

Abstract
Interleukin (IL)-17A exists as a homodimer (A/A) or as a heterodimer (A/F) with IL-17F. IL-17A is expressed by a subset of T-cells, called Th17 cells, at inflammatory sites. Most cell types can respond to the local production of IL-17A because of the near ubiquitous expression of IL-17A receptors, IL-17RA and IL-17RC. IL-17A stimulates the release of cytokines and chemokines designed to recruit and activate both neutrophils and memory T-cells to the site of injury or inflammation and maintain a proinflammatory state. IL-17A-producing pathogenic T-cells contribute to the pathogenesis of autoimmune diseases, including psoriasis, psoriatic arthritis, rheumatoid arthritis, and ankylosing spondylitis. This study describes the generation and characterization of ixekizumab, a humanized IgG4 variant IL-17A-neutralizing antibody. Ixekizumab binds human and cynomolgus monkey IL-17A with high affinity and binds rabbit IL-17A weakly but does not bind to rodent IL-17A or other IL-17 family members. Ixekizumab effectively inhibits the interaction between IL-17A and its receptor in binding assays and potently blocks IL-17A-induced GRO or KC secretion in cell-based assays. In an in vivo mouse pharmcodynamic model, ixekizumab blocks human IL-17A-induced mouse KC secretion. These data provide a comprehensive preclinical characterization of ixekizumab, for which the efficacy and safety have been demonstrated in human clinical trials in psoriasis and psoriatic arthritis.
Keywords: ixekizumab, IL-17A monoclonal antibody, anti-IL-17A
Video abstract
Introduction
Interleukin (IL)-17A (also known as IL-17) is the eponymous member of a new six-member family of cytokines.1 Biologically active IL-17A exists as a homodimer (A/A) or as a heterodimer (A/F) with IL-17F. Almost all cell types can respond to the local production of IL-17A because of the near ubiquitous expression of the heterodimeric receptor subunits, IL-17RA and IL-17RC. By virtue of the unique signaling properties of the IL-17R complex and the differential affinities for IL-17A homodimer, IL-17A/F heterodimer, and IL-17F homodimer, there are common but distinct responses culminating in the release of proinflammatory mediators.2 The cellular response to IL-17A is highlighted by the release of cytokines and chemokines that are designed to recruit and activate both neutrophils and memory T-cells to the site of injury or inflammation thereby maintaining a proinflammatory state. While both IL-17A and IL-17F are important in the defense against extracellular bacteria and fungi, a variety of data suggest that IL-17A has a dominant role in chronic inflammation and autoimmunity.
Current literature suggests that IL-17A has three main roles: 1) host defense against extracellular bacteria and fungi, 2) neutrophil homeostasis, and 3) chronic pathogenic inflammation.3,4 IL-17A is produced by a subset of CD4+ T-cells, called Th17 cells, that represent a third subset of CD4+ helper lymphocytes, distinct from the two classically described CD4+ Th1 and Th2 populations.5 More recently, γ/δ T-cells, subsets of innate lymphoid cells (ILC3), and mast cells have also been reported to produce IL-17A.4 Cells of the innate immune system influence the adaptive immune response, and conversely, IL-17A-secreting Th17 cells, part of the adaptive immune system, have a profound impact on the ability to generate an effective innate immune response to certain pathogens. IL-17A appears to be critical for host defense against extracellular pathogens by regulating the chemokine gradients required for neutrophil emigration into infected tissue sites, as well as granulopoiesis. IL-17A is also instrumental in inducing the production of IL-6, a potent activator of neutrophils. In addition, IL-17A can also induce the production of antibacterial peptides and mucus secretion. Many of the cell types identified that respond strongly to IL-17A stimulation, such as keratinocytes, bronchial epithelial cells, and intestinal epithelial cells, are at the interface of the internal and external environment.5 There is recent evidence in humans for increased susceptibility to recurrent staphylococcal and mucocutaneous Candida infections in the absence of normal IL-17A responses.6,7
An optimally regulated T-cell subset response results in the clearance of a pathogen and the generation of memory T-cells. Inappropriate or continuous activation of these T-cell subsets can lead to disease that is autoimmune or allergic in nature. It is now clear that IL-17A-producing pathogenic T-cells are responsible for many of the inflammatory autoimmune responses once attributed to Th1 cells, and multiple reports link aberrant Th17 response and increased IL-17A production in a variety of autoimmune diseases, including rheumatoid arthritis, psoriasis, psoriatic arthritis, and ankylosing spondylitis.5,8
Because IL-17A is produced and acts locally at the sites of inflammation, its neutralization may have the potential for an enhanced efficacy and an improved safety profile in patients with autoimmune diseases.9,10 In addition, the specific neutralization of IL-17A and not IL-17F leaves intact a vital component of host defense against extracellular pathogens. Therefore, the development of a neutralizing antibody recognizing human IL-17A was undertaken resulting in LY2439821, which began a first-in-man study in November 2006. LY2439821 is hereafter referred to as ixekizumab (pronounced as “ik see kiz oo mab”).
