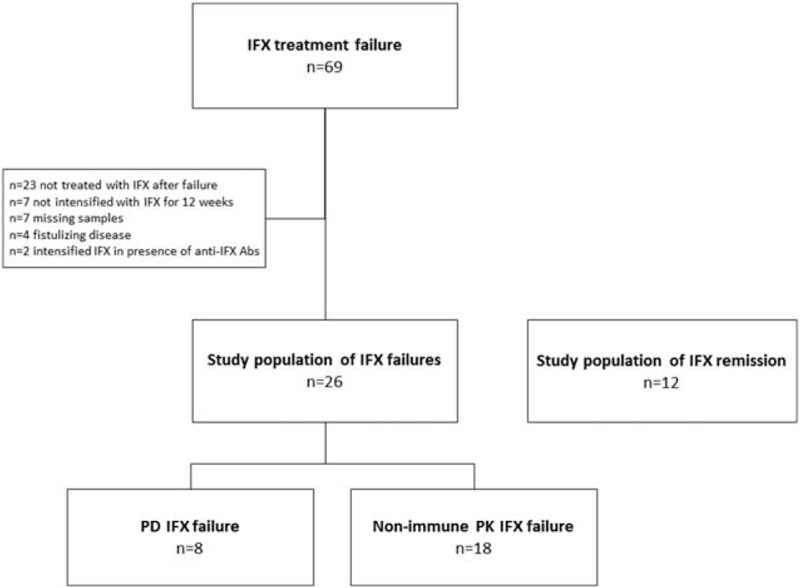
Study flow. The study population consisted of Crohn disease patients with infliximab (IFX) treatment failure due to a pharmacodynamic (PD) problem or a nonimmune pharmacokinetic (PK) problem. In addition, a cohort of patients in remission on IFX was included.
The study population also included a cohort of patients with luminal Crohn disease (n = 12) treated with IFX for minimum 1 year, and in clinical remission (CDAI <150), biochemical remission (normal values of CRP, white blood cells, hemoglobin, and albumin), endoscopic remission (Simple Endoscopic Score for Crohn disease [SES-CD] ≤1), and deep remission (histologically inactive disease) (Figure 2).
The study was approved by the Danish Medicines Agency (EudraCT 2009–009926–94; 2012–002702–51), the Danish regional ethics committees (HA-2009–009; H-4–2012–099), and the Danish Data Protection Agency (2007–58–0015; 750.89–27), and was registered at ClinicalTrials.gov (NCT00851565; NCT01817426). Informed oral and written consent was obtained from all patients.
Blood Samples
Blood samples were collected as trough levels. Serum and plasma were collected after centrifugation of 10 mL venous blood (5 minutes at 3500 rpm), and stored at −80°C. All analyses were performed under blinded conditions.
Analyses of IFX and Anti-IFX Abs Concentrations
Serum samples were analyzed for concentrations of IFX and anti-IFX Abs by a functional cell-based reporter gene assay (RGA) (Eurodiagnostica, Malmö, Sweden).
Cytokine Assessments
Plasma levels of interleukin (IL)-6 were measured by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN) (lower limit of quantification [LLOQ] 3.1 pg/mL). Plasma levels of the following cytokines were examined by Bio-Plex Pro Human Inflammation multiplex ELISA: IFN (interferon)-γ (LLOQ 6.3 pg/mL), IL-10 (1.7 pg/mL), IL-12 p70 (1.3 pg/mL), soluble TNF receptor 1 (sTNF-R1) (26.8 pg/mL), and sTNF-R2 (30.3 pg/mL), and by Bio-Pex Pro Human group I multiplex ELISA: granulocyte-macrophage colony-stimulating factor (GM-CSF) (63.3 pg/mL), IL-1α (1.4 pg/mL), IL-1β (3.2 pg/mL), IL-1 receptor antagonist (Ra) (81.1 pg/mL), IL-17A (4.9 pg/mL), and monocyte chemotactic protein 1 (MCP-1) (2.1 pg/mL). Cytokines assessed in the study were selected based on of their putative involvement in the processes underlying inflammatory bowel disease (IBD).14,21–28 All measurements were performed in duplicate and with coefficient of variations <20%. Concentrations were given in pg/mL and with 1 decimal in case of a very high sensitivity (LLOQ <10 pg/mL).
Statistics
Descriptive statistics were given as percentages for discrete variables, and as median with interquartile range (IQR) for continuous variables. Comparisons of patient characteristics were done by Fisher exact test or chi-square test (discrete variables), or by Mann–Whitney U test (continuous variables). Comparisons of systemic cytokine expression in patients with IFX treatment failure and in remission on IFX were done by Mann–Whitney U test. Comparisons of cytokine levels, and IFX levels, in patients with PD or nonimmune PK failure at baseline and week 12 were done by Mann–Whitney U test, and changes across time in each subgroup were evaluated by Wilcoxon signed-rank test. Values below LLOQ were considered to be null. Statistical analyses were done in SPSS version 22 (IBM, Somer, NY). Two-sided P values <0.05 were considered statistically significant.
