

Abstract
MSI is a molecular imaging technique that allows for the generation of topographic 2D maps for various endogenous and some exogenous molecules without prior specification of the molecule. In this paper, we start with the premise that a region of interest (ROI) is given to us based on preselected morphological criteria. Given an ROI, we develop a pipeline, first to determine mass values with distinct expression signatures, localized to the ROI, and second to identify the peptides corresponding to these mass values. To identify spatially differentiated masses, we implement a statistic that allows us to estimate, for each spectral peak, the probability that it is over- or under-expressed within the ROI versus outside. To identify peptides corresponding to these masses, we apply LC-MS/MS to fragment endogenous (nonprotease digested) peptides. A novel pipeline based on constructing sequence tags de novo from both original and decharged spectra and a subsequent database search is used to identify peptides. As the MSI signal and the identified peptide are only related by a single mass value, we isolate the corresponding transcript and perform a second validation via in situ hybridization of the transcript. We tested our approach, MSI-Query, on a number of ROIs in the medicinal leech, Hirudo medicinalis, including the central nervous system (CNS). The Hirudo CNS is capable of regenerating itself after injury, thus forming an important model system for neuropeptide identification. The pipeline helps identify a number of novel peptides. Specifically, we identify a gene that we name HmIF4, which is a member of the intermediate filament family involved in neural development and a second novel, uncharacterized peptide. A third peptide, derived from the histone H2B, is also identified, in agreement with the previously suggested role of histone H2B in axon targeting.
Keywords: mass spectrometry imaging, MALDI imaging, targeted peptide identification
Graphical abstract

1. INTRODUCTION
The use of multiple imaging techniques to assess the presence and location of specific proteins in tissues and cells is central to the study of biological systems. The prevailing approach is to label one or several proteins at a time either by attaching a fluorescent domain genetically or by treating a biological sample with labeled antibodies, and then to record two-dimensional (2D) micrographs of the sample, possibly reconstructing them into a three-dimensional (3D) object or movie. Such imaging techniques are low-to-medium throughput approaches and give the biologist insight into just a small number of biological samples, limited to known proteins for which antibodies or tagged forms exist. In contrast to the low throughput of imaging technologies, some available genomic, transcriptomic, and proteomic (particularly via mass spectrometry) technologies allow for the sampling and exploration of the entire complement of active molecules in the cell.
An exciting and innovative recent advance in mass spectrometry is mass spectrometric imaging (MSI). MSI is a molecular imaging technique that allows for the generation of topographic 2D maps for various endogenous and some exogenous molecules (e.g., drugs and their metabolites) involving the application of mass spectrometry directly on tissue. In the matrix-assisted laser desorption/ionization (MALDI) MSI workflow, thin tissue sections (10–15 μm) from organs or even whole body animals are mounted onto a conductive glass slide allowing microscopic observation of the tissue prior to MS analysis. Important preparative steps include appropriate tissue treatments1 and ionic matrix deposition, which must be optimized to reach the highest analytical performance.2 By incorporating a target scanning capability within the mass spectrometer itself, it is then possible to obtain mass spectra at a series of specified locations on the target. After its introduction,3 direct MALDI analysis of tissue sections was developed by various groups.4–8 The studies performed by these groups demonstrated that acquisition of tissue expression profiles while maintaining cellular and molecular integrity was feasible. With automation and new analysis software, it also became possible to produce multiplex imaging maps of selected biomolecules within tissue sections.9–11
While the true abundance of a molecule cannot be measured using this approach, the intensity of its corresponding spectral peak (or its expression level) often correlates with its abundance, albeit in a complicated manner (e.g., compounds with poorer ionization efficiency display lower intensity peaks than would be expected purely on their abundance). Molecules that are preferentially expressed in a region of the sample will show higher intensity in the image corresponding to a specific m/z value when represented with the intensity encoded by a color map. It is also important to note that when looking at these m/z images, it is possible to be looking at the combined intensity of several compounds with similar m/z values. Most bioinformatics approaches have focused on using MALDI MSI as a tool for the discovery of signature markers of particular physiological stages. One approach is to distinguish regions of the tissue presenting very different mass spectral signatures. This has been addressed by a number of researchers, who use unsupervised clustering methods to characterize a region of interest (ROI).12,13 While unsupervised clustering is essential to the analysis of large data sets without user input, it ignores prior knowledge about tissue morphology. In many cases, a more targeted, or supervised, approach is desirable, allowing the user to pull out the molecular signature for a specific area of interest. In this paper, we start with the premise that a region of interest (ROI) is given to us based on preselected morphological criteria. As an example of an ROI, consider the central nervous system (CNS) of the medicinal leech, Hirudo medicinalis, one of the best-studied representatives of the phylum Annelida (segmented worms). Given a particular ROI, we ask (a) which masses have a distinct expression signature, localized to the ROI and (b) to which peptides do these masses correspond? The answer to these questions, in the context of, for example, several embryonic stages, can help us identify key peptides and proteins in leech neuronal development. As the leech CNS has a demonstrated capacity to repair itself after injury and to restore function,14–16 the discovery of peptides involved in neuronal development and regeneration could have therapeutic implications.
To answer our first question, that is, what are the masses that are specifically expressed in a region of interest, we developed a statistical method that operates on MALDI MSI data. While some recently published methods seek to differentiate molecules between two regions (e.g., cancerous vs noncancerous),17–20 we provide a publicly available tool that allows for the analysis of noncontiguous regions, using various methods. We also validate our method using simulations. An interactive tool allows the user to define the ROI on a histological image of a leech embryo. We defined several different ROI corresponding to CNS, the lateral/ventral regions, and the nephridia. We implemented a statistic that allows us to estimate, for each spectral peak, the probability that it is more highly or less highly expressed within the ROI versus outside. The m/z values that we find include some whose expressions are very low relative to other peaks (see Figure 1) but strongly localized to the region of interest. The method was validated, using both simulated perturbations of the original intensities as well as visual inspection of MALDI images restricted to the peaks of interest. All selected peak images displayed localization in the area of interest and the signal became visually weaker and less localized to the ROI as the score decreases. The statistic was used to identify peak masses specifically, expressed in the different ROIs.
Figure 1.
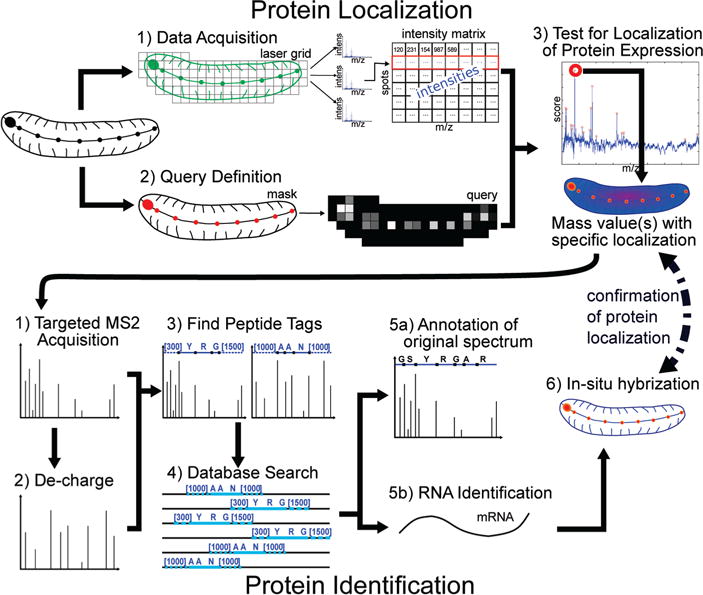
Overall process for detecting and identifying masses specifically expressed within an ROI or specific morphological feature. The process consists of two major parts: protein localization (top) and protein identification (bottom). Protein localization: We developed a pipeline to detect proteins that are preferably expressed in a given ROI using MALDI imaging data. This pipeline consists of 3 parts: (1) data acquisition and processing, (2) query definition, and (3) analysis. (1) We acquire spectra across a raster of the entire animal or section; thus we obtain a list of spectra with spatial coordinates. These can be viewed as a matrix of intensities for each spot and each m/z value. (2) We manually create a mask for the ROI by adding a binary image layer onto the optical image using a standard image editing tool. The mask is converted into a MALDI spot resolution query as described in the Experimental Section. An example of a mask and query for the central nervous system is shown in Supplemental Figure 3 (Supporting Information). (3) We run our algorithm against the obtained MS data to find m/z values which are localized in the defined region of interest. Protein identification: We developed a pipeline to identify peptides specifically expressed in our ROI. This pipeline consists of 6 steps: (1) targeted MS2 acquisition, (2) decharging of acquired spectra, (3) finding peptide tags, (4) database search, (5) annotation of spectrum and RNA identification, and (6) in situ hybridization. (1) We acquire MS2 spectra specifically targeting the lists of masses which were found to be specifically localized in the ROI in the first part of our method. This is done in a data-dependent or semidata-dependent manner. (2) We decharge the spectra to help tag identification. (3) We generate tags on both the original and the decharged spectra. (4) We search the generated tags against a protein database. The search allows for modifications. (5) We annotate the original spectrum and identify the corresponding mRNA. (6) We perform in situ hybridization by synthesizing a probe from this mRNA to test colocalization to the ROI.
