

ABSTRACT
Gemcitabine is the standard-of-care for chemotherapy in patients with pancreatic adenocarcinoma and it can directly incorporate into DNA or inhibit ribonucleotide reductase to prevent DNA replication and, thus, tumor cell growth. Most pancreatic tumors, however, develop resistance to gemcitabine. Polo-like kinase 1 (Plk1), a critical regulator in many cell cycle events, is significantly elevated in human pancreatic cancer. In this study, we show that Plk1 is required for the G1/S transition and that inhibition of Plk1 significantly reduces the DNA synthesis rate in human pancreatic cancer cells. Furthermore, the combined effect of a specific Plk1 inhibitor GSK461364A with gemcitabine was examined. We show that inhibition of Plk1 significantly potentiates the anti-neoplastic activity of gemcitabine in both cultured pancreatic cancer cells and Panc1-derived orthotopic pancreatic cancer xenograft tumors. Overall, our study demonstrates that co-targeting Plk1 can significantly enhance the efficacy of gemcitabine, offering a promising new therapeutic option for the treatment of gemcitabine-resistant human pancreatic cancer.
KEYWORDS: combination therapy, gemcitabine resistance, pancreatic cancer, Plk1
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has the highest mortality rate of all cancers, is the fourth leading cause of cancer deaths in the US,1 and is estimated to cost the health care system $2.4 billion each year.2 Despite significant research efforts in treating primary and metastatic tumors, only 7% of patients survive 5 y.1 The standard therapeutic for advanced PDAC is the nucleoside analog gemcitabine, which induces stalled DNA replication forks and subsequent cell death.3 Despite its unique molecular properties and its prevalent use in the clinic, a major contributing factor for the dismal prognosis of patients is that they develop gemcitabine resistance where DNA replication and cell survival continue, even in the presence of the drug. Clearly, we need to identify novel targets whose inhibition can enhance therapeutic efficacy in PDAC cases.
Polo-like kinases (Plks) are a family of serine-threonine kinases that regulate multiple intracellular processes including DNA replication, mitosis, and stress response.4,5 Plk1, the most well understood family member, regulates numerous stages of mitosis and is overexpressed in various types of cancer and high levels of Plk1 correlate with unfavorable patient outcomes. Therefore, Plk1 serves as a strong candidate target for the development of novel approaches to manage many cancers. Of note, we and others have shown that the majority of PDAC tumors exhibit elevated Plk1 and that high Plk1 expression is a significant prognostic indicator of diminished patient survival.6,7 Significantly, several potent and selective ATP competitive inhibitors of Plk1, including GSK461364A, have been shown to effectively inhibit tumor growth in in vivo studies.8
In the current study, using biochemical fractionation and RNA interference approaches, we found that Plk1 was required for both G1/S and G2/M phases in human pancreatic cancer cells and that inhibition of Plk1 significantly reduced the DNA synthesis rate. We also demonstrated that combination therapy with GSK461364A and gemcitabine is a novel and therapeutically effective approach to treat pancreatic cancer in both cultured cells and orthotopic pancreatic cancer xenograft tumors.
Results
Nuclear localization of Plk1 at interphase in human pancreatic cancer cells
It has been well documented that Plk1 is a critical regulator of many mitotic events and that overexpression of Plk1 has been found in many types of tumors.4,5 However, increasing evidence suggests that Plk1 also has multiple functions beyond mitosis.9,10 To investigate whether Plk1 also functions in interphase in human pancreas cells, we first examined the expression pattern of Plk1 in several human pancreas derived cell lines. While HPDE6 is a non-transformed and immortalized human pancreatic epithelial cell line,11 Pa03C, Panc-1 and Panc10.05 are 3 human pancreatic cancer cell lines. As expected, overexpression of Plk1 was detected in pancreatic cancer cell lines but not in HPDE6 (Fig. 1A). Upon arresting cells in G2/M phase by nocodazole, Plk1 expression levels increased in all 4 cell lines. The difference of Plk1 protein expression between HPDE6 and the 3 pancreatic cancer cell lines is statistically significant, suggesting that Plk1 expression in cancerous cells in comparison to normal cells is fundamentally different.
Figure 1.