RESULTS
Study Population
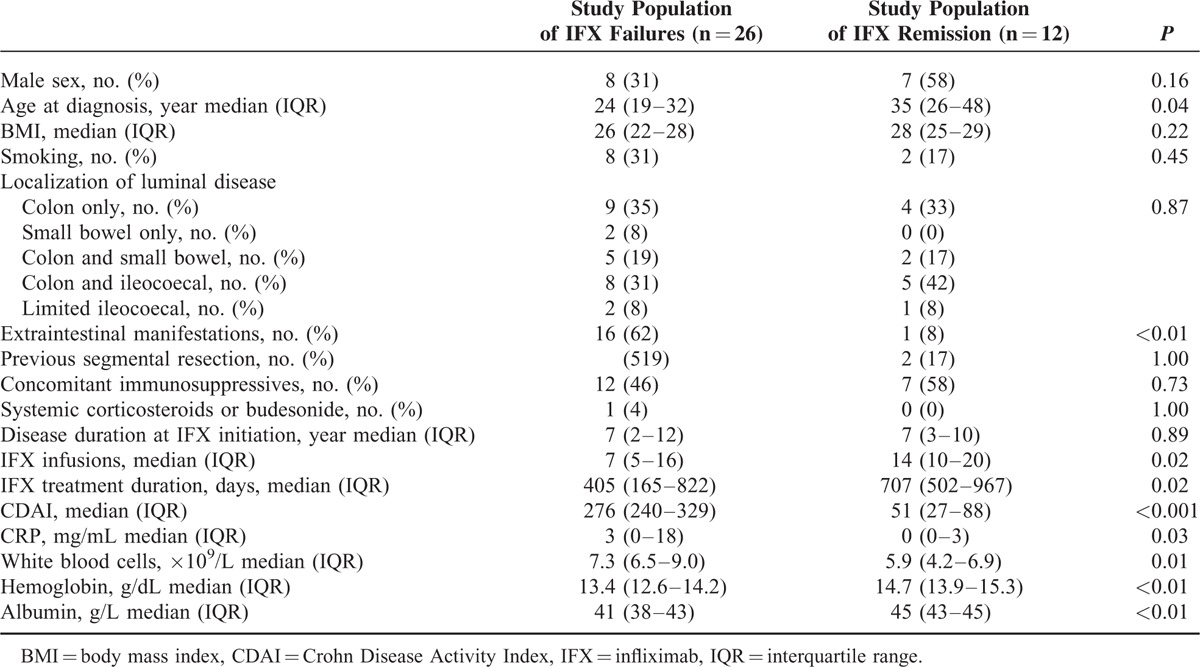
The study population comprised patients in remission on IFX, and also patients with IFX treatment failure due to presumed PD or nonimmune PK issues (Figure 2). As shown in Table 1, characteristics of included patients reflected the different clinical response types to IFX therapy.
TABLE 1.
Patient Characteristics
Cytokine and Cytokine Receptor Levels at IFX Treatment Failure
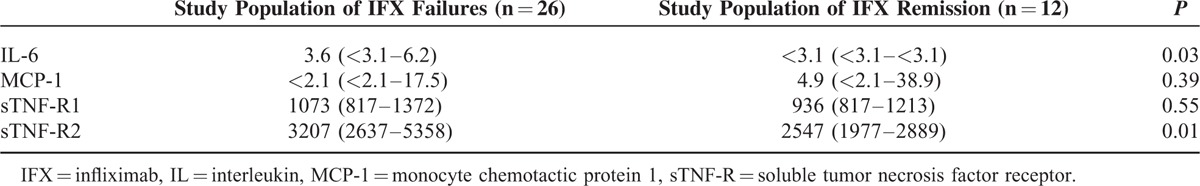
As detailed in Table 2, circulating levels of IL-6 and sTNF-R2 were significantly higher at the time of IFX treatment failure as compared with levels in patients on IFX with quiescent disease. Levels of sTNF-R1 and MCP-1 were not significantly different between patients with IFX failure and those with an IFX-induced remission. The remaining cytokines and cytokine receptors assessed were generally below LLOQ (not shown).
TABLE 2.
Systemic Inflammatory Response at IFX Treatment Failure
Inflammatory Characteristics at PD and Nonimmune PK IFX Treatment Failure
Characteristics of patients with IFX treatment failure due to PD or nonimmune PK are shown in Table 3. Circulating anti-TNF activities in these subgroups at treatment failure were median 1.4 versus 2.4 μg/mL (P = 0.52); after 12 weeks of intensified IFX regimen 8.8 versus 7.7 (P = 0.93); and the increase in anti-TNF activity during the 12-week period of treatment intensification was 8.1 versus 5.6 (P = 0.85) (Figure 3).
TABLE 3.