Materials and methods
Immunizations and screening mouse antibodies
All animal studies were conducted in accordance with, and approved by, the research guidelines of Eli Lilly and Company (Indianapolis, IN, USA) Animal Care and Use Committee. Mice were immunized with carrier-free recombinant human IL-17A (R&D Systems, Minneapolis, MN, USA), and splenic B-cells were isolated to generate a fragment antigen-binding (Fab)-expressing phage display library using standard DNA techniques. Recombinant Fabs were screened for binding and neutralization of human IL-17A. The Fab genes were sequenced, and a subset was expressed, purified, and characterized for their affinity, selectivity, and neutralization. None of the Fabs bound to mouse or rat IL-17A. Four Fabs were converted to chimeric monoclonal antibodies (mAbs) using the human IgG4 backbone and further tested for selectivity, cell-based neutralization, affinity, and binding to human and cynomolgus monkey IL-17A. Clone 2321 was chosen for humanization and optimization; the detailed methods are described in the Supplementary materials.
Expression and purification
Ixekizumab heavy-chain and light-chain variable regions were cloned into expression plasmids, pEE6.4 and pEE12.4, respectively. The vector backbones are used in the GS Gene Expression System™ (Lonza Biologics, Slough, UK). Ixekizumab was expressed in Chinese hamster ovary cells. Antibodies used in these studies were purified by Protein A affinity chromatography and size exclusion chromatography.
Affinity determination
Affinity of ixekizumab for IL-17A for human, cynomolgus monkey, mouse, rat, and rabbit was determined with the surface plasmon resonance (SPR) using a Biacore biosensor 2000 (Biacore Life Sciences, Pittsburgh, PA, USA). Mouse IL-17A was obtained from R&D Systems, and all other species of IL-17A were made in-house. Ixekizumab was captured by Protein A using a CM4 biosensor chip with the amine-coupling chemistry to yield at least 400 response units (GE Healthcare Bio-sciences Corp., Piscataway, NJ, USA). Biacore experiments were performed at 25°C in HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.005% P20, and 3 mM EDTA). The fitting parameters to determine on- and off-rates were obtained from global fit of duplicate runs using seven different concentrations of IL-17A with a 1:1 binding with mass transfer model using the BIAevaluation software. The concentration range for human and cynomolgus monkey IL-17A started from 10 nM in twofold serial dilutions. Seven different concentrations of rabbit IL-17A were used, starting from 100 nM in twofold dilutions. The binding of rabbit IL-17A is biphasic; therefore, a heterogeneous ligand model was used to fit the data. For mouse and rat IL-17A, the concentrations of 200 nM or 2 µM were used, respectively. At least three independent experiments were performed to calculate the standard deviation for the fitting parameters.
The affinity of ixekizumab to human IL-17A or human IL-17A/F (KD = koff/kon) was also measured at 37°C using the similar methodology. Ixekizumab was captured by Protein A using a CM5 biosensor chip with the amine-coupling method to yield a total of 120–135 response units (GE Healthcare Bio-sciences Corp.). Multiple cycles of increasing concentrations of IL-17A or IL-17A/F were then injected over flow cells (0.625–10 nM) followed by a regeneration step using glycine HCl (pH 1.5) between each cycle. Each cycle includes an antibody capture step followed by the injection of IL-17A or IL-17A/F at a single concentration with a 25–30-minute dissociation period, followed by regeneration (flow rate =100 µL/min). The assay was performed in triplicate with both IL-17A and IL-17A/F.