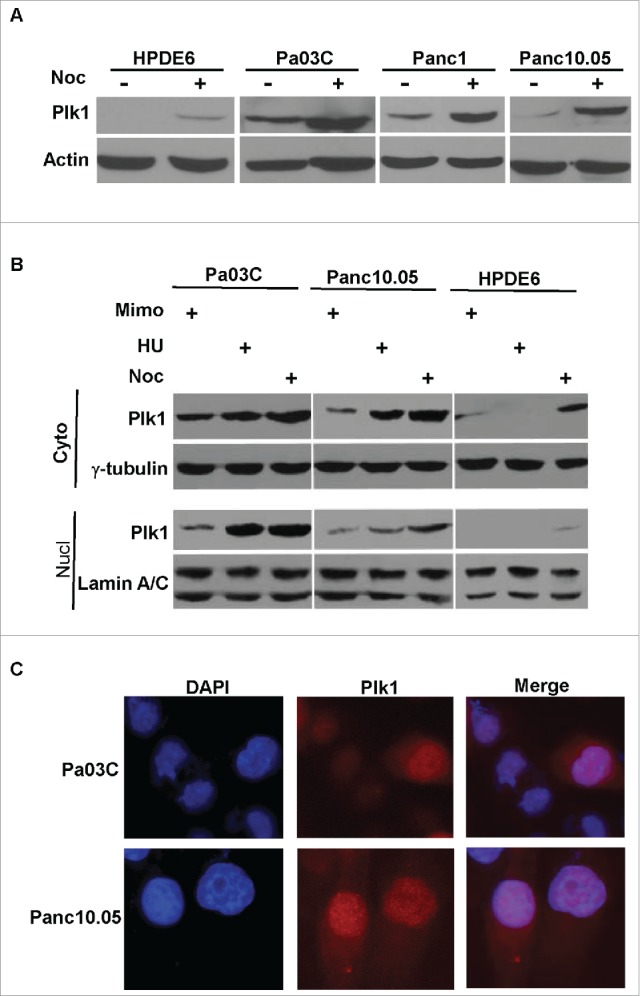
Nuclear localization of Plk1 in human pancreas cells. A, HPDE6, Pa03C, Panc1 and Panc10.05 were treated with or without 200 ng/ml nocodazole (Noc) for 12 h and harvested for IB. B, Pa03C, Panc10.05 and HPDE6 cells were treated with 0.3 mM mimosine (Mimo) for 20h, 4 mM hydroxyurea (HU) for 24 h, or 200 ng/ml nocodazole for 12 h to block cells at G1, S, or M phase, respectively. Cytoplasmic and nuclear fractions were isolated and analyzed by Western blot. C, Pa03C or Panc10.05 cells randomly growing on coverslips were stained with a Plk1 antibody and analyzed by IF microscopy. DNA is visualized with DAPI staining.
To determine whether nuclear localization of Plk1 was a general phenomenon in human pancreas cells, HPDE6, Pa03C and Panc10.05 cells were utilized to analyze Plk1 localization and expression. Accordingly, cells were treated with mimosine, hydroxyurea, or nocodazole to block at G1, S, or M phase, respectively. Cytoplasmic and nuclear fractions were prepared for anti-Plk1 immunoblot (IB) analysis. Nuclear localization of Plk1 during interphase was clearly detected in Pa03C and Panc10.05 cells but not in HPDE6 cells (Fig. 1B), indicating that the nuclear localization of Plk1 might be tumor cell-specific. Moreover, 2 tumor cell lines showed much higher overall levels of Plk1 than that of HPDE6 cells, in agreement with the notion that Plk1 overexpression correlates with transformation.4,5 Finally, nuclear localization of Plk1 in Pa03C and Panc10.05 cells was also detected by IF staining, using a modified permeabilization/fixation protocol (Fig. 1C).
Plk1 is required for G1/S phase transition in human pancreatic cancer cells
Next, RNAi was used to test whether Plk1 is required for cell cycle progression in the early stages, such as G1 and S phases, in human pancreatic cancer cells. Plk1 was depleted by using a lentivirus-based RNAi approach in randomly growing cells as previously described.12 Pa03C cells were infected with lentivirus for 36 h to deplete Plk1, followed by treatment with puromycin for an additional 36 h to select infection-positive cells. Upon removal of floating cells, the remaining attached cells were treated with 200 ng/ml nocodazole for different times and harvested for IB. We examined the degradation rate of cyclin E, a G1/S marker protein, upon nocodazole addition. In control cells, cyclin E was almost completely degraded after 12 h of treatment with nocodazole. In striking contrast, a significant amount of cyclin E was still detected in Plk1-depleted cells even after 12 h of nocodazole treatment (Fig. 2A), indicating that Plk1 is required for the G1/S transition. To confirm this novel observation, we also performed fluorescence-activated cell sorting (FACS) analysis to follow any cell-cycle defect upon Plk1 depletion in pancreatic cancer cells. Plk1 in Panc1 cells was depleted by using a vector-based RNAi approach in randomly growing cells as previously described (Fig. 2B).13 To be consistent with the stabilization of Cyclin E upon Plk1 depletion, FACS profiles of Plk1-depleted cells showed 2 populations (2n and 4n), suggesting that Plk1 depletion leads to G1/S arrest as well as G2/M block (Fig. 2C, left panels). To further confirm the G1/S arrest induced by Plk1 depletion, we compared FACS profiles of cells following nocodazole treatment. Upon nocodazole treatment, control cells quickly accumulated at the G2/M phase, indicated by the increase in cell population with 4n DNA content by FACS. In contrast, Plk1-depleted Panc1 cells were much more resistant to nocodazole treatment (Fig. 2C, compare the FACS profiles at 12 h after nocodazole treatment), supporting the notion that Plk1 is indeed required for cell cycle progression in the early stage and also required for mitotic entry in pancreatic cancer cells.
Figure 2.