The second question we pursued is the identification of the peptides associated with those mass values. In fact, identification of the species showing interesting spatial distributions remains one of the most challenging problems in MSI. Many recent studies have focused on this problem and some of these approaches obtained sequence information by performing MS/MS directly from the tissue. In the case of identifying larger peptides or proteins, these approaches have favored a bottom-up method comprising of in situ tissue digestion by applying a proteolytic enzyme with a spotter or sprayer, followed by MS/MS on tissue.21–23 While these methods are powerful in that they give us a broader overview of the proteome while combining imaging and identification in one step, they have several limitations. First, identification from on-tissue MS/MS has been restricted to high-abundance molecules and remains a challenge for low abundance molecules.21,24 Moreover, digestion greatly increases the complexity of the spectrum, especially for lower masses, although one proposed solution is to couple an ion mobility mass spectrometer to the MALDI-TOF instrument thus using drift time as an additional separating dimension.25 Finally, enzymatic product diffusion,24 variation in peptide intensities,26 and the fact that many parent masses will have similar distributions, all increase uncertainties in the correlation between parent image and trypsin product images. It is worth noting that Chen et al.27 opted for another bottom-up approach from the sample in the identification of neuropeptides in the lobster. While they successfully sequenced many neuropeptides from extracts, MALDI imaging was used independently only as a second step to visualize the localization of these identified neuropeptides, using mass value to correlate the images to the peptides. Thus, the approach does not necessarily identify specific molecules of interest.
In contrast, we focus on LC–MS/MS identification of endogenously processed peptides (2000–5000 Da). By not using a protease digestion step, we maintain the link between the observed mass and the identified peptide. The identification is challenging, as the fragmentation patterns of high-charge, nontryptic peptides are poorly understood.28 Currently, while top-down mass spectrometry allows for the identification of spectra of larger proteins, it requires either a) labor-intensive sample purification to isolate the protein of interest or b) a highly abundant protein in order to obtain spectra with good isotope resolution which is necessary for identification. In order to use complex sample and identify less abundant peptides, we limit the identification here to intermediate sized peptides despite a larger range of interesting m/z values. We developed a custom peptide identification pipeline based on constructing sequence tags de novo from both original and decharged spectra, and performing a database search including modifications (see Figure 1). As the MSI signal and the identified peptide are only related by a single mass value, we isolate the corresponding transcript, and perform a second validation via in situ hybridization of the transcript. Using this method, we successfully identified a number of peptides (see Supplemental Figure 12, Supporting Information). One of these peptides belongs to a novel gene that we call HmIF4; it is a member of the family of intermediate filaments (IF), with strong sequence similarity to gliarin, macrolin and filarin, three previously characterized IFs in Hirudo medicinalis, which were known to be expressed in the CNS. Whole mount in situ hybridization (see Experimental Section) with a probe to the corresponding RNA matched well with MALDI imaging data, supporting the identification. A second identified peptide corresponds to a segment of histone H2B, and showed consistent localization via in situ hybridization as well. A third identified peptide is completely novel, and not currently represented in the leech genomic databases (NCBI nr, Helobdella proteins and Hirudo EST, see Experimental Section).
2. EXPERIMENTAL SECTION
Figure 1 provides an overview of the method, which has two subprocesses: MSI based peptide/protein localization and MS/MS based peptide/protein identification.
2.1. MALDI Imaging Data Acquisition
In brief, the MSI data used to test the computational methods reported here were acquired from two 12-day old leech embryo specimens, herein referred to as LeechE12a and LeechE12b to reflect their embryonic age (12 days at 24 °C). The specimens were opened along the dorsal midline, pinned flat and the yolk sack and endoderm removed to expose the central nervous system. Next they were exposed briefly (1–2 min) to methanol in order to lightly fix and permeabilize the tissues, then placed on glass slides coated with indium tin oxide (ITO) and immediately dried. Methanol was selected because it provided a quick, onestep fixation (noncross-linking) and permeabilization that works well with leech embryos. We also found that it aids efficient peptide extraction following matrix application, though this was not assayed against other possible lipid solvents, as this was not the principal goal of the work reported here. The embryos were mounted so the internal surface of the body wall faced the laser beam. After recording optical images of the mounted embryos, they were coated with several layers of special solid ionic matrices (CHCA/Aniline), using a manual pneumatic TLC sprayer (VWR, Strasbourg, France). Such matrices have proved to be quite efficient for peptide/protein analysis directly from tissue sections (increased signal intensity, increased number of detected peptides/proteins, higher stability under vacuum conditions, lower ablation rate).1 MALDI Imaging was performed on a MALDI-TOF/TOF instrument (Ultraflex II, Bruker Daltonics, Germany) at the University of Lille. While only MS1 spectra were acquired in the mass spectrometric imaging stage, it is worth noting that a TOF/TOF acquisition could be useful to help correlate the sequenced peptides with the original imaging data by using the TOF/TOF partial fragmentation. However, because many of the interesting molecules are of lower abundance (see Supplemental Figure 11, Supporting Information), it is likely that even partial fragmentation may be hard to obtain straight on tissue without protein concentration. Spectra were acquired over 38 837 m/z values from 12 115 locations in a rectangular raster of points 60 μm apart on LeechE12a and 37 199 m/z values from 22 230 locations at raster in a rectangular raster of points 35 μm apart on LeechE12b. Data was acquired on a wide range of m/z values to ensure that our software could detect spatially localized molecules on a large scale of values with different noise levels. Because the data was acquired on a wide m/z range, the m/z resolution did not allow us to detect isotopic patterns on the imaging data. However, peaks were well matched across spectra and across samples. The complete data set is a collection of spectra, each associated with a “spot” on the leech surface. Conceptually, the data can be represented as a collection of triplets <m,s,Im,s> describing the spectral intensity Im,s at each spot s, and m/z value m.
2.2. MALDI Imaging Data Normalization
Each spectrum was normalized to correct for systematic biases, including an m/z dependent bias, and a region specific bias. The spatial bias is clearly seen in Supplemental Figure 2 (Supporting Information), with an order of magnitude difference in total intensity across different regions. A median baseline correction (flexAnalysis) was employed to correct for the m/z bias. Note that baseline correction causes some intensity values to become negative. To correct for spatial bias, we performed normalization after baseline correction. The average intensity for positive intensities after baseline correction at each spot was computed as
The data was normalized by recomputing the intensities as
This data was written into a custom compressed lossless format to facilitate data analysis.
2.3. Query Definition
We defined an ROI by manually creating a mask, which is an image that can be superposed onto the histological image. Formally, a mask M maps each high-resolution pixel to a binary value Mp ∈ {0,1}, indicating whether or not the pixel is part of the ROI. These masks are easily created by adding layers onto the image using any standard editing tool with layer capabilities. These layers can then be exported as separate images. We developed a plug-in that extends the open-source GNU Image Manipulation Program (GIMP, http://www.gimp.org/) to facilitate the exportation process, allowing the user to export any combination of layers into new images.
Because the MALDI spots are at much lower resolution than the mask, they can be partially inside and partially outside the ROI. Thus, we define our query to designate how much of each MALDI spot belongs to the ROI. Thus, the query Q maps each low-resolution laser spots s onto a real value Qs ∈ [0;1]. For each low-resolution laser spot s, we can define the collection of pixels p ∈ s on the light-transmitted image which belong to the laser spot. We assign the following value to each spot:
Figure 3 shows the user-defined mask for the leech CNS, and the resulting gray-image query Qs for all spots s.
Figure 3.
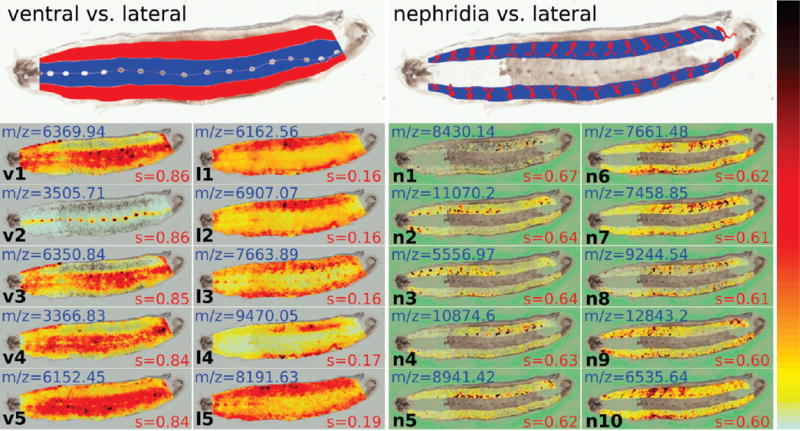
Mask and top results for LeechE12a with other ROIs: (left) ventral vs lateral query and (right) nephridia vs lateral. For ventral vs lateral query, the top 5 high-coring images (v1–v5) and the top 5 low-scoring images (l1–l5) are shown. A high score indicates strong expression in the ventral region against the lateral region, while a low score indicates the inverse. In all images, there is a clear division between the two sections (ventral and lateral). For nephridia vs lateral, the top 10 results are shown. Scores are lower than for the other queries. The expression pattern is noisier and nonhomogeneous.