Specificity enzyme-linked immunosorbent assay
IL-17A family member recombinant proteins and human recombinant IL-22 (R&D Systems) were coated into individual enzyme-linked immunosorbent assay (ELISA) plate wells at 0.5 µg/mL (50 ng per well) in duplicate at 4°C for 16 hours. Ixekizumab was then added at 0 µg/mL, 0.016 µg/mL, 0.08 µg/mL, 0.4 µg/mL, 2 µg/mL, or 10 µg/mL. The plates were incubated at room temperature for 2 hours and washed four times with the washing buffer. The plates were then incubated with HRP-conjugated secondary antibody. The colorimetric signal was developed and measured using an ELISA plate reader. The degree of ixekizumab binding to the tested proteins was proportional to the color signal production. The assay was repeated two times.
SPR for ligand/receptor interactions
Binding of human IL-17A to ixekizumab or human IL-17RA/Fc fusion (R&D Systems) was measured using a Biacore T100 system (Biacore Life Sciences) with a CM5 chip immobilized with human IL-17A using the amine-coupling chemistry (GE Healthcare Bio-sciences Corp.). All experiments were performed at 25°C with a flow rate of 5 µL/min. Binding of human IL-17RA/Fc to human IL-17A on the chip surface was demonstrated by injecting 50 µL of human IL-17RA/Fc at 1 µM. After regeneration, 50 µL of ixekizumab at 500 nM was added nearly saturating the human IL-17A on the surface. To determine whether ixekizumab blocks human IL-17A binding to human IL-17RA, ixekizumab at 500 nM was first injected to nearly saturate the human IL-17A surface followed by an injection of human IL-17RA/Fc at 1 µM.
Cell-based neutralization assays
IL-17A is a cytokine that can stimulate fibroblasts and other cells to secrete human growth-regulated oncogene (GRO)α or keratinocyte chemoattractant (KC) human or mouse CXCL1, respectively. The human colorectal adenocarcinoma epithelial cell line HT-29 was used for testing ixekizumab neutralization of human IL-17A, human IL-17A/F heterodimer, and cynomolgus monkey IL-17A. HT-29 cells (ATCC #HTB-38) were treated with a constant amount of human IL-17A (60 ng/mL =1,875 pM), cynomolgus monkey IL-17A (60 ng/mL =1,618 pM), or human IL-17A/F heterodimer (1,000 ng/mL =32,573 pM) in the presence of either ixekizumab or control human IgG4 at the indicated concentrations. A dose range of ixekizumab or control antibody from 2.56 pM to 40,000 pM was evaluated. After ∼48 hours, GROα in the culture media was measured using a commercial ELISA kit (#DY275; R&D Systems). Ixekizumab was tested in the HT-29 assay seven times for the ability to neutralize human IL-17-induced GROα and twice for the ability to neutralize cynomolgus monkey IL-17-induced GROα. Medium alone treatments were included in every experiment to assess the basal levels of GROα. Percent inhibition was calculated by subtracting the mean IL-17A or F alone GROα values and then dividing by the IL-17A or F alone value (subtracted from the basal GROα level); 100% neutralization of IL-17 reflects the ability of ixekizumab to bring the GROα values down to the medium alone-treated GROα values.
Mouse 4T1 cells (BALB/cfC3H mammary gland epithelial tumor cells, ATCC #CRL-2539) were maintained in culture/assay medium in tissue culture-treated flasks using the standard techniques. Recombinant mouse IL-17A (R&D Systems) was used as a positive control for these experiments. The supernatants of mutant IL-17 clones from transient transfections of 293 cells were diluted in assay/culture medium to a final concentration of 20 ng/mL based on the concentration determined by an IL-17A ELISA (#DY317; R&D Systems). Plates containing IL-17A and antibody mixtures were incubated for 2.5 hours at 37°C, 95% relative humidity, and 5% CO2, before adding the 4T1 cells. The plates were placed in a tissue culture incubator (37°C, 95% relative humidity, and 5% CO2) for ∼72 hours. KC levels were measured in media using a mouse KC sandwich ELISA (#DY453; R&D Systems), as per the manufacturer’s instructions. All samples were run in duplicate.