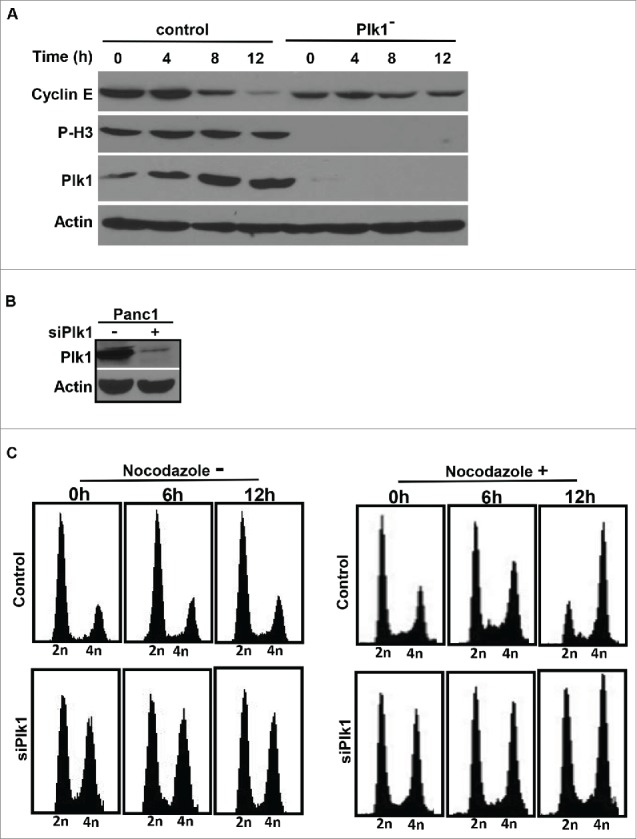
Plk1 depletion in human pancreatic cancer cells. A, Pa03C cells were infected with lentivirus to deplete Plk1. 36h after lentivirus infection, puromycin was added for an additional 36 h to select for infection-positive cells. After floating cells were removed, the remaining attached cells were treated with 200 ng/ml nocodazole for different times as indicated and harvested for IB. B, Panc1 cells were co-transfected with pBS/U6-Plk1 and pBabe-puro at a ratio of 8:1. At 1 d post-transfection, puromycin was added for an additional 36 h to select for transfection-positive cells. After floating cells were removed, the remaining attached cells were harvested for IB. C, Randomly growing Panc1 cells were depleted of Plk1 using the protocol described in B, cells were treated with or without 200 ng/ml nocodazole and harvested at different time points for FACS.
Inhibition of Plk1 reduces DNA synthesis in human pancreatic cancer cells
To uncover the function of Plk1 at the G1/S phase in human pancreatic cancer cells, we aimed to investigate whether Plk1 inhibition affects DNA synthesis. Accordingly, GSK461364A, a Plk1 inhibitor currently in clinical trials14 was used in a synchronized culture. After Pa03C cells were subjected to a double thymidine block protocol to arrest cells at the G1/S boundary, GSK461364A was added to the medium 1 h prior to release to ensure that the cells would enter S phase with inhibited Plk1 activity. After the second thymidine block, cells were released into GSK461364A-containing medium (25 nM or 100 nM) and harvested at different times for various analyses. FACS was used to monitor cell cycle progression. As indicated, we observed an early delay in cell cycle progression using either GSK461364A concentration (Fig. 3A, compare FACS profiles at 3 h after release) and an arrest at G2/M phase with the higher concentration (100 nM) and at the later time point (13 h). In line with the FACS profiles, DNA synthesis, indicated by 5-bromo-2′-deoxyuridine (BrdU) incorporation, was reduced in GSK461364A-treated cells relative to control cells (Fig. 3B and C).
Figure 3.