2.4. Query Shift
Mapping of the MALDI spots to the histological image is done here by manually defining teaching points, which are a set of spots with coordinates on both images, in flexImaging software (Bruker Daltonics) prior to acquisition. By using these matching sets of coordinates, it is possible to calculate the relative scale ratios and establish a correspondence between the coordinates of both images. However, because of the lack of precision in the definition of the teaching points, the mapping and/or scaling of MALDI images to the histological image may be slightly off. Because the ROI is defined on the optical image, which shows the morphological features, it is important to minimize mapping errors between the two types of images. In order to correct for imprecision in defining the teaching points, we introduced the possibility of manually setting shifts by translation and/or scaling of the mapping from the MALDI image to the optical image. Both shifts are done independently on the x-axis and y-axis, as teaching points can have lack of precision on either axis.
2.5. Testing for Localization of Protein Expression
The input to this process is the set of normalized intensities Im,s and one or two query(ies). If only one query is specified, the given ROI is compared to the rest of the spots, as done in Figure 2. If two queries are specified, then the intensities from the first ROI are compared to the intensities from the second ROI, ignoring the rest of the data, as done in Figure 3. Depending on the statistical test, it may be necessary to define two sets of spots from the query: those in the ROI and those outside the ROI. For a query Q, we can define two thresholds t1 and t2, such that t1 ≥ t2. Then, the set of spots such that Q1 ≥ t1 are within the ROI and the set of spots such that Qs ≤ t2 and Qs > t1 are outside the ROI. Note that there is no overlap between the two sets but that there may be spots not belonging to either set. If the input is two queries, one threshold t1 is given for each query and spots belonging to both query are arbitrarily assigned to the first ROI. While the user has the option to define those query thresholds, all values are defaulted to 0.5.
Figure 2.
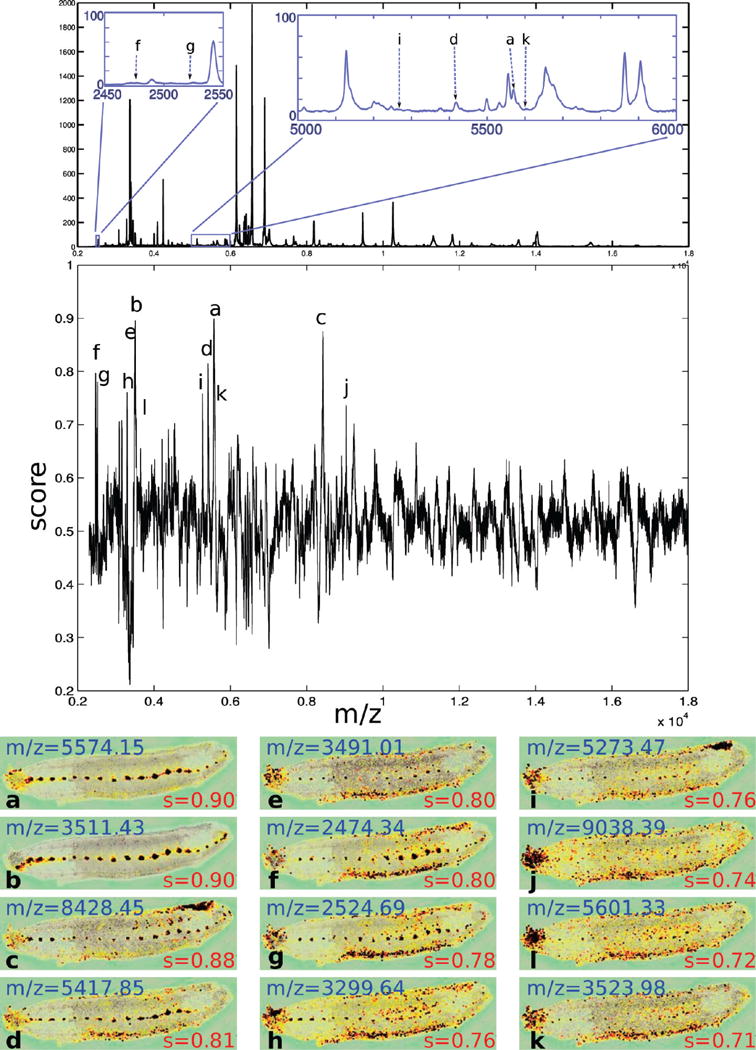
Results of the ρ-statistical test for the CNS query in the leech. We identified 43 m/z values that were significantly present in the CNS of LeechE12a (score ≥0.65), which are listed in Supplemental Figure 1 (Supporting Information). Visual inspection clearly demonstrates the power of the method as illustrated by the images for the top 12 most significant m/z values. Score decreases along with quality of CNS localization.
A set of intensities for all spots exhibits the same pattern as the given ROI if the intensities are distributed such that there is a separation between those within the ROI and those outside the ROI. While our software allows the user to choose between several statistics, we use the ρ statistic, which is the Mann – Whitney U statistic normalized by its maximum possible value. For each m/z, we calculate the Mann – Whitney U statistic for the average intensity over a range of ±2 Da as U = RROI − (nROI(nROI+1)/2) where nROI is the number of spots in the ROI and RROI is the sum of the ranks of the intensities in the ROI. The rho statistic is calculate as ρ = U/(nROInbg), where nbg is the number of spots outside the ROI. High-scoring peaks for m/z < 2200 were discarded because many spectra did not have any peaks in that region causing a bias in the localization.
2.6. Simulations
In order to assess the performance of our method, we generate some simulated data. The first simulated data aims to see how our method performs when the ROI signal decreases in terms of area, that is we want to see how the statistic behaves as less spots in the ROI show higher expression. Let n1 and n2 be the number of spots inside and outside the ROI respectively, and let and be the corresponding sets of intensities. We sort the ROI spots by location and the background spots by intensities. In order to generate a random background intensity, we randomly select a background spot bi such that 1 ≤ i ≤ (n2 − 1) and we generate a random intensity Irand sampled uniformly in [Ibg(bi),Ibg(bi+1)]. To generate the simulated data, we incrementally set k ROI spots r1,…,rk to a random background intensity. Thus, the ROI spots now have intensities Isim assigned to them such that Isim(r1,…,rk) are random background intensities, and Isim(ri) = IROI(ri) for k + 1 ≤ i ≤ n1). In the case of rebalancing the total intensities, we distribute the subtracted intensity by setting
Our second simulation aims to see how our method performs when the ROI signal decreases over the entire region. In this case, we simply decrease the intensities of each ROI spots by a certain percentage until the average intensity inside the ROI is the same as the non-ROI (or background) intensities. This means that for each spot ri within the ROI, we assign the intensity Isim(ri) = x*IROI(ri) where x∈ [mean(Ibg)/mean(IROI),1].
2.7. MS/MS Sample Preparation and Data Acquisition
Identification of the molecules with particular m/z values selected from the MALDI-TOF imaging required the acquisition of high-resolution MS/MS spectra. We therefore tested several extraction procedures for obtaining intact proteins/peptides without enzymatic digestion that yielded good MS and MS/MS results with either MALDI or ESI methods. These included extraction with 1N acetic acid, 1N HCL, TCA, basic extraction with ammonia, 50:50:1 Methanol:water:FA, and PBS. For the purposes of the work described here, the most consistent results were obtained with a simple PBS extraction (a comparative study of these methods will be published elsewhere). Peptides were extracted from leech embryos of embryonic ages between 6 and 12 days old. Forty embryos with or without yolk were snap frozen in liquid nitrogen and then pulverized in a Dounce homogenizer. The homogenized tissue was stored at −80 °C after thorough lyophilization. The homogenized tissue was then dissolved in 200 μL of ice cold 100 mM PBS (pH 7.4) containing protease inhibitor cocktail and 0.1 M PMSF and vortexed. Tissue was dissolved by stirring at 4 °C for another 4 h. Samples were sonicated for 5 min with 10 s pulses at constant voltage. The extract was then centrifugated at 12 000 rpm for 30 min at 4 °C. The supernatant was separated from the pellet and was reextracted using PBS. Supernatants from all the extractions were pooled and samples were desalted and then dried using a Speedvac before proceeding to mass spectral identification.