Characteristics of the Study Population According to Mechanism for IFX Failure
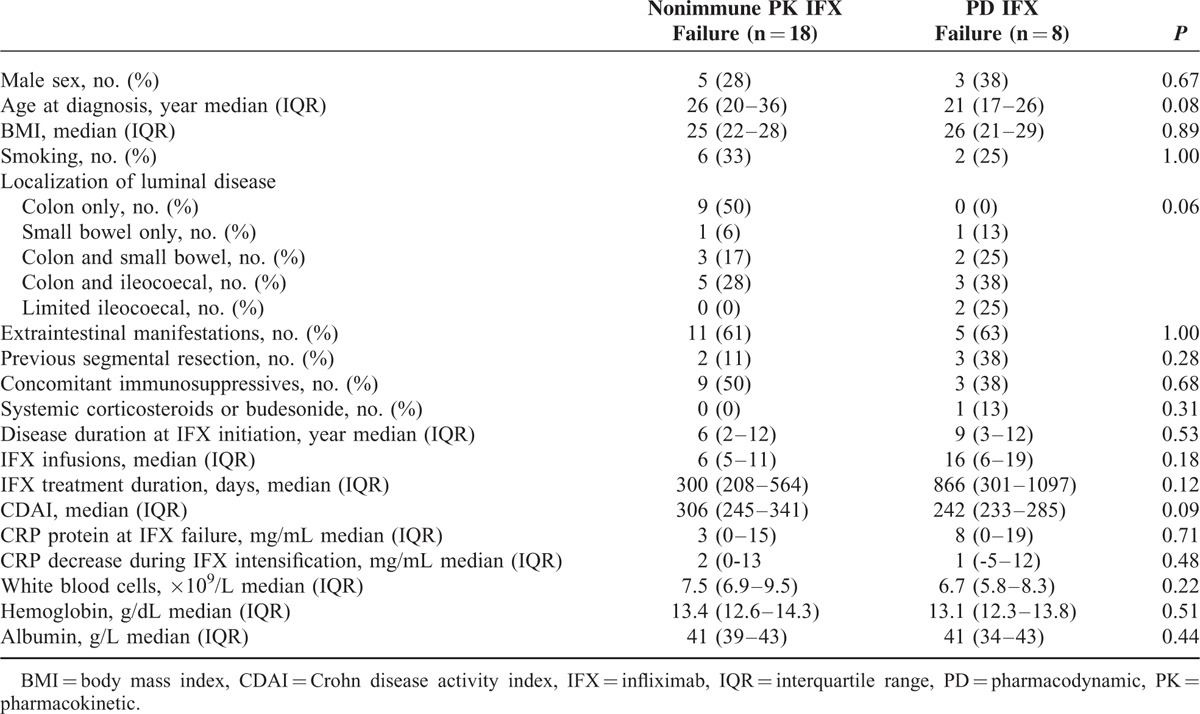
FIGURE 3.
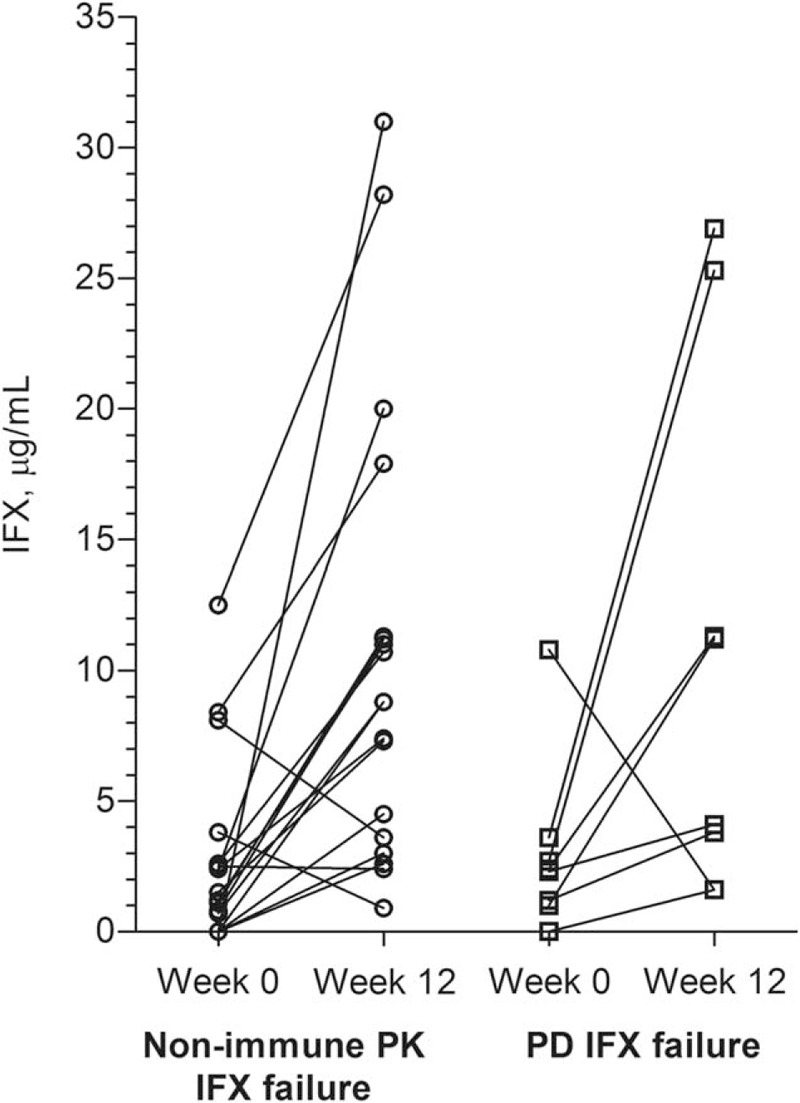
Infliximab levels. Circulating infliximab (IFX) trough levels measured by functional reporter gene assay (RGA) in patients with IFX treatment failure due to nonimmune pharmacokinetics (PK) (n = 18) or pharmacodynamics (PD) (n = 8) at time of treatment failure (week 0) and after 12 weeks of intensified IFX regimen.