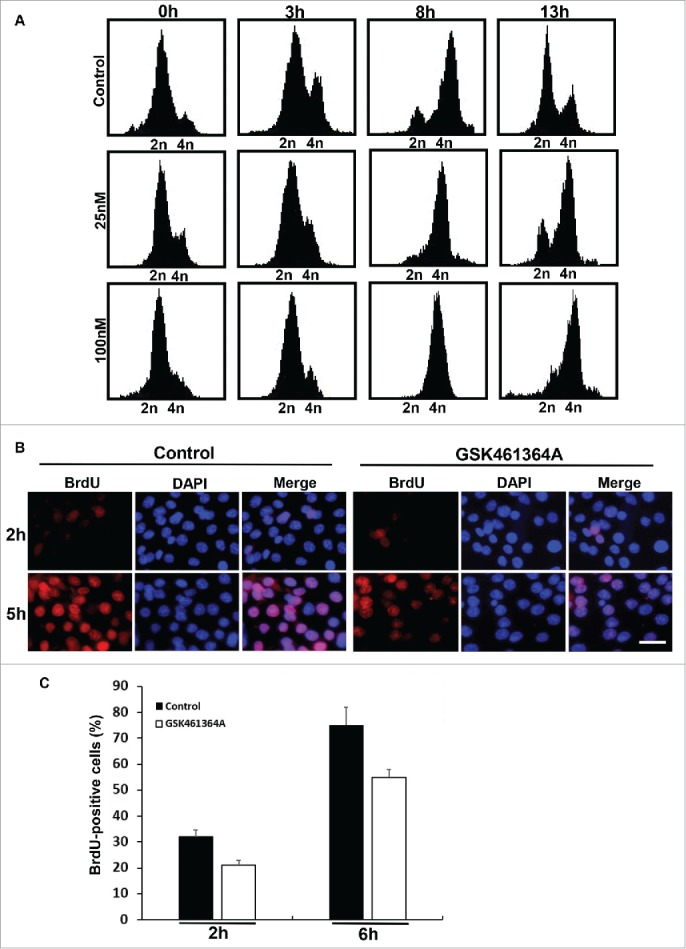
Effects of inhibition of Plk1 on DNA replication. A, FACS analysis of cell cycle progression in Pa03C cells. After Pa03C cells were synchronized with a double thymidine block (DTB; 16 h treatment with thymidine, 8 h release, and a second thymidine block for 16 h), synchronized cells were released into medium in the absence or presence of GSK461364A (25nM or 100nM) and harvested at different time points for FACS. GSK461364A was added into the medium1 h prior to release. B, Representative images of BrdU labeling in control and Plk1-inhibited cells. Pa03C cells were synchronized with the DTB, released into medium with or without GSK461364A (added 1 h prior to release) for 2h or 5h, labeled with BrdU for 30 min, and stained with anti-BrdU antibodies. DNA was stained with DAPI. Scale bar, 100 µm. C, Quantification of BrdU-positive cells in B.
Inhibition of Plk1 enhances the efficacy of gemcitabine in Panc1-derived orthotopic xenograft tumors
Because gemcitabine can directly incorporate into DNA or inhibit ribonucleotide reductase to prevent DNA replication and, thus, tumor cell growth and in this study we found that inhibition of Plk1 significantly reduced DNA synthesis in pancreatic cancer cells, we asked whether gemcitabine and the Plk1 inhibitor GSK461364A could act synergistically to inhibit growth of pancreatic cancer. First, Panc1 cells were treated with GSK461364A, gemcitabine or GSK461364A in combination with gemcitabine (combo), and then harvested for analysis of cleaved-PARP, a marker of apoptosis. As predicted, the combination of gemcitabine and GSK461364A led to significantly increased cellular apoptotic responses when compared to GSK461364A or gemcitabine alone (Fig. 4A).
Figure 4.
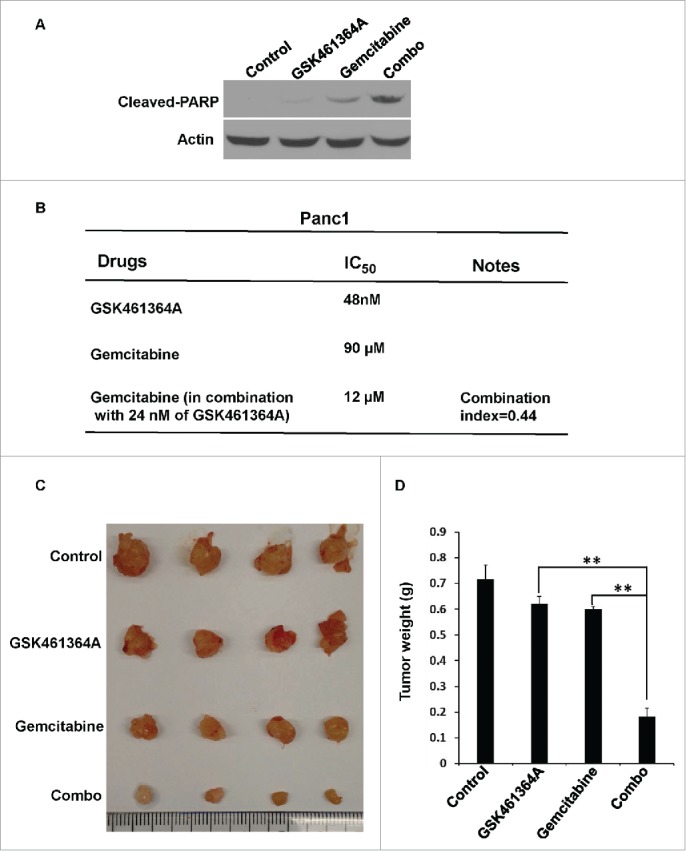
GSK461463A and gemcitabine synergistically inhibit growth of Panc1-derived orthotopic xenograft tumors. A, Panc-1 cells were treated with GSK461364 (10 nmol/L), gemcitabine (100 nmol/L), or both for 72 h, followed by Western blot. B, The combination index of GSK461364A, gemcitabine in Panc1 cells. C, Panc1-derived orthotopic xenograft tumors that had been treated with GSK461364A (12 mg/kg), gemcitabine (40 mg/kg) or a combination of both drugs for 6 weeks. NSG mice were inoculated with Panc1 cells (5 × 105) for 2 weeks, intravenously injected with GSK461364A, gemcitabine or a combination of both drugs. D, Quantification of tumor weight in C. Data are expressed as mean ± SEM, n = 6; **, p < 0.01.