The samples were analyzed by liquid chromatography (LC) coupled with tandem mass spectrometry with electrospray ionization. All nanospray ionization experiments were performed by using a QSTAR-Elite hybrid mass spectrometer (AB/MDS Sciex) interfaced to a nanoscale reversed-phase high-pressure liquid chromatograph (Tempo) using a 10 cm-180 ID glass capillary column packed with 5-μm C18 ZorbaxTM beads (Agilent). The buffer compositions were as follows: buffer A was composed of 98% H2O, 2% ACN, 0.2% formic acid, and 0.005% TFA; buffer B was composed of 100% ACN, 0.2% formic acid, and 0.005% TFA. Peptides were eluted from the C-18 column into the mass spectrometer using a linear gradient of 5–80% buffer B over 140 min at 400 μL/min. Time-of-flight MS were acquired at m/z 400 to 2000 Da for 0.5 s with 12 time bins to sum. MS/MS data were acquired from m/z 50 to 2000 Da by using “enhance all” and 24 time bins to sum, dynamic background subtract, automatic collision energy, and automatic MS/MS accumulation with the fragment intensity multiplier set to 6 and maximum accumulation set to 2 s before returning to the survey scan. LC–MS/MS data were acquired in a data-dependent fashion by selecting the 5 most intense peaks with charge state of 2–5 that exceeds 20 counts, with exclusion of former target ions set to “360 seconds” and the mass tolerance for exclusion set to 100 ppm. The data dependent acquisition was also operated with inclusion and exclusion lists to include an ion selection list for MS/MS analysis and exclusion of ions already analyzed.
2.8. MS/MS Identification
The Hirudo genome has not been sequenced, so we create a custom database, LeechProtsDB, comprised of Hirudo EST sequences29 (http://genomes.sdsc.edu/leechmaster/database/), the predicted Helobdella robusta proteins (JGI, v1.0, http://genome.jgi-psf.org/Helro1/Helro1.download.ftp.html) and all Hirudo protein sequences from the NCBI nr database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/). We cluster and decharge the raw MS/MS spectra and predict peptide tags on both the original and the decharged spectra using PepNovo30 version 20090907. Peptide tags were predicted using no enzyme, fragment tolerance of 0.5 Da, parent mass tolerance of 2.5 Da, considering the post-translational modifications M+16, Q+1 and N+1. A peptide tag is a string of residues with flanking masses on the C-terminus and the N-terminus (see Figure 1, Database Search). The fragmentions comprising the top scoring tags were manually investigated for charge confirmation. Tags that passed this validation were searched against our custom database. A database peptide was considered a candidate if it matched the tag perfectly, and the residues on either end had masses that matched the tag masses. All candidate peptides were scored according to the fraction of explained intensity, which is the proportion of the total spectrum intensity which can be explained by annotated peaks.
2.9. In situ Hybridization
A complementary coding strand probe was obtained by in vitro transcription of the PCR product using T3 polymerase (Invitrogen), and subsequently hydrolyzed into shorter fragments. In situ hybridization was then performed with a digoxigenin-labeled RNA probe as described previously.31 In brief, embryos of various ages were fixed in paraformaldehyde and further by Pronase E digestion. Day 6–8 embryos were digested for 20–25 min, older embryos for longer times. Digested embryos were then hybridized overnight at 59 °C in 50% formamide with approximately 1 ng/mL digoxigenin labeled RNA. Washed embryos were treated with RNAase A (Sigma) to degrade unhybridized probe. Hybridized probe was visualized immunohistologically with an alkaline phosphatase (AP)- conjugated antidigoxigenin (Roche) reacted for periods ranging from 15 h to 3 days, using NBT and X-phosphate color reagents-(Roche). Intact embryos were cleared in 80% glycerol, mounted under a coverslip, and photographed.
3. RESULTS AND DISCUSSIONS
3.1. Overview of the Pipeline
In Figure 1, we show our customized pipeline for detecting and identifying masses specifically expressed in a given morphological feature. It consists of two major subprocesses: protein localization (top) and peptide identification (bottom). The first subprocess allows us to detect peptides or proteins that are preferentially expressed in a given ROI using MALDI imaging data and consists of 3 parts: (1) data acquisition and processing, (2) query definition, and (3) analysis. First, we acquire spectra across a raster of locations across the entire tissue or specimen, obtaining a list of spectra associated with specific spatial coordinates. A histological image of the specimen taken prior to matrix deposition serves to localize raster points, or MALDI spots, on the specimen. We use the histological image to manually define a mask of the ROI, which is stored as a transparent layer with black pixels only within the ROI. Because MALDI spots are generally at lower resolution than the mask, the mask must be converted into a query as described in the Experimental Section. The query define which MALDI spots are in the ROI, or more precisely, how much of each MALDI spot belongs to the ROI. Queries can also be searched against each other in order to find molecules that are differentially expressed from one ROI to another. An example of a mask and query for the central nervous system of a leech embryo specimen (LeechE12a) is shown in Supplemental Figure 3 (Supporting Information). For each m/z, we apply a statistical test to decide if it is preferentially expressed in the ROI.
The critical performance issue is to rank results correctly. We use the ρ statistic, which is the Mann–Whitney U statistic normalized by its maximum possible value. Conceptually, it represents the probability that, given two random spots, one in ROI and one outside the ROI (or in the second ROI), the intensity of the spot inside the ROI is higher than that of the spot outside the ROI. Thus, a ρ statistic value of 0.5 indicates that the expression is not specific to the ROI. As the statistic approaches 1, it becomes increasingly likely that the intensity of a random ROI spot is greater than that of a random non-ROI spot. This means that the expression becomes more localized to the ROI. Conversely, as the statistics approaches 0, it becomes increasingly likely that the expression is inversely localized to the ROI. There is no hard line between “good” and “bad” localizations, but rather gradual decrease in specificifity of the localization to the ROI. Therefore, we leave it up to the user to decide a probability threshold based on the desired quality of results. For our purpose, we often used a threshold of 0.65. The performance of the statistic for simulated and actual leech data is discussed in detail in the following sections.
The second stage of the pipeline is aimed at identifying the peptides specifically expressed in our ROI. MS/MS spectra are acquired by specifically targeting the list of masses detected by the first stage (Figure 1). This is done in a data-dependent or semidata-dependent manner (see Experimental Section). To maintain the connection with the m/z values, the MS/MS spectra are acquired using a nonproteolytically digested sample. We use a high-accuracy QTOF instrument (sub-3ppm), with multiple collision energies to provide a high-quality fragmentation. Multiply charged fragment ions are decharged using isotopic peaks. Next, we generate peptide sequence tags on both the original and the decharged spectra. We define a tag as a short string of aminoacids flanked by mass values (see Figure 1, Database Search). We search all tags in a modification-tolerant manner against a custom protein database and annotate and score the resulting candidate peptides from the search. At this stage, we have a top-scoring candidate peptide identified through MS/MS, with a parent mass value that shows preferential expression in the ROI. As a test of this identification, we synthesize a probe from the corresponding mRNA with embryonic cDNA as a template, which is then used in in situ hybridization assays to verify that mRNA and peptide colocalize to our ROI. We would expect peptide colocalization to the mRNA, but in some instances the mRNA could have a wider distribution, suggesting post-translational regulation. Conversely, it is also possible that the peptide is transported to a subcellular location that is different from the region of synthesis. Hence, colocalization is supportive of identification, but lack of it cannot be taken as proof of mis-identification.
3.2. Data Normalization
Similar to other studies,32 we observe significant and systematic bias in the distribution of the intensities, both on the m/z axis and spatially. Reasons for the spatial bias include differing tissue composition or thickness and heterogeneity of ionic matrix crystallization.1 To eliminate this bias, we must first do a baseline correction on all spectra (m/z-dependent bias), then we must normalize the intensities across all spectra (see Experimental Section). In Supplemental Figure 2a (Supporting Information), we see that there is large variation in the average spot intensities. In panel b, we see the distributions of the average spot intensities across the leech surface before (top) and after (bottom) normalization. After normalization, all spot spectra have the same average peak intensity. Not correcting the bias can lead to erroneous conclusions on the localization of expression for many molecular species. As an example, the leech brain appears to have overall significantly lower total intensity compared to other regions. In panel c, correcting for this bias reveals the species m/z = 10357.1 (right) as being higher in the brain (top vs bottom panels). On the other hand, the species at m/z = 8563.04 (left) appears to have significantly localized expression initially. However, the significance is diminished after correction.
3.3. Defining Regions of Interest
As described in Experimental Procedures, MSI-Query was applied to two samples denoted as LeechE12a and LeechE12b. A total of 12115 and 22230 MALDI MS spectra were acquired on LeechE12a, and LeechE12b respectively. We created masks for three distinct ROIs (CNS, lateral-ventral, and nephridia) onto the histological images and converted them into queries (see Experimental Section). These masks correspond to the following embryonic tissues.