As shown in Table 4, cytokine levels did not differ significantly between patients with IFX treatment failure due to PD or nonimmune PK at time of manifestation of IFX treatment failure. However, IL-6 levels decreased significantly during intensification of IFX treatment in individual patients with nonimmune PK failure, but not in those with PD failure (Table 4 and Figure 4). Furthermore, IL-6 and sTNF-R2 levels were significantly lower after 12 weeks of treatment in patients with nonimmune PK failure (Table 4), and at levels comparable with those in patients in remission on IFX (IL-6: <3.1 vs <3.1 pg/mL; P = 0.85; sTNF-R2: 3209 vs 2547 pg/mL; P = 0.19) (Table 2).
TABLE 4.
Systemic Inflammatory Response According to Mechanism for IFX Failure
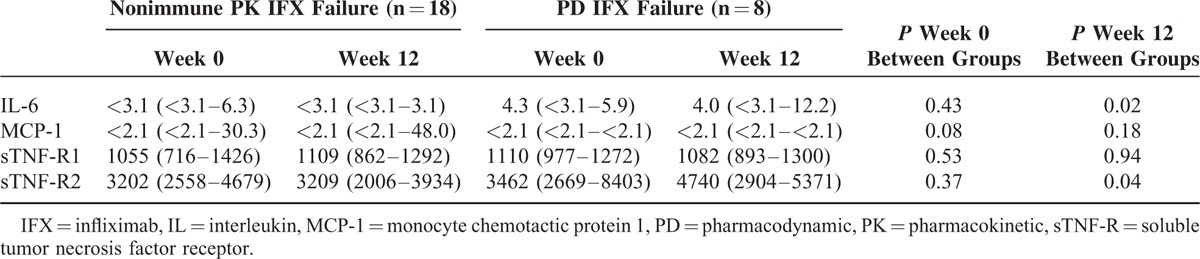
FIGURE 4.
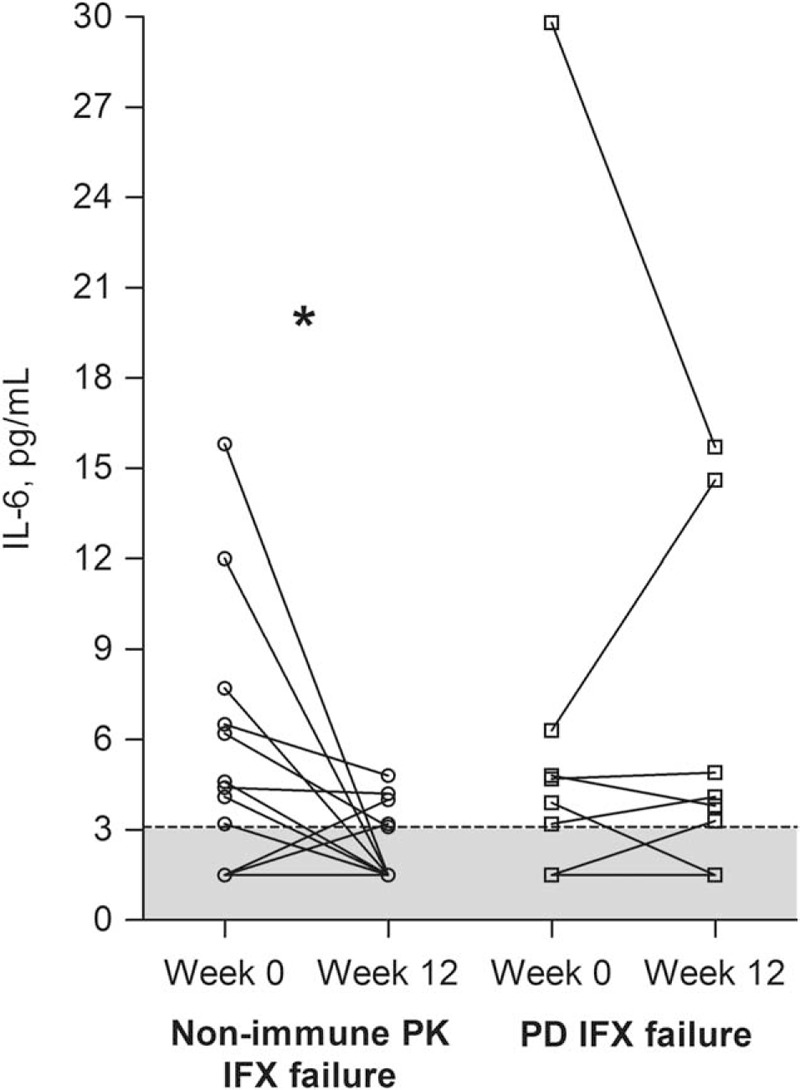
Interleukin (IL)-6 levels. Circulating IL-6 levels in patients with infliximab (IFX) treatment failure due to nonimmune pharmacokinetics (PK) (n = 18) or pharmacodynamics (PD) (n = 8) at treatment failure (week 0) and after 12 weeks of intensified IFX regimen. (∗) P < 0.05 by Wilcoxon signed-rank test. Lower limit of IL-6 quantification (LLOQ) was 3.1 pg/mL (dotted line), and values below LLOQ are arbitrarily set to 1.5 pg/mL and shown as gray (n = 9 and n = 2, respectively).