We also asked whether gemcitabine and GSK461364A act synergistically in Panc1 cells by measuring the combination index (CI) with the following equation: combination index = (Am)50/(As)50 + (Bm)50/(Bs)50, where (Am)50 is the concentration of gemcitabine necessary to achieve a 50% inhibitory effect in the combination with half of the concentration of the GSK461364A IC50; (As)50 is the concentration of gemcitabine that will produce the identical level of effect by itself; (Bm)50 is the concentration of GSK461364A that will produce a 50% inhibitory effect in the combination with half of the concentration of gemcitabine IC50; and (Bs)50 is the concentration of GSK461364A that will produce the same level of effect by itself. Antagonism is indicated when CI > 1, CI = 1 indicates an additive effect and CI < 1 indicates synergy.15
Measurements of IC50 values revealed an IC50 value of 90 µM for gemcitabine-treated Panc1 cells (Fig. 4B). However, the IC50 value of gemcitabine was reduced to 12 µM when cells were treated in combination with 24 nM GSK461364A. The combination index of the 2 drugs was calculated to be 0.44, suggesting a strong synergistic effect between gemcitabine and GSK461364.
Finally, to better assess this synergistic activity, we next tested the effect of combination treatment in a Panc1-derived orthotopic pancreatic cancer xenograft mouse model. As shown in Figure 4C and D, the combination of gemcitabine and GSK461364 led to a significantly greater tumor inhibitory effect than monotherapy with either gemcitabine or GSK461364.
Histological analyses of these Panc1 cell-derived orthotopic tumors under different treatment conditions revealed that tumors from the control group exhibited sheets of malignant cells with marked nuclear pleomorphism and numerous mitotic figures (Fig. 5A) and high Ki67 levels (Fig. 5B and C). These histopathologic features are similar to high-grade pancreatic cancer in vivo, which is typically associated with poor clinical outcomes. Gemcitabine-treated tumors also showed similar cytomorphologic features although the vascular network was more prominent and the tumors had a more nested appearance. Treatment with GSK461364A altered the tumor growth pattern so that there was a significant decrease in skeletal muscle invasion (Fig. 5A). Remarkably, the gemcitabine plus GSK461364A-treated tumors were very tiny and surrounded by normal pancreas tissues, showing marked apoptotic bodies with condensed cytoplasm and pyknotic nuclei. Immunostaining for Ki67 also confirmed that tumors from the combination therapy had a significant reduction in overall proliferation (Fig. 5B and C), suggesting that the combination of gemcitabine and GSK461364A is a more effective approach to killing pancreatic cancer cells compared to monotherapy with gemcitabine. These results are consistent with our observation in the cell-based study, providing additional evidence of a strong synergistic effect between gemcitabine and GSK461364A in vitro and in vivo. In summary, these studies support the notion that the Plk1 inhibitor GSK461364A and gemcitabine act synergistically in human pancreatic cancer cells, providing a novel and promising therapeutic option to treat pancreatic cancer patients.
Figure 5.
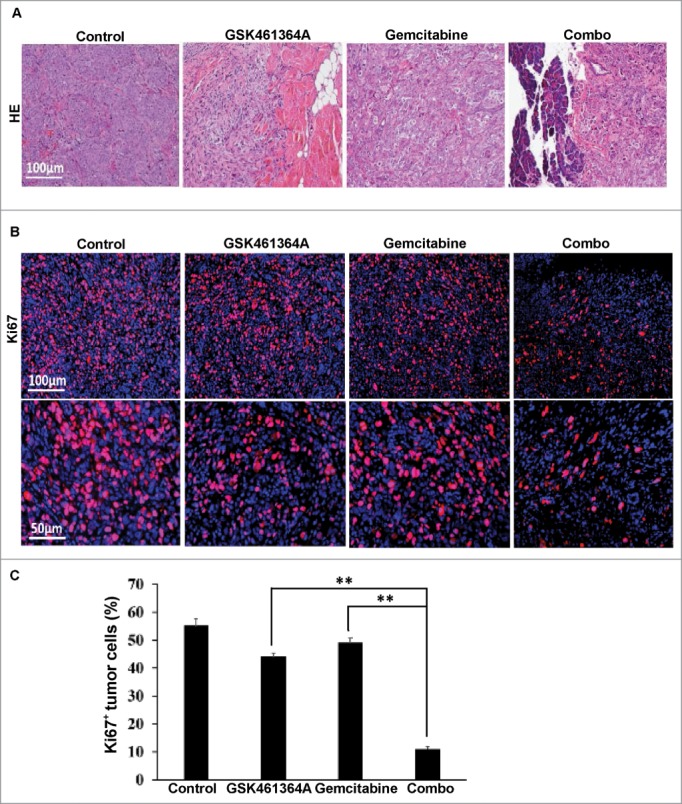
Histological analysis of Panc1-derived orthotopic xenograft tumors. A, Representative images of H&E on formaldehyde-fixed, paraffin-embedded Panc1-derived orthotopic xenograft tumors sections from different treatment groups. B, Representative images of IFC staining for Ki67 on formaldehyde-fixed, paraffin-embedded Panc1-derived orthotopic xenograft tumors sections from different treatment groups. C, Microscopic quantification of Ki67 as percentages of Ki67-positive cells to total numbers of cells. Multiple tumor sections were calculated (mean ± SEM; n = 4). **, p < 0.01.