CNS
The leech segmental ganglia are very similar to each other and comprise of about 400 neurons, ~180–190 pairs and ~30 unpaired,33 many of which are very well characterized developmentally, anatomically, physiologically and neurochemically.34,35 Furthermore, unlike the mammalian CNS, the leech CNS has a demonstrated capacity to repair itself after injury and to restore function.14–16 We defined the central nervous system (CNS) as the segmental ganglia, the head ganglion, and the tail ganglion in each specimen.
Lateral-Ventral
As in other animals, mechanosensory neurons in the leech innervate the skin in a regular pattern of domains also known as tiles. One example of these types of cells are those that respond to light touch on the skin surface (TV, TL and TD cells). These cells have an interesting difference in how they set up their sensory arbors, subdividing each segment: the TV cells innervate left and right ventral tiles, the TL cells innervate left and right lateral tiles, and the TD cells innervate left and right dorsal tiles. A very interesting problem is to identify what peptides/proteins (or other molecules) might mark the boundaries or areas of each tile and signal to the sensory cells where to locate their arbors. We defined the lateral domains as those extending from head to tail between two lines drawn along the ventral-most and dorsal-most boundaries of the laterally positioned nephridia (see Figure 3 top left). The ventral domains were then defined as lying between the lateral domains and the ventral midline. The area of the CNS was subtracted from the ventral domains in order to examine ventral domain information without the CNS contribution and in order to reduce noise.
Nephridia
The nephridia are the segmentally iterated excretory organs of the leech and as such serve the purposes of ridding the animal of waste products and maintaining water and electrolyte balance. Both the structure and transport mechanisms of the leech nephridia have been studied in some detail.36,37 Because the nephridia connect to the outside of the animal (through the nephridiopores), they can serve as pathways for bacterial or other microbial invasion, and some preliminary data suggests that cells in the nephridia may be expressing and releasing antibacterial peptides. We created a mask of the nephridia as shown in Figure 3 (top right).
3.4. Simulations
Currently, there are no standard data sets to assess performance, or standards to generate simulated data. To assess performance, we chose a mass value (m/z = 5574.15) that was significantly localized in the leech CNS. Our first simulation tests the performance of our method when the ROI signal decreases over the entire region. In this case, we simply decrease the intensities in the ROI spots by a certain percentage until the average intensity inside the ROI is the same as the non-ROI (or background) intensities. In Supplemental Figure 6 (Supporting Information), we see that the score decreases slowly at first, and starts dropping drastically once the average ROI intensity is less than about twice the average background intensity. Visually, we can see that the signal also starts dropping more rapidly around the same point. In the original image, the average ROI intensity is about 6.8 times the non-ROI average intensity, and the signal is very clear. In the second image, the ratio of average ROI intensity to average background intensity are approximately 2.74. While the signal is visually not as pronounced as in the original image, especially in the posterior ganglia, we can still see clear CNS expression and the score is still high (s = 0.75). However, in the third and fourth images (intensity ratios 1.87 and 1.58, scores 0.66 and 0.62), we observe a lower signal. We can also see a decrease of signal in the anterior ganglia between the two images. Finally, the last two images (ratios 1.29 and 1.0, scores 0.57 and 0.5) show almost no CNS localization. Again, similar intensities in the ROI and in the background lead to a score close to 0.5 as expected.
Our second simulation tests our method when degrading the signal in the ROI. In this case, we set a proportion of the ROI spots to have random non-ROI (or background) intensities (see Experimental Section). It is then possible to balance the total ROI intensities by distributing the subtracted intensity to the remaining spots; that way, the total intensities in ROI and outside ROI remain the same throughout the simulation. In Supplemental Figure 7 (Supporting Information), we show the results for two simulated runs for the two cases described above: with and without balancing the ROI intensities. In both cases, the score linearly decreases as more ROI spots are set to background intensity. In the balancing case, the intensities of the remaining spots increase to compensate for the other spots; consequently, the score remains higher in the balanced case than in the unbalanced case, as expected. Note that because the background intensities are set in a random manner, the results differ slightly for each run. This explains why the ending scores are different in the two runs. Visually, we can see that around s = 0.65, which indicates a 65% chance that a random ROI spot has higher intensity than a random non-ROI spot, the signal is still CNS-specific, but is targeted to a subregion. When all spots are set to background intensities, the signal is lost and the probability score decreases to 0.5 as expected. The results show that the ρ statistic provides a direct interpretation of the strength of the ROI signal.
3.5. Overexpressed Molecules in the Leech
CNS
Any MALDI spot partially hitting the CNS is considered in the ROI. Even so, only 2.66% and 3.28% of the spots in LeechE12a and LeechE12b respectively were in our ROIs, and each ganglion had at most 2–3 laser spots 50% or more coverage by the ROI (see Supplemental Figure 3, Supporting Information). Despite this challenge, we find that our method performs extremely well. We identified 43 m/z values that were significantly present in the CNS of LeechE12a (score ≥0.65), which are listed in Supplemental Figure 1 (Supporting Information). Visual inspection clearly demonstrates the power of the method as illustrated by the images for the top 12 most significant m/z values shown in Figure 2. We can see from these images that the ions corresponding to these m/z values are clearly more highly represented in the CNS and that specific expression can be detected even in the presence of other signal. For example, mass 8428.45 (case c) is detected as having significantly higher expression in the CNS even though it is expressed in another area in a dorsal posterior region. Likewise, we can see signal in other areas for cases e, f, g, h (m/z = 2474.11, 2524.06, 9240.11, 3299.11). Panels i–k are marked by lower (albeit significant) scores which corresponds to the decrease in CNS specificity. In those cases, not only is the noise higher in the rest of the leech, but the uniformity of the expression inside the CNS decreases. Specifically, we can see that fewer spots within the ganglia display strong expression while expression in the head ganglion increases compared to the rest of the ganglia. However, even in those cases, the CNS intensities are uniformly higher than non-CNS intensities.
The correlation between decreasing score and decreasing quality of CNS localization is also evident in the lower ranked masses. In Supplemental Figure 4 (Supporting Information), we show the expression pattern for 3 representative m/z values. The first image is taken for m/z = 2797.28 which was assigned a score of s = 0.62, just below our cutoff of 0.65. It still shows regional distribution specific to the CNS, but signal in other regions significantly impairs the ROI signal compared to the top-scoring images in Figure 2. In the second panel, the intensities outside the nervous system almost perfectly balance out the intensities within the CNS, and thus we get a score of 0.51, which represents no CNS localization. Finally, at the other end of the range, we can detect ions which have specific expression to outside the ROI. In the third image, at score s = 0.23, the molecule is highly expressed in the ventral region of the leech but shows distinct underexpression in the ganglia and the brain, displaying the inversed CNS expression pattern expected by such a low score.
Lateral-Ventral
To detect some of the potential signaling peptides that might be involved in the development of mechanosensory arbors, we looked for m/z values expressed differentially between the ventral and the lateral regions by using our algorithm. Given that there are more spots in these ROIs than in the CNS ROI, and that they are more evenly distributed, we expected the algorithm to perform well for these masks. Indeed, we had many high-scored results for both ventral and lateral regions. Figure 3 shows the images for the top 5 highest scores (v1–v5) and for the top 5 lowest scores (l1–l5) when running the algorithm for the ventral region against the lateral region. A high score indicates strong expression in the ventral region against the lateral region, while a low score indicates the inverse. In all images, there is a clear division between the two sections (ventral and lateral) on the left side of the leech. The right side of the leech seems to have more noise, but the demarcation between the two sections is maintained throughout the results. Interestingly, we pick out a nervous system signal at m/z = 3505. However, when looking at the image we can see that there is also a clear separation in signal between the ventral and lateral regions. This signal could be from a molecule that is expressed in both the ventral region and the CNS yielding a higher intensity in the CNS, possibly due to higher abundance there. Alternatively, the different signal intensities might reflect a mixture of two molecules with similar m/z values, one molecule producing a high intensity signal in the CNS and the other a lower intensity signal in the ventral region. When looking at the localization of the lower signal molecule, we can see the same intensity separation between the ventral and lateral regions.
Nephridia
When querying the nephridia against the rest of the leech, the top results were clearly in the nephridia alone; however, the fact that many masses are expressed more strongly in the lateral section than the ventral section caused some of the lower scoring results to be noisy. As a consequence, we queried the nephridia against the lateral section of the leech. The top 10 results are shown in Figure 3. It is important to note that the scores for nephridia are much lower than for the previous queries. In fact, only the top score s = 0.67 is above our threshold of s = 0.65. When looking at the images, we can see that even though there is nephridia localization, the expression pattern is noisy and nonhomogeneous. For example, m/z = 5557 and m/z = 10875.6 are expressed more in the anterior and posterior sections of the leech, respectively. Interestingly, several masses show a colocalization with the nervous system (m/z = 8429, 7663, 6535) (data not shown), reflecting perhaps the accumulation of strongly expressed and secreted neuropeptides in the CNS and the secretory organs.38 Besides detecting interesting masses for the ROI, we can also see regional differences in the expression of the molecules. Corresponding molecules may represent segmental functional differentiation.