Assessment of conditions for IL-6 in patients with detectable IL-6 at baseline only revealed comparable findings to the above: baseline IL-6 levels were not significantly different between nonimmune PK or PD IFX failures (median 6.2 vs 4.8 pg/mL; P = 0.72), IL-6 decreased significantly solely in individual patients with nonimmune PK failure (P < 0.01 and P = 0.69, respectively), and IL-6 levels were significantly lower after 12 weeks in patients with nonimmune PK failure (<3.1 vs 4.5 pg/mL; P < 0.05).
DISCUSSION
It is known that PK or PD mechanisms are involved in loss of response to anti-TNF therapy as outlined in Figure 1.3,7,8,12,20 Although previous studies have focused mainly on PK reasons for anti-TNF treatment failure due to immunogenicity or nonimmune-mediated reasons for insufficient drug bioavailability, this study, however, explored characteristics of the systemic inflammatory response in patients with IFX treatment failure presumably originating from an underlying PD problem in which inhibition of TNF-α per se is ineffective.13,14 The main findings of the present investigation are that a subgroup of Crohn disease patients refractory to an intensified IFX regimen—presumably due to PD-related treatment failure—exhibited a maintained detectable systemic expression of IL-6 during the course of IFX intensification as opposed to patients with nonimmune PK-related treatment failure, or patients in remission on IFX. Furthermore, sTNF-R2 levels were found to be higher among patients with IFX failure as compared with those with clinically quiescent disease, and generally higher at PD-related treatment failure than at nonimmune PK failure. Taken together, these observations indicate that patients with PD-related IFX treatment failure exhibit a predominantly non-TNF-α-mediated inflammatory disease phenotype that may involve IL-6 and/or sTNF-R2.
Transcriptional data have suggested that anti-TNF therapy associates with a diminished mucosal IL-6 expression among primary anti-TNF nonresponsive Crohn disease patients.26 On the contrary, as IL-6 was elevated before IFX intensification, IL-6 may simply be a confounder due to unspecific inflammatory activity.29 However, maintained expression of increased circulating IL-6 in patients with ulcerative colitis and primary IFX treatment failure supports involvement of IL-6 in TNF-α-independent inflammation.27 Several studies have earlier proposed IL-17A to be a central mediator of anti-TNF-α-independent inflammation.24–26,28,30,31 Although IL-17A was below LLOQ in the current study, involvement of IL-6, amongst other cytokines, in the induction of a Th17 inflammatory response might indicate that IL-6 plays a role in predominantly TNF-α-independent inflammation among patients with a presumed PD-related treatment failure. Hence, even though our data do not provide direct evidence, they do, however, support the concept that IL-6, the IL-6 receptor, or IL-6 intracellular signaling might prove useful as therapeutic targets in the subgroup of patients with IFX treatment failure characterized by an underlying PD mechanism. Thus, clinical trials are obviously needed to assess this hypothesis. Interestingly, a monoclonal Ab targeting the IL-6 receptor was found effective for treatment of Crohn disease in a pilot study.32–34 Furthermore, a recently published phase II trial only available in abstract form reported significantly higher clinical response rates among Crohn disease patients with previous anti-TNF treatment failure and treated with anti-IL-6 monoclonal Ab (PF-0423921) than with placebo.35
Our findings concur with observations that outcome of IFX therapy is associated with variations in the gene encoding TNF-R2, and with reported characteristics of the mucosal gene expression profile of TNF-R2 during ongoing IFX therapy.23,36–38 Although it cannot be ruled out that maintained elevated sTNF-R2 level observed in patients with PD-related IFX treatment failure was a result of diminished TNF-α expression imposed by IFX intensification followed by a relative increase in non-TNF-bound sTNF-R2, the corresponding constant level of sTNF-R1 in both nonimmune PK and PD treatment failures renders this unlikely. In contrast to the conditions in Crohn disease, systemic sTNF-R2 levels are lower in patients with active than inactive rheumatoid arthritis.39 Thus, it is possible that the opposing trends in sTNF-R2 levels in active versus quiescent Crohn disease and rheumatoid arthritis, respectively, may contribute to observed differences in efficacy of the sTNF-R2/human IgG construct, etanercept, in these diseases.40,41
Several notable limitations of this study, however, need to be attended. The sample size was restricted as relevant samples were available only in some patients enrolled in an earlier clinical trial.7,8 Further, the assessed cytokines and cytokine receptors were selected based on their putative involvement in IBD and did not cover all potentially involved inflammatory mediators. Circulating IL-6 levels were only marginally higher than LLOQ, and the inflammatory response was assessed in the circulation where conditions may diverge from those of the inflamed gut. As this was an exploratory study, correction for multiple comparisons was not performed. Taken together, our new observations described based on available knowledge of IFX response in IBD are hypothesis-generating and need to be validated in independent and larger cohorts.