Pharmacodynamic assay in mice
Ixekizumab was administered intravenously to C57BL/6 NHsd mice (n=5 per group, 8–12-week old; Harlan, Indianapolis, IN, USA) 1 hour prior to a subcutaneous (SC) injection of human IL-17A. At 2 hours post-IL-17A administration, blood samples were collected, and CXCL1 chemokine level in the plasma was determined by ELISA. Human IgG4 was used as an isotype control antibody. The pairwise comparison of KC chemokine levels between treatments was performed using the one-way analysis of variance. The unadjusted P-values were reported. The study was repeated twice.
Pharmacokinetic profile of ixekizumab in male cynomolgus monkeys
Serum Pharmacokinetics (PK) of ixekizumab in cynomolgus monkeys was determined after intravenous (IV) and SC administrations in male cynomolgus monkeys. Concentrations of ixekizumab were determined using an antigen-capture ELISA assay, in which plates were coated with human IL-17A, and bound antibody was detected using an anti-IgG4 secondary antibody.
Results
Ixekizumab generation
IL-17A is a potent cytokine and acts locally at the sites of inflammation. A therapeutic antibody against such a target needs high specificity, optimal affinity, low immunogenicity, and good physical/chemical properties to achieve clinical efficacy. The initial murine antibodies were generated from immunizations and screened for the neutralization of human IL-17A, for affinity by SPR, and for no binding to other IL-17 family members as well as rodent IL-17A by ELISA. The top murine mAb, clone 2321 with high affinity and specificity for human IL-17A, was humanized and optimized for affinity and biophysical properties as described in detail in the Supplementary materials.11 Briefly, the engineering process led to a panel of high-affinity antibodies (KD <10 pM and off-rate <2×10−5/s). The amino acid sequences of the variable regions for the mouse parent antibody (clone 2321) and ixekizumab are summarized in Figure S1A. During the humanization process, the variable region frameworks and entire constant regions were converted to human germline sequences.11 Comparison of the complementarity-determining region (CDR) residues within the 51 known human variable heavy and 40 human variable kappa germline sequences revealed that only 12 amino acids are of nongermline origin. The percentage of human germline sequence present in ixekizumab is 98.2% (Figure S1B).
Human IgG4 can form two half-antibodies (half-mers), each with a single heavy and light chain that can recombine, resulting in Fab-arm exchange in vivo.12 To prevent half-antibodies, a serine-to-proline mutation was engineered into the hinge region connecting the CH1 and CH2 of the heavy chain.13 This specific mutation makes the hinge more similar to that of the human IgG1 subclass and minimizes the antibody heterogeneity.
Binding affinity of ixekizumab to human and cynomolgus monkey IL-17A
The binding affinity and kinetics of ixekizumab were determined by SPR at 25°C. Ixekizumab bound human and cynomolgus monkey IL-17A with comparable affinity. The equilibrium dissociation constants (KD) of ixekizumab for human and cynomolgus monkey IL-17A were 1.8 pM and 0.8 pM, respectively (Table 1). Ixekizumab also bound to rabbit IL-17A, but the affinity was lower, and the binding was heterogeneous (KD of 1.3 nM and 14 nM; Table 1). However, ixekizumab showed no binding to either mouse or rat IL-17A.
Table 1.
In vitro binding parameters of ixekizumab to human, cynomolgus monkey, and rabbit IL-17A determined using surface plasmon resonance
| IL-17A | kona (1/Ms) | koffa (1/s) | KDa,b (M) |
|---|---|---|---|
| Human | 7.5 (±1.4) ×106 | 1.3 (±0.8) ×10−5 | 1.8 (±1.1) ×10−12 |
| Cynomolgus monkey | 7.9 (±0.3) ×106 | 0.7 (±0.9) ×10−5 | 0.8 (±1.1) ×10−12 |
| Rabbitc | 1.5 (±0.6) ×105 | 1.7 (±0.5) ×10−4 | 1.3 (±0.6) ×10−9 |
| 9 (±3) ×106 | 1.1 (±0.2) ×10−1 | 14 (±4) ×10−9 |
Notes:
The dissociation constants for human and cynomolgus monkey are comparable within the error of measurements from Biacore. The values reported were averages ± SD calculated from at least three independent measurements.
KD were calculated using koff/kon for each measurement, and the final values were averages of independent measurements. koff, dissociation rate constant or off-rate constant; kon, association rate constant or on-rate constant.
Binding was biphasic, and data were fit with heterogeneous ligand models. KD, equilibrium dissociation constant.
Abbreviations: IL, interleukin; SD, standard deviation.