Comparative Analysis and Reproducibility
To address the reproducibility of CNS specific mass values, we repeated the experiment in another leech. LeechE12b, like LeechE12a, is a 12 day embryo and expected to show similar distribution of peptides. While the two samples were prepared using the same methods on comparable samples, spectra were acquired on a slightly greater m/z range and on a much tighter raster for LeechE12b. In Supplemental Figure 5 (Supporting Information), we display the top scoring masses for the two samples. All high-scoring masses in LeechE12b were found in LeechE12a, but the inverse is not true. This is attributed to a lower overall intensity of the data in LeechE12b, as can be seen by the high-scoring images from LeechE12b. This difference in data quality is attributed to the difference in data acquisition parameters as mentioned above. Notwithstanding the low overall quality of LeechE12b, the scores between the samples are comparable and reflect the true quality of the localization.
3.6. Peptide Identification
Initial experiments were performed on extracts from 39 whole embryos. The samples were subjected to nano-LC–ESI–QTOF–MS. We selected peaks corresponding to interesting imaging masses from the MS1 data and acquired MS/MS spectra in a data-dependent manner or semidata-dependent manner (see Experimental Section). We increased acquisition time in order to acquire high-quality spectra of lower abundance peptides. In Supplemental Figure 11 (Supporting Information), we can see that two of the identified spectra had lower MS1 intensity counts than many of the other molecules present in the sample. No trypsin digestion was performed so we could use the peptides’ parent masses to link the MS/MS data to the MALDI imaging data, which captures endogenously processed peptides. Identification of intermediate sized endogenous peptides is difficult due to a limited understanding of fragmentation chemistry and high charge on fragment ions. To overcome these issues, we developed a novel peptide identification pipeline, based on construction of sequence tags from both original and deconvoluted spectra, and a database search including modifications. We identified a number of peptides (see Supplemental Figure 12, Supporting Information). A few are described below.
We first identified a candidate sequence for parent mass ~2474 Da that was specifically expressed in the CNS. In Figure 4, we present the annotated spectrum for the peptide that was derived from an EST transcript in our LeechProtsDB database, as well as the corresponding MALDI image showing CNS specific expression. The annotation explains 78.41% of the total peak intensity, with strong b and a fragment-ion series. As expected, we observe a very strong peak for fragments at the N-terminus of the prolines. A large part of the b ion series, namely b15–b22, is built on validated charge 3 fragment ions, which makes identification of this peptide difficult using standard tools.
Figure 4.
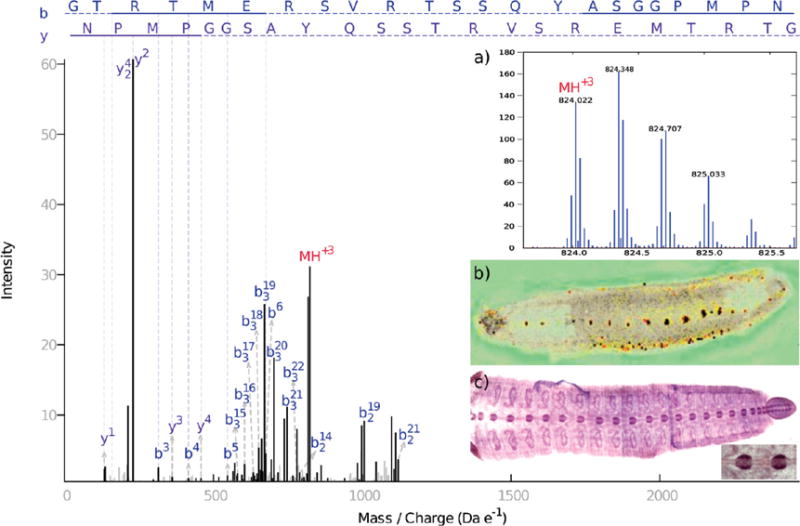
Annotated MS/MS spectrum for HmIF4, a novel peptide in the family of glial intermediate filament. The annotation explains 78.41% of the total peak intensity, with strong b and a fragment-ion series. Corresponding MALDI and in situ hybridization images (panels b and c respectively) both show CNS localization. MS1 (panel a) has good isotope resolution. Charges for all fragments were manually verified by examining isotope patterns. Text annotation is available by request.
A BLAST search of the EST sequence against the NCBI nr database established that the peptide came from a previously unreported protein. The strongest similarities (but not identity) are all to intermediate filament proteins, and specifically to gliarin, macrolin and filarin, the three known intermediate filaments in Hirudo medicinalis,39 which have been described as important in neuronal development in leech. Thus, we believe to have found a novel member of the family of intermediate filaments, which we call HmIF4. We aligned the four protein sequences using ClustalW 2.0.12,40 as shown in Supplemental Figure 8 (Supporting Information). The EST open reading frame aligned particularly well in the conserved rod domain, and has more variability outside of that domain. The peptide we identified is located in a variable region in the 5′ end of the rod domain where the sequences are quite dissimilar, thus confirming the discovery of a novel protein. Finally, we examined the distribution of the HmIF4 transcript using in situ hybridization (see Experimental Section). In Figure 4c, we can clearly see that the mRNA is indeed preferentially expressed in the CNS. The concordance of the spatial distributions at the peptide and corresponding mRNA strongly support the specificity of expression, and also suggests that differential gene expression, not protein targeting, is the reason for the spatial distribution.
A second peptide, with parent mass ~3666 Da, was targeted for being specifically expressed in the lateral/dorsal region. The identified sequence is noted as
and explains 74.45% of the total intensity. The annotated spectrum is shown in Figure 5. Moreover, most of the rest of the intensity can be explained by internal ions from breaks at the dominant peaks. Out of this long annotation, we have a very strong 7-residue tag, [1089.62]VTSDVLV[1863.22], with a complete doubly charged b ion series b10–b17 and a complete y ion series y17–y24. There is also a partial a-ion ladder supporting the direction of this tag. On its own, the tag explains 67.3% of the spectral intensity. A BLAST search of the protein sequence did not get any good hits, suggesting that this is a novel peptide. Another spectrum of the same peptide, unmodified and 2 amino acids longer, confirms the identification. The parent mass of this peptide, ~3841 Da, also shows dorsal localization (see Supplemental Figure 10, Supporting Information).
Figure 5.
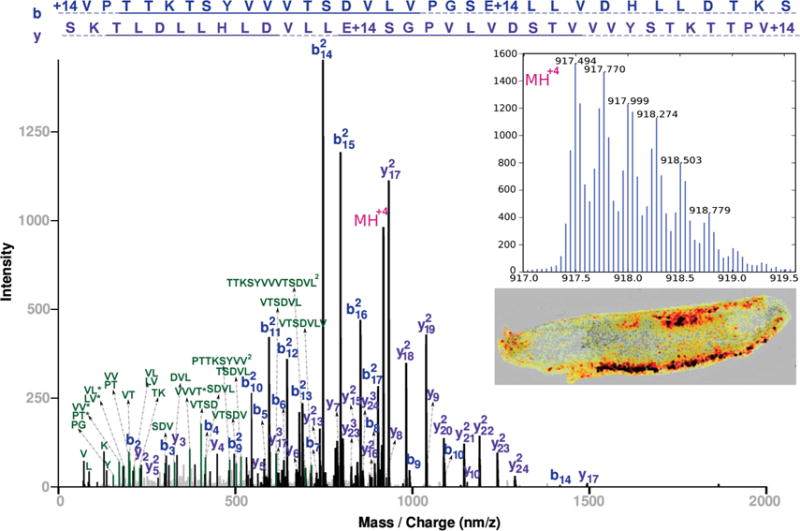
Annotated MS/MS spectrum for novel peptide with parent mass ~3666 Da, targeted for being specifically expressed in the lateral/dorsal region. The entire annotation explains 74.45% of the total intensity, including a very strong 7-residue tag, [1089.62]VTSDVLV[1863.22], with a complete doubly charged b ion series b10–b17 and a complete y ion series y17–y24. A BLAST search of the protein sequence did not get any good hits suggesting that this is a novel peptide. Another spectrum of the same peptide, unmodified and 2 amino acids longer, confirms the identification. The parent mass of this peptide, ~3841 Da, also shows dorsal localization (see Supplemental Figure 10, Supporting Information).
A third molecule (parent mass ~2500 Da) was shown to have a CNS specific expression. A database search identified the peptide LPGELAKHAVSEGTKAVKTYTSSK, which is part of the histone H2B in a related species, Helobdella robusta (Supplemental Figure 9, Supporting Information). Histones, which are part of the DNA packaging complex, are highly conserved. Indeed, a BLAST search41 of the translated EST sequence against the NCBI nr database, returned complete perfect matches to 179 sequences in many different species. Therefore, it is is very likely that the peptide is conserved between the sequences in Helobdella robusta and Hirudo medicinalis. Regarding CNS localization, Shimma et al.23 have previously identified a histone H2B expressed in the mouse brain using MALDI imaging. In situ hybridization of the mRNA again shows a preferential location in the CNS, but with a relatively weaker signal (see Supplemental Figure 9).