It is currently impossible to discriminate between TNF-α and non-TNF-α-driven pathways. It has been assumed that patients refractory to IFX intensification, and without functional anti-IFX Abs, had an underlying PD-related problem caused by a non-TNF-α-driven disease, whereas responders had treatment failure due to nonimmune PK problems because of TNF-α-mediated disease activity.3,12,19 Although this was supported by a similar anti-TNF activity in responders and nonresponders, and a trend of more pronounced CRP decrease among responders than nonresponders, these patients with IBD might have experienced treatment failure due to other reasons as well.20 This is rather crucial because lack of routine endoscopy to verify inflammatory active disease at treatment failure introduces a potential bias, that some patients may have entered the trial without having genuine inflammatory loss of response, but rather symptoms mimicking IFX treatment failure.42 Nevertheless, another research group has used a similar approach.26 Furthermore, exploratory analyses with exclusion of the minority with undetectable IL-6 at time of manifestation of treatment failure did not change our findings, but they actually revealed more significant results. Assessment of IFX and anti-IFX Abs by a high-sensitivity homogeneous mobility shift binding assay revealed results coherent to those obtained by the functional RGA primarily used here adding further support to our findings.43 Finally, patients with functional anti-IFX Abs present in the circulation at treatment failure were excluded because the contribution of TNF-α in the inflammatory response could not be defined based on the outcome of intensified IFX treatments. Thus, anti-IFX Abs might a priory bias this outcome. Although this is also a potential bias, only 2 patients had anti-IFX Abs, and an explorative inclusion of these patients did not impose changes to our findings.
In conclusion, the present study indicates systemic alterations of IL-6 and sTNF-R2 in Crohn disease refractory to IFX caused by a TNF-α-independent disease pathway. If reproduced and validated, these findings may lead to potential targets of importance for the subgroup of patients with a PD IFX treatment failure.
Acknowledgments
We thank the following people for their assistance: Yvonne Krogager, Charlotte Kühnel, Lene Neergaard, Lise Olsen, (Dept. of Gastroenterology, Herlev University Hospital, DK); Lars K Munck and Sussi Holbæk (Dept. of Medical Gastroenterology, Køge University Hospital, DK); Lisbet A Christensen, Rikke Charlotte Andersen, Lisbet Gerdes, Catriona Nairn Marcussen, Birgitte Sperling Wilms Nielsen, and Inger Schjødt (Dept. of Hepatology and Gastroenterology V, Aarhus University Hospital, DK), Gitte Pedersen and Carina Blixt (Dept. of Gastroenterology, Hvidovre University Hospital, DK); Jens Kjeldsen and Anne Berg (Dept. of Medical Gastroenterology S, Odense University Hospital, DK); Jan Fallingborg, Bent Jacobsen, and Tove Nygaard (Dept. of Medical Gastroenterology, Aalborg University Hospital, DK). We thank the Danish Colitis Crohn Foundation for financial support.
Footnotes
Abbreviations: Abs = antibodies, CDAI = Crohn Disease Activity Index, ELISA = enzyme-linked immunosorbent assay, GM-CSF = granulocyte-macrophage colony-stimulating factor, IFN = interferon, IFX = infliximab, IL = interleukin, IQR = interquartile range, LLOQ = lower limit of quantification, MCP-1 = monocyte chemotactic protein 1, PD = pharmacodynamic, PK = pharmacokinetic, q = frequency of IFX dosing in weeks, ra = receptor antagonist, RGA = reporter gene assay, SES-CD = Simple Endoscopic Score for Crohn disease, sTNF-R = soluble TNF receptor, TNF = tumor necrosis factor.
This study has been awarded poster of distinction at the DDW 2015, Washington, D.C.
Authors’ contributions: Study design—CS, MC, KB, OHN; collection of data—CS, MC, SB, JB, MAA; analysis of data—CS; interpretation of data—all authors; drafting the manuscript—CS; revising the manuscript and approval of final manuscript—all authors.
Disclosures: Within the past year, CS has served as speaker for Abbvie and Takeda Pharmaceutical Company Ltd, and consultant for Pfizer and Takeda Pharmaceutical Company Ltd. KB has served as a speaker for Pfizer and Hospira, and owns stocks in Novo-Nordisk and Eurodiagnostica. JB has served as advisory board member for Abbvie and Takeda Pharmaceuticals, and primary investigator for Abbvie, and Janssen. MC, SB, MAA, and OHN have no interests to declare.