Discussion
For many years, Plk1 has been described as a cytoplasmic protein in interphase. This concept has been challenged recently due to a series of findings supporting a critical role of Plk1 in many nuclear events, such as DNA synthesis16,17 and DNA damage repair.18 To provide direct evidence that Plk1 localizes to the nucleus, we previously isolated nuclear and cytoplasmic fractions of 3 human cell lines: a non-transformed hTERT-RPE1 cell line, and 2 cancer cell lines, HeLa and U2OS. Our analysis led to the conclusion that Plk1 is significantly elevated in interphase of cancer cells and that a significant portion of Plk1 localizes in the nucleus in the interphase of cancer cells.19
To provide direct evidence that Plk1 is indeed involved in the G1/S transition of cancer cells, the Spankuch lab systematically analyzed the effect of a more selective type II Plk1 inhibitor, SBE13. These authors showed for the first time the delayed progression through the cell cycle in lower SBE13 concentrations and a G2/M arrest using higher SBE13 concentrations followed by apoptosis in various cancer cell lines.20 In perfect agreement with the data generated by the Spankuch lab with SBE13, we observed a similar defect during cell cycle progression in pancreatic cancer cells with an early delay in cell cycle in Pa03C cells using lower or higher GSK461364A concentration (Fig. 3A, compare FACS profiles at 3 h after release) and an arrest at G2/M phase with higher concentration (100 nM) and at the later time point (13 h). Considering that the Plk1 level begins to rise significantly in the G1/S boundary and peaks in mitosis, one would predict that a much higher concentration of Plk1 inhibitor is needed to inhibit its multiple mitotic functions but that a relatively low concentration of Plk1 inhibitor would be sufficient to induce the observed defect during G1/S transition. Therefore, we conclude that a relative lower activity of Plk1 is sufficient for cells to go through G1/S transition but a maximum Plk1 activity is needed for pancreatic cancer cells to traverse through mitosis.
Increasing evidence supports that notion that Plk1 has multiple non-mitotic functions, in particular, in cancer cells. Importantly, these newly identified functions of Plk1 strongly suggest that inhibition of Plk1 is a powerful approach to overcome resistance of many treatments. For example, we recently showed that Plk1 phosphorylates Axin2, a negative regulator of the WNT/β-catenin pathway, whose expression is significantly elevated in late stage prostate cancer, and that a combination of inhibition of Plk1 and the WNT/β-catenin pathway is a novel approach to treat late stage prostate cancer.21 In a separate line of study, we reported that Plk1 regulates androgen receptor signaling, whose elevation is essential for prostate cancer growth. Furthermore, we found that inhibition of Plk1 overcomes the resistance to androgen signaling inhibitors in castration-resistant prostate cancer.22,23 More recently, we described that Plk1 also regulates cancer metabolism as its elevation leads to a tumor-promoting metabolic state.24,25 Based on this finding, we further showed that inhibition of Plk1 enhances the efficacy of metformin in prostate cancer.26 In this work, we provide evidence to show that a combination of GSK461364A, a very specific Plk1 inhibitor, with gemcitabine offers an effective approach to treat pancreatic cancer. In summary, elevation of Plk1 in cancer might contribute to development of resistance to multiple treatments.
Materials and methods
Cell culture
Pa03C, Panc1 and Panc10.05 cells were cultured in in Dulbecco's Modified Eagle Medium supplemented with 10% (vol/vol) fetal bovine serum (FBS) at 37°C in 5% CO2. HPDE6 cells were cultured in keratinocyte medium (Invitrogen).
Reagents
Nocodazole, gemcitabine and GSK461364A were purchased from Sigma, Tocris Bioscience (Cat. 3259) and Selleckchem (Cat# S2193), respectively.
Immunoblotting (IB)
Harvested cells were lysed in TBSN buffer (20 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1.5 mmol/L EDTA, 5 mmol/L EGTA, 0.5% Nonidet P-40, and 0.5 mmol/L Na3VO4) supplemented with proteinase inhibitors. The lysates were resolved by SDS-PAGE and transferred to Whatman Westran polyvinylidenedi-fluoride (PVDF) membrane (Sigma; Z671088), followed by incubation with antibodies against Plk1 (sc-17783; Santa Cruz), β-actin (A5441; Sigma), γ-tubulin (T3559, Sigma), Lamin A/C (#4777; Cell Signaling), cleaved-PARP (AB3620; EMD Millipore), Cyclin E (sc247, Santa Cruz), and phosphor-histone H3 antibody (06-570; Upstate Biotechnology).