4. CONCLUSIONS
Recent years have seen a tremendous improvement in instrumentation for mass spectrometric imaging employing MALDI, DESI and SIMS techniques.42 MALDI MSI is particularly useful for the study of the tissue distribution of biologically interesting molecules because it affords both access to large range of intact molecules and a relatively higher spatial resolution when compared to other ion sources. MALDI MSI is therefore the approach of choice when studying tissue distributions of larger molecules, such as peptides or proteins.
While MALDI imaging offers great advantages for detecting and mapping unknown molecules in their native, processed state, it does have some important physical limitations. For example, despite recent and potential further improvements, MSI cannot achieve either the level of detectability, single or a few molecules, or the spatial resolution of conventional light microscopic cell imaging techniques. The pixel resolution obtained with MSI, as reported in most published studies, is ~50–300 μm, though a possible lower limit of ~5 μm for more abundant species has been reported.42 In comparison, resolution of 200 nm is achievable using laser scanning confocal microscopy of cells immunostained with fluorescent tags.43
The advantages of MSI, then, are 2-fold: first, a pixel is not simply a pixel but a complex array of mass values that can be resolved to high-accuracy. Second, MSI allows for an unsupervised (label-free) interrogation of the sample, allowing for the discovery of previously unknown species that are active in specific spatiotemporal contexts. The approach we report, referred as MSI-Query, addresses and exploits these two facets, developing a novel analysis methodology.
As noted above, a hurdle in the assessment and analysis of the large amounts of data inherent in MSI is to determine which of the many masses represented in the spectra are worth pursuing, given that the identification of the corresponding protein requires a great investment in time and effort. In our approach, we have started with the premise that the topographic distribution of a particular m/z value can be a first order filter for selecting those molecules of particular interest. Thus, we first developed a statistical technique to identify mass values that are specifically expressed in a morphological region specified by the user. Using this constraint, we then obtained a collection of mass values (presumably endogenously processed peptides or proteins) that are specifically expressed in the CNS, nephridia, and ventral/dorsal segments of the medicinal leech embryo, the model system we used to test our technique. To obtain amino acid sequence information for less abundant species, we decided to use a procedure for identifying the peptides/proteins corresponding to these interesting masses from secondary fragmentation (MS/MS) data that required decoupling the imaging and MS/MS44 performed with LC–MS separation of nondigested proteome extracts. While other approaches obtain sequence information by performing MS/MS directly from the tissue while maintaining spatial information, our method allows for the concentration of peptides, helping identify peptides and proteins with intermediate abundance.28
Our approach does have some shortcomings that should be noted. The identification of endogenous peptides based on fragmentation of intermediate-sized, highly charged precursors is challenging given available tools. We developed a customized pipeline for identification. As a second issue, the link between MS/MS and MSI parent masses is tenuous due to the lower accuracy of mass resolution in MSI. However, we test our results by using a second independent validation through in situ hybridization of the identified mRNA. While colocalization of the ISH and MSI signals can provide only a measure of consistency, it may also lead us to interesting differences between mRNA and protein localization that can be further explored.
Initial tests of our methods on the leech embryo MSI data have thus far resulted in the identification of a few novel proteins, including a member of the intermediate filament (IF) family and a completely novel peptide sequence. The discovery of a new IF expressed by neurons in the leech CNS is also of significant biological interest. IFs form a diverse family of proteins important for cytoskeletal architecture. Invertebrate IF proteins are relatively less analyzed and might be evolutionarily and functionally distinct from their vertebrate counterparts. The three known IFs in leech have distinct patterns of expression. The expression of macrolin is limited to macroglias, gliarin is expressed in both glial, and macroglial cells, and filarin is selectively expressed in neurons.39,45 While their function is poorly understood, the neuronal IFs are suggested to be developmentally regulated and may be involved in stabilizing the neural cytoskeleton. Our discovery of a novel IF protein adds to the diversity of invertebrate neuronal IFs.
Our results also include the detection of CNS expression of a fragment of histone H2B in early leech development. Interestingly, although histones are mainly known for their essential roles in chromosome packaging, histone H2B has been reported previously to be localized to the mouse brain.23 Moreover, recent studies in Drosophila have suggested that the specific targeting of some axons (R1–R6) in the optic ganglia is mediated by the selective deubiquitination of the fly ortholog of histone H2B.46,47 Further, the deubiquitination is mediated by the SAGA complex, which has analogs from yeast to human.46,47 The discovery of these and other peptides using MSI shows the power of mass spectrometric imaging in a label-free identification of spatially differentiated proteins.
Supplementary Material
Acknowledgments
This research was supported by the National Center for Research Resources of NIH via grant P-41-RR24851, by NSF via grant DBI-0852081, by the Centre National de la Recherche Scientifique (CNRS), the Ministère de l’Enseignement, de la Recherche et des Technologies (MERT), the Genoscope, and the Agence Nationale de la Recherche (ANR). Software source code and installation and usage guide are available at http://bix.ucsd.edu/MSI-Query/.
Footnotes
Supporting Information
Supplemental figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lemaire R, Tabet JC, Ducoroy P, Hendra JB, Salzet M, Fournier I. Solid ionic matrixes for direct tissue analysis and MALDI imaging. Anal Chem. 2006;78:809–819. doi: 10.1021/ac0514669. [DOI] [PubMed] [Google Scholar]
- 2.Franck J, Arafah K, Barnes A, Wisztorski M, Salzet M, Fournier I. Improving tissue preparation for matrix-assisted laser desorption ionization mass spectrometry imaging. Part 1: using micro-spotting. Anal Chem. 2009;81:8193–8202. doi: 10.1021/ac901328p. [DOI] [PubMed] [Google Scholar]
- 3.Spengler B, Hubert M, Kaufmann R. MALDI ion imaging and biological ion imaging with new scanning UV-laser microprobe. Proceedings of the 42nd ASMS Conference on Mass Spectrometry and Allied Topics; ASMS: Santa Fe, NM. 2004. [Google Scholar]
- 4.Chaurand P, Stoeckli M, Caprioli RM. Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Anal Chem. 1999;71:5263–5270. doi: 10.1021/ac990781q. [DOI] [PubMed] [Google Scholar]
- 5.Fournier I, Day R, Salzet M. Direct analysis of neuropeptides by in situ MALDI-TOF mass spectrometry in the rat brain. Neuro Endocrinol Lett. 2003;24:9–14. [PubMed] [Google Scholar]
- 6.Dreisewerd K, Kingston R, Geraerts WPM, Li KW. Direct mass spectrometric peptide profiling and sequencing of nervous tissues to identify peptides involved in male copulatory behavior in lymnaea stagnalis. Int J Mass Spectrom Ion Processes. 1997;169–170:291–299. Matrix-Assisted Laser Desorption Ionization Mass Spectrometry. [Google Scholar]
- 7.Jiménez CR, Li KW, Dreisewerd K, Spijker S, Kingston R, Bateman RH, Burlingame AL, Smit AB, van Minnen J, Geraerts WP. Direct mass spectrometric peptide profiling and sequencing of single neurons reveals differential peptide patterns in a small neuronal network. Biochemistry. 1998;37:2070–2076. doi: 10.1021/bi971848b. [DOI] [PubMed] [Google Scholar]
- 8.Li KW, Hoek RM, Smith F, Jiménez CR, van der Schors RC, van Veelen PA, Chen S, van der Greef J, Parish DC, Benjamin PR. Direct peptide profiling by mass spectrometry of single identified neurons reveals complex neuropeptide-processing pattern. J Biol Chem. 1994;269:30288–30292. [PubMed] [Google Scholar]
- 9.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 10.Stoeckli M, Farmer TB, Caprioli RM. Automated mass spectrometry imaging with a matrix-assisted laser desorption ionization time-of-flight instrument. J Am Soc Mass Spectrom. 1999;10:67–71. doi: 10.1016/S1044-0305(98)00126-3. [DOI] [PubMed] [Google Scholar]