REFERENCES
- 1.Ford AC, Sandborn WJ, Khan KJ, et al. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol 2011; 106:644–659. [DOI] [PubMed] [Google Scholar]
- 2.Gisbert JP, Panes J. Loss of response and requirement of infliximab dose intensification in Crohn's disease: a review. Am J Gastroenterol 2009; 104:760–767. [DOI] [PubMed] [Google Scholar]
- 3.Bendtzen K, Ainsworth M, Steenholdt C, et al. Individual medicine in inflammatory bowel disease: monitoring bioavailability, pharmacokinetics and immunogenicity of anti-tumour necrosis factor-alpha antibodies. Scand J Gastroenterol 2009; 44:774–781. [DOI] [PubMed] [Google Scholar]
- 4.Afif W, Loftus EV, Jr, Faubion WA, et al. Clinical utility of measuring infliximab and human anti-chimeric antibody concentrations in patients with inflammatory bowel disease. Am J Gastroenterol 2010; 105:1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velayos FS, Kahn JG, Sandborn WJ, et al. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn's disease who lose responsiveness to infliximab. Clin Gastroenterol Hepatol 2013; 11:654–666. [DOI] [PubMed] [Google Scholar]
- 6.Vande Casteele N, Gils A, Singh S, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol 2013; 108:962–971. [DOI] [PubMed] [Google Scholar]
- 7.Steenholdt C, Brynskov J, Thomsen OO, et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn's disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut 2014; 63:919–927. [DOI] [PubMed] [Google Scholar]
- 8.Steenholdt C, Brynskov J, Thomsen OO, et al. Individualized therapy is a long-term cost-effective method compared to dose intensification in Crohn's disease patients failing infliximab. Dig Dis Sci 2015; 60:2762–2770. [DOI] [PubMed] [Google Scholar]
- 9.Roblin X, Rinaudo M, Del TE, et al. Development of an algorithm incorporating pharmacokinetics of adalimumab in inflammatory bowel diseases. Am J Gastroenterol 2014; 109:1250–1256. [DOI] [PubMed] [Google Scholar]
- 10.Yanai H, lichtenstein L, Assa A, et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin Gastroenterol Hepatol 2015; 13:522–530. [DOI] [PubMed] [Google Scholar]
- 11.Steenholdt C, Brynskov J, Thomsen OO, et al. Implications of infliximab treatment failure and influence of personalized treatment on patient-reported health-related quality of life and productivity outcomes in Crohn's disease. J Crohns Colitis 2015; 9:1032–1042. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Horin S, Chowers Y. Tailoring anti-TNF therapy in IBD: drug levels and disease activity. Nat Rev Gastroenterol Hepatol 2014; 11:243–255. [DOI] [PubMed] [Google Scholar]
- 13.Nanda KS, Cheifetz AS, Moss AC. Impact of antibodies to infliximab on clinical outcomes and serum infliximab levels in patients with inflammatory bowel disease (IBD): a meta-analysis. Am J Gastroenterol 2013; 108:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yarur AJ, Jain A, Sussman DA, et al. The association of tissue anti-TNF drug levels with serological and endoscopic disease activity in inflammatory bowel disease: the ATLAS study. Gut 2016; 65:249–255. [DOI] [PubMed] [Google Scholar]
- 15.Biancheri P, Brezski RJ, Di SA, et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology 2015; 149:1564–1574. [DOI] [PubMed] [Google Scholar]
- 16.Brandse JF, van den Brink GR, Wildenberg ME, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology 2015; 149:350–355. [DOI] [PubMed] [Google Scholar]
- 17.Yarur AJ, Rubin DT. Therapeutic drug monitoring of anti-tumor necrosis factor agents in patients with inflammatory bowel diseases. Inflamm Bowel Dis 2015; 21:1709–1718. [DOI] [PubMed] [Google Scholar]
- 18.Slevin SM, Egan LJ. New insights into the mechanisms of action of anti-tumor necrosis factor-alpha monoclonal antibodies in inflammatory bowel disease. Inflamm Bowel Dis 2015; 21:2909–2920. [DOI] [PubMed] [Google Scholar]
- 19.Steenholdt C, Bendtzen K, Brynskov J, et al. Cut-off levels and diagnostic accuracy of infliximab trough levels and anti-infliximab antibodies in Crohn's disease. Scand J Gastroenterol 2011; 46:310–318. [DOI] [PubMed] [Google Scholar]
- 20.Steenholdt C, Bendtzen K, Brynskov J, et al. Changes in serum trough levels of infliximab during treatment intensification but not in anti-infliximab antibody detection are associated with clinical outcomes after therapeutic failure in Crohn's disease. J Crohns Colitis 2015; 9:238–245. [DOI] [PubMed] [Google Scholar]
- 21.Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009; 58:1612–1619. [DOI] [PubMed] [Google Scholar]