Cytoplasmic and nuclear protein extract preparations
Cytoplasmic and nuclear protein extracts were prepared with a kit from Active Motif (catalog no., 40410) according to the manufacturer's instructions.
FACS analysis
Cells were harvested by trypsinization, fixed in 75% ethanol, stained with propidium iodide solution at a final concentration of 50 μg/ml, and subjected to fluorescence-activated cell sorting (FACS) analysis.
BrdU labeling assay
BrdU labeling assays were performed with a kit from Roche (Cat.11170376001) according to the manufacturer's instructions.
Immunofluorescent staining (IF)
IF was performed as described previously.22 Antibody against Plk1 (sc-17783; Santa Cruz) was incubated for 1 h at room temperature, followed by incubation secondary antibody and 4,6-diamidino-2-phenylindole (DAPI, Sigma) for 1 h.
Depletion of Plk1
HEK293T cells in 10-cm dishes were co-transfected with 4 µg of pHR’-CMV-△R 8.20vpr, 2 µg of pHR’-CMV-VSV-G, and 4 µg of pLKO.1-Plk1 (nucleotide 1424 relative to the starting codon) for depletion of Plk1.12 Supernatants were collected every 12 h after 24 h post-transfection. Viruses were filtered through a 0.45-µm poresize filter, concentrated by spin at 20,000 rpm for 2 h, re-suspended in TNE buffer (50 mM Tris-HCl, pH7.8, 130 mM NaCl, 1 mM EDTA), and rotated overnight at 4°C. Infections were carried out in the presence of 10 µg/ml of polybrene and 10 mM of HEPES, followed by selection with 1 µg/ml of puromycin for at least 36 h. Alternatively, Panc1 cells were co-transfected with pBS/U6-Plk1 (the targeting sequence of human Plk1 is GGGCGGCTTTGCCAAGT-GCTT, corresponding to the coding region 183-203 relative to the first nucleotide of the starting codon) and pBabe-puro at a ratio of 10:1 using Lipofectamine 2000 reagents.13 Plasmid pBS/U6-Plk1-1st half (sense strand) was used as a control vector. This control vector produces RNA that cannot form a hairpin structure to generate interfering RNA. At 24 h post-transfection, the medium was changed, and 2 µg/ml puromycin was added to select the transfection-positive cells. After 2 d of drug selection, floating cells were washed away, and the remaining attached cells were used for phenotype analysis.
Panc1-derived orthotopic xenograft model
Panc1 cells (5 × 105 cells per mouse) were inoculated into pancreas of NSG mice. Two weeks later, animals were randomized into treatment and control groups of 6 mice each. GSK461364A was dissolved in 0.1N HCl, diluted with 0.9% NaCl, and injected into the tail vein twice weekly for 6 weeks. Gemcitabine was dissolved in 0.9% NaCl, diluted with 0.9% NaCl, and injected into the tail vein twice weekly for 6 weeks.
Histology and immunohistochemistry
Xenograft tumors were fixed in 10% neutral buffered formalin, paraffin embedded, sectioned to 5 µM, and stained using conventional hematoxylin and eosin (H&E) staining. Immunofluorescence (IFC) staining for Ki67 was accomplished with Elite Vectastain ABC kit.
Statistical analysis
The statistical significance of the results was analyzed using an unpaired Student t test (StatView I, Abacus Concepts Inc., Berkeley, CA). A P value of less than 0.05 indicates statistical significance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sandra Torregrosa-Allen for help with the mouse xenograft study.
Funding
This work was supported by NIH grant R01 CA157429 (X.L.), NIH grant R01 CA124586 (S.F.K), NIH grant R01 AR059130 (N.A.), and ACS grant RSG-13-073 (X.L.). Xenograft data were acquired by a Purdue Center for Cancer Research facility supported by P30 CA023168.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2015. CA: a cancer journal for clinicians, 2015. 65(1): 5–29; PMID:25559415 [DOI] [PubMed] [Google Scholar]
- [2].Mariotto AB, Yabroff KR, Shao Y, Feuer EJ, Brown ML. Projections of the cost of cancer care in the United States: 2010–2020. J Natl Cancer Inst 2011. 103(2): 117–28; PMID:21228314; http://dx.doi.org/ 10.1093/jnci/djq495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ewald B, Sampath D, Plunkett W. ATM and the Mre11-Rad50-Nbs1 complex respond to nucleoside analogue-induced stalled replication forks and contribute to drug resistance. Cancer Res, 2008. 68(19):7947–55; PMID:18829552; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl Oncol 2015. 8(3): p. 185–95; PMID; http://dx.doi.org/ 10.1016/j.tranon.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9(8):643–60; PMID:20671765; http://dx.doi.org/ 10.1038/nrd3184 [DOI] [PubMed] [Google Scholar]