- 11.Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry a new technology for the analysis of protein expression in mammalian tissues. Nat Med. 2001;7:493–496. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]
- 12.McCombie G, Staab D, Stoeckli M, Knochenmuss R. Spatial and spectral correlations in MALDI mass spectrometry images by clustering and multivariate analysis. Anal Chem. 2005;77:6118–6124. doi: 10.1021/ac051081q. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrov T, Becker M, Deininger SO, Ernst G, Wehder L, Grasmair M, von Eggeling F, Thiele H, Maass P. Spatial segmentation of imaging mass spectrometry data with edge-preserving image denoising and clustering. J Proteome Res. 2010;12:6535–46. doi: 10.1021/pr100734z. [DOI] [PubMed] [Google Scholar]
- 14.Blackshaw SE, Henderson LP, Malek J, Porter DM, Gross RH, Angstadt JD, Levasseur SM, Maue RA. Single-cell analysis reveals cell-specific patterns of expression of a family of putative voltage-gated sodium channel genes in the leech. J Neurobiol. 2003;55:355–371. doi: 10.1002/neu.10214. [DOI] [PubMed] [Google Scholar]
- 15.Burrell BD, Sahley CL, Muller KJ. Progressive recovery of learning during regeneration of a single synapse in the medicinal leech. J Comp Neurol. 2003;457:67–74. doi: 10.1002/cne.10530. [DOI] [PubMed] [Google Scholar]
- 16.Skierczynski BA, Wilson RJ, Kristan WB, Skalak R. A model of the hydrostatic skeleton of the leech. J Theor Biol. 1996;181:329–342. doi: 10.1006/jtbi.1996.0135. [DOI] [PubMed] [Google Scholar]
- 17.Cazares LH, Troyer D, Mendrinos S, Lamce RA, Nyalwidhe JO, Beydoun HA, Clements MA, Drake RR, Semmes OJ. Imaging Mass Spectrometry of a Specific Fragment of Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase Kinase 2 Discriminates Cancer from Uninvolved Prostate Tissue. Clin Cancer Res. 2009;15:5541–5551. doi: 10.1158/1078-0432.CCR-08-2892. [DOI] [PubMed] [Google Scholar]
- 18.Rauser S, Marquardt C, Balluff B, Deininger SO, Albers C, Belau E, Hartmer R, Suckau D, Specht K, Ebert M, Schmitt M, Aubele M, Höfler H, Walch A. Classification of HER2 receptor status in breast cancer tissues by MALDI imaging mass spectrometry. J Proteome Res. 2010;9:1854–1863. doi: 10.1021/pr901008d. [DOI] [PubMed] [Google Scholar]
- 19.Schwamborn K, Caprioli RM. Molecular imaging by mass spectrometry–looking beyond classical histology. Nat Rev Cancer. 2010;10:639–646. doi: 10.1038/nrc2917. [DOI] [PubMed] [Google Scholar]
- 20.El Ayed M, Bonnel D, Longuespée R, Castelier C, Franck J, Vergara D, Desmons A, Tasiemski A, Kenani A, Vinatier D, Day R, Fournier I, Salzet M. MALDI imaging mass spectrometry in ovarian cancer for tracking, identifying, and validating biomarkers. Med Sci Monit. 2010;16:233–245. [PubMed] [Google Scholar]
- 21.Groseclose MR, Andersson M, Hardesty WM, Caprioli RM. Identification of proteins directly from tissue in situ tryptic digestions coupled with imaging mass spectrometry. J Mass Spectrom. 2007;42:254–262. doi: 10.1002/jms.1177. [DOI] [PubMed] [Google Scholar]
- 22.Seeley EH, Caprioli RM. Molecular imaging of proteins in tissues by mass spectrometry. Proc Natl Acad Sci USA. 2008;105:18126–18131. doi: 10.1073/pnas.0801374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimma S, Sugiura Y, Hayasaka T, Zaima N, Matsumoto M, Setou M. Mass imaging and identification of biomolecules with MALDI-QIT-TOF-based system. Anal Chem. 2008;80:878–885. doi: 10.1021/ac071301v. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin RJ, Pennington SR, Pitt AR. Protein and peptides in pictures imaging with MALDI mass spectrometry. Proteomics. 2008;8:3785–3800. doi: 10.1002/pmic.200800320. [DOI] [PubMed] [Google Scholar]
- 25.Stauber J, Macaleese L, Franck J, Claude E, Snel M, Küuyeakrer Kaletas B, Wiel IM, Wisztorski M, Fournier I, Heeren RM. On-Tissue Protein Identification and Imaging by MALDI-Ion Mobility Mass Spectrometry. J Am Soc Mass Spectrom. 2010;21(3):338–347. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Mallick P, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 27.Chen R, Jiang X, Prieto Conaway MC, Mohtashemi I, Hui L, Viner R, Li L. Mass Spectral Analysis of Neuropeptide Expression and Distribution in the Nervous System of the Lobster Homarus americanus. J Proteome Res. 2010;9(2):818–832. doi: 10.1021/pr900736t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JE, Atkins N, Hatcher NG, Zamdborg L, Gillette MU, Sweedler JV, Kelleher NL. Endogenous peptide discovery of the rat circadian clock a focused study of the suprachiasmatic nucleus by ultrahigh performance tandem mass spectrometry. Mol Cell Proteomics. 2010;9:285–297. doi: 10.1074/mcp.M900362-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macagno ER, Gaasterland T, Edsall L, Bafna V, Soares MB, Scheetz T, Casavant T, Da Silva C, Wincker P, Tasiemski A, Salzet M. Construction of a Medicinal Leech Transcriptome Database and Its Application to the Identification of Leech Homologs of Neural and Innate Immune Genes. BMC Genomics. 2010;11:407. doi: 10.1186/1471-2164-11-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank A, Tanner S, Bafna V, Pevzner P. Peptide sequence tags for fast database search in mass-spectrometry. J Proteome Res. 2005;4:1287–1295. doi: 10.1021/pr050011x. [DOI] [PubMed] [Google Scholar]
- 31.Nardelli-Haefliger D, Shankland M. Lox10, a member of the NK-2 homeobox gene class, is expressed in a segmental pattern in the endoderm and in the cephalic nervous system of the leech Helobdella. Development. 1993;118:877–892. doi: 10.1242/dev.118.3.877. [DOI] [PubMed] [Google Scholar]
- 32.Norris JL, Cornett DS, Mobley JA, Andersson M, Seeley EH, Chaurand P, Caprioli RM. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int J Mass Spectrom. 2007;260:212–221. doi: 10.1016/j.ijms.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macagno ER. Number and distribution of neurons in leech segmental ganglia. J Comp Neurol. 1980;190:283–302. doi: 10.1002/cne.901900206. [DOI] [PubMed] [Google Scholar]
- 34.Lefebvre C, Salzet M. Annelid neuroimmune system. Curr Pharm Des. 2003;9:149–158. doi: 10.2174/1381612033392251. [DOI] [PubMed] [Google Scholar]
- 35.Sawyer R. Leech biology and behaviour. Oxford University Press; New York: 1986. [Google Scholar]
- 36.Zerbst-Boroffka I, Wenning A. Mechanism of regulatory salt and water excretion in the leech, Hirudo medicinalis. L Zool Beitr N F. 1986;30:359–377. [Google Scholar]
- 37.Zerbst-Boroffka I, Bazin B, Wenning A. Chloride secretion drives urine formation in leech nephridia. J Exp Biol. 1997;200:2217–2227. doi: 10.1242/jeb.200.16.2217. [DOI] [PubMed] [Google Scholar]
- 38.Schikorski D, Cuvillier-Hot V, Leippe M, Boidin-Wichlacz C, Slomianny C, Macagno E, Salzet M, Tasiemski A. Microbial challenge promotes the regenerative process of the injured central nervous system of the medicinal leech by inducing the synthesis of antimicrobial peptides in neurons and microglia. J Immunol. 2008;181:1083–1095. doi: 10.4049/jimmunol.181.2.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Bolton B, Zipser B, Jellies J, Johansen KM, Johansen J. Gliarin and macrolin, two novel intermediate filament proteins specifically expressed in sets and subsets of glial cells in leech central nervous system. J Neurobiol. 1999;40:244–253. doi: 10.1002/(sici)1097-4695(199908)40:2<244::aid-neu10>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Madden TL, Schãffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amstalden van Hove ER, Smith DF, Heeren RM. A concise review of mass spectrometry imaging. J Chromatogr A. 2010;1217(25):3946–3954. doi: 10.1016/j.chroma.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 43.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 44.Lemaire R, Desmons A, Tabet JC, Day R, Salzet M, Fournier I. Direct analysis and MALDI imaging of formalin-fixed, paraffin-embedded tissue sections. J Proteome Res. 2007;6:1295–1305. doi: 10.1021/pr060549i. [DOI] [PubMed] [Google Scholar]
- 45.Johansen KM, Johansen J. Filarin, a novel invertebrate intermediate filament protein present in axons and perikarya of developing and mature leech neurons. J Neurobiol. 1995;27:227–239. doi: 10.1002/neu.480270209. [DOI] [PubMed] [Google Scholar]
- 46.Weake VM, Lee KK, Guelman S, Lin CH, Seidel C, Abmayr SM, Workman JL. SAGA-mediated H2B deubiquitination controls the development of neuronal connectivity in the Drosophila visual system. EMBO J. 2008;27:394–405. doi: 10.1038/sj.emboj.7601966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weake VM, Workman JL. Histone ubiquitination triggering gene activity. Mol Cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.