- 22.Olsen T, Goll R, Cui G, et al. TNF-alpha gene expression in colorectal mucosa as a predictor of remission after induction therapy with infliximab in ulcerative colitis. Cytokine 2009; 46:222–227. [DOI] [PubMed] [Google Scholar]
- 23.Arijs I, Quintens R, Van LL, et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn's disease. Inflamm Bowel Dis 2010; 16:2090–2098. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa K, Matsumoto T, Esaki M, et al. Profiles of circulating cytokines in patients with Crohn's disease under maintenance therapy with infliximab. J Crohns Colitis 2012; 6:529–535. [DOI] [PubMed] [Google Scholar]
- 25.Rismo R, Olsen T, Cui G, et al. Mucosal cytokine gene expression profiles as biomarkers of response to infliximab in ulcerative colitis. Scand J Gastroenterol 2012; 47:538–547. [DOI] [PubMed] [Google Scholar]
- 26.Leal RF, Planell N, Kajekar R, et al. Identification of inflammatory mediators in patients with Crohn's disease unresponsive to anti-TNFalpha therapy. Gut 2015; 64:233–242. [DOI] [PubMed] [Google Scholar]
- 27.Dahlen R, Magnusson MK, Bajor A, et al. Global mucosal and serum cytokine profile in patients with ulcerative colitis undergoing anti-TNF therapy. Scand J Gastroenterol 2015; 1–9. [DOI] [PubMed] [Google Scholar]
- 28.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011; 140:1756–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nancey S, Hamzaoui N, Moussata D, et al. Serum interleukin-6, soluble interleukin-6 receptor and Crohn's disease activity. Dig Dis Sci 2008; 53:242–247. [DOI] [PubMed] [Google Scholar]
- 30.Mudter J, Neurath MF. Il-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm Bowel Dis 2007; 13:1016–1023. [DOI] [PubMed] [Google Scholar]
- 31.Toedter G, Li K, Marano C, et al. Gene expression profiling and response signatures associated with differential responses to infliximab treatment in ulcerative colitis. Am J Gastroenterol 2011; 106:1272–1280. [DOI] [PubMed] [Google Scholar]
- 32.Ito H, Takazoe M, Fukuda Y, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 2004; 126:989–996. [DOI] [PubMed] [Google Scholar]
- 33.Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther 2014; 141:125–139. [DOI] [PubMed] [Google Scholar]
- 34.Allocca M, Jovani M, Fiorino G, et al. Anti-IL-6 treatment for inflammatory bowel diseases: next cytokine, next target. Curr Drug Targets 2013; 14:1508–1521. [DOI] [PubMed] [Google Scholar]
- 35.Danese S, Vermeire S, Hellstern P, et al. Results of ANDANTE, a randomised clinical study with an anti-IL-6 antibody (PF-04236921) in subjects with Crohn's disease who are anti-tumour necrosis factor inadequate responders. J Crohn Colitis 2016; O015.[Epub ahead of print]. [Google Scholar]
- 36.Sveaas SH, Berg IJ, Provan SA, et al. Circulating levels of inflammatory cytokines and cytokine receptors in patients with ankylosing spondylitis: a cross-sectional comparative study. Scand J Rheumatol 2015; 44:118–124. [DOI] [PubMed] [Google Scholar]
- 37.Matsukura H, Ikeda S, Yoshimura N, et al. Genetic polymorphisms of tumour necrosis factor receptor superfamily 1A and 1B affect responses to infliximab in Japanese patients with Crohn's disease. Aliment Pharmacol Ther 2008; 27:765–770. [DOI] [PubMed] [Google Scholar]
- 38.Steenholdt C, Enevold C, Ainsworth MA, et al. Genetic polymorphisms of tumour necrosis factor receptor superfamily 1b and fas ligand are associated with clinical efficacy and/or acute severe infusion reactions to infliximab in Crohn's disease. Aliment Pharmacol Ther 2012; 36:650–659. [DOI] [PubMed] [Google Scholar]
- 39.Sivalingam SP, Thumboo J, Vasoo S, et al. In vivo pro- and anti-inflammatory cytokines in normal and patients with rheumatoid arthritis. Ann Acad Med Singapore 2007; 36:96–99. [PubMed] [Google Scholar]
- 40.Akobeng AK, Zachos M. Tumor necrosis factor-alpha antibody for induction of remission in Crohn's disease. Cochrane Database Syst Rev 2004; CD003574.DOI:10.1002/14651858.CD003574.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh JA, Christensen R, Wells GA, et al. Biologics for rheumatoid arthritis: an overview of Cochrane reviews. Cochrane Database Syst Rev 2009; CD007848.DOI:10.1002/14651858.CD007848.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steenholdt C, Ainsworth MA. Authors’ response: importance of defining loss of response before therapeutic drug monitoring. Gut 2015; 64:1340–1341. [DOI] [PubMed] [Google Scholar]
- 43.Wang SL, Ohrmund L, Hauenstein S, et al. Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods 2012; 382:177–188. [DOI] [PubMed] [Google Scholar]