- [6].Song B, Liu XS, Rice SJ, Kuang S, Elzey BD, Konieczny SF, Ratliff TL, Hazbun T, Chiorean EG, Liu X. Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Molecular cancer therapeutics 2013; 12(1):58–68; PMID:23188630; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Weichert W, Schmidt M, Jacob J, Gekeler V, Langrehr J, Neuhaus P, Bahra M, Denkert C, Dietel M, Kristiansen G. Overexpression of Polo-like kinase 1 is a common and early event in pancreatic cancer. Pancreatol : Off J Int Asso Pancreatol 2005; 5(2–3):259–65; PMID:15855824; http://dx.doi.org/ 10.1159/000085280 [DOI] [PubMed] [Google Scholar]
- [8].Russo MA, Kang KS, Di A. Cristofano, The PLK1 inhibitor GSK461364A is effective in poorly differentiated and anaplastic thyroid carcinoma cells, independent of the nature of their driver mutations. Thyroid 2013; 23(10):1284–93; PMID; http://dx.doi.org/ 10.1089/thy.2013.0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Song B, Liu XS, Liu X. Polo-like kinase 1 (Plk1): an Unexpected Player in DNA Replication. Cell Div 2012; 7:3; PMID:22309699; http://dx.doi.org/ 10.1186/1747-1028-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu XS, Song B, Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein Cell 2010; 1(11):999–1010; PMID:21153517; http://dx.doi.org/ 10.1007/s13238-010-0131-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ouyang H, Mou LJ, Luk C, Liu N, Karaskova J, Squire J, Tsao MS. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol 2000; 157(5):1623–31; PMID:11073822; http://dx.doi.org/ 10.1016/S0002-9440(10)64800-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26(6):2093–108; PMID:16507989; http://dx.doi.org/ 10.1128/MCB.26.6.2093-2108.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A 2003; 100(10):5789–94; PMID:12732729; http://dx.doi.org/ 10.1073/pnas.1031523100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res 2011; 17(10):3420–30; PMID:21459796; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2946 [DOI] [PubMed] [Google Scholar]
- [15].Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27–55; PMID: 6382953; http://dx.doi.org/ 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
- [16].Song B, Liu XS, Rice SJ, Kuang S, Elzey BD, Konieczny SF, Ratliff TL, Hazbun T, Chiorean EG, Liu X. Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Mol Cancer Ther 2013; 12(1):58–68; PMID:23188630; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu ZQ, Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A 2008; 105(6):1919–24; PMID:18250300; http://dx.doi.org/ 10.1073/pnas.0712063105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yata K, Lloyd J, Maslen S, Bleuyard JY, Skehel M, Smerdon SJ, Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol Cell 2012; 45(3):371–83; PMID:22325354; http://dx.doi.org/ 10.1016/j.molcel.2011.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li H, Wang Y, Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem 2008; 283(10):6209–21; PMID:18171681; http://dx.doi.org/ 10.1074/jbc.M709007200 [DOI] [PubMed] [Google Scholar]
- [20].Keppner S, Proschak E, Kaufmann M, Strebhardt K, Schneider G, Spänkuch B. Biological impact of freezing Plk1 in its inactive conformation in cancer cells. Cell Cycle 2010; 9(4):761–73; PMID:20139717; http://dx.doi.org/ 10.4161/cc.9.4.10644 [DOI] [PubMed] [Google Scholar]
- [21].Li J, Karki A, Hodges KB, Ahmad N, Zoubeidi A, Strebhardt K, Ratliff TL, Konieczny SF, Liu X. Co-targeting Polo-like kinase 1(Plk1) and the Wnt/beta-catenin signaling pathway in castration-resistant prostate cancer. Mol Cell Biol 2015; 35:4185–98; PMID:26438599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, Ahmad N, Ratliff T, Liu X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res 2014; 74(22):6635–47; PMID:25252916; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang Z, Chen L, Wang H, Ahmad N, Liu X. Inhibition of Plk1 represses androgen signaling pathway in castration-resistant prostate cancer. Cell Cycle 2015; 14(13):2142–8; PMID:25927139; http://dx.doi.org/ 10.1080/15384101.2015.1041689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li Z, Li J, Bi P, Lu Y, Burcham G, Elzey BD, Ratliff T, Konieczny SF, Ahmad N, Kuang S, et al.. Plk1 Phosphorylation of PTEN Causes a Tumor-Promoting Metabolic State. Mol Cell Biol 2014; 34:3642–61; PMID:2504783; http://dx.doi.org/ 10.1128/MCB.00814-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Z, Lu Y, Ahmad N, Strebhardt K, Liu X. Low-dose arsenic-mediated metabolic shift is associated with activation of Polo-like kinase 1 (Plk1). Cell Cycle 2015; 14:3030–9; PMID:26709750; http://dx.doi.org/ 10.1080/15384101.2015.1120924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shao C, Ahmad N, Hodges K, Kuang S, Ratliff T, Liu X. Inhibition of polo-like kinase 1 (Plk1) enhances the antineoplastic activity of metformin in prostate cancer. J Biol Chem 2015; 290(4):2024–33; PMID:25505174; http://dx.doi.org/ 10.1074/jbc.M114.596817 [DOI] [PMC free article] [PubMed] [Google Scholar]