In addition, at 37°C, ixekizumab produced a concentration-dependent binding response with human IL-17A or IL-17A/F and saturation of binding occurred at a concentration of 10 nM. Under the conditions tested, the binding affinity (KD) of human IL-17A or IL-17A/F to ixekizumab was <3 pM.
Ixekizumab is specific for IL-17A
IL-17A belongs to the IL-17 family of cytokines along with IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F. These secreted homodimeric proteins differ in both amino acid sequence and structure. At the amino acid level, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F share 20%–50% homology to IL-17A. A study was designed to determine the binding of ixekizumab to human IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, IL-17F, mouse IL-17A, and a structurally unrelated cytokine human IL-22. Using an ELISA plate coated with the various cytokines, ixekizumab bound to human IL-17A but not to human IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F, mouse IL-17A, or human IL-22 (Figure 1). These data demonstrated that ixekizumab is highly specific for IL-17A.
Figure 1.

Ixekizumab binds specifically to IL-17A.
Notes: Human IL-17 family member proteins, mouse IL-17A, and human IL-22 were coated into individual ELISA plate wells. Ixekizumab was then added at varying concentration up to 10 µg/mL. The absorbance at 450 nm represents the average values and standard error obtained from duplicate determinations at each concentration of ixekizumab bound to the test proteins.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IL, interleukin.
Ixekizumab blocks IL-17A binding to IL-17RA
SPR was used to determine if ixekizumab can block the binding of human IL-17A to the human IL-17RA subunit. As shown in Figure 2, ixekizumab or human IL-17RA/Fc bound immobilized human IL-17A on the chip surface, independently. However, once human IL-17A was bound to ixekizumab, binding of human IL-17A to human IL-17RA was completely blocked. This confirms that ixekizumab prevents human IL-17A binding to human IL-17RA.
Figure 2.

Ixekizumab competes with IL-17RA binding to immobilized human IL-17A.
Notes: Human IL-17A was immobilized on a Biacore chip surface. Binding of IL-17RA/Fc to immobilized IL-17A was shown as increase in response units upon injection of IL-17RA/Fc (A). Ixekizumab bound to immobilized IL-17A as shown by the increase in response units following injection (B). Upon binding of ixekizumab to IL-17A, IL-17RA/Fc can no longer bind (B).
Abbreviations: IL, interleukin; RU, response units; Resp Diff, the difference in RU between active flow cell and reference flow cell.
Ixekizumab neutralizes IL-17A homodimers and IL-17A/F heterodimers in a cell-based assay
IL-17A can stimulate epithelial cells to secrete the chemokine GROα. HT-29 is a human colorectal adenocarcinoma epithelial cell line. To determine if HT-29 cells secrete GROα in response to IL-17A or IL-17A/F, dose–response titrations of recombinant human IL-17A or IL-17A/F were tested. Figure 3A shows that human IL-17A and IL-17A/F induced GROα secretion from HT-29 cells. Ixekizumab inhibited human IL-17A- or human IL-17A/F heterodimer-induced GROα secretion from HT-29 cells in a dose-dependent fashion, whereas control human IgG4 did not (Figure 3B). In addition, ixekizumab inhibited cynomolgus monkey IL-17A-induced GROα secretion from HT-29 cells in a dose-dependent fashion, whereas control human IgG4 did not cause a decrease in IL-17-induced GROα secretion (Figure 3C). Both human and cynomolgus monkeys are neutralized 100% by ixekizumab. The data shown in Figure 3B and C are from separate experiments; consistent with these data in additional head-to-head experiments, human and cynomolgus monkey IL-17A were neutralized equivalently.
Figure 3.

Ixekizumab neutralizes human IL-17A, human IL-17A/F heterodimer, and cynomolgus monkey IL-17A.
Notes: (A) HT-29 cells were treated with a dose range of human IL-17A or IL-17A/F for ∼48 hours. GROα was measured in the culture medium by ELISA. (B) HT-29 cells were treated with a constant amount of either human IL-17A (60 ng/mL =1,875 pM) or human IL-17A/F (1,000 ng/mL =32,573 pM), in the presence of either ixekizumab or control human IgG4 at the indicated concentrations. After ∼48 hours, GROα in the culture media was measured by ELISA. (C) HT-29 colon cancer cells were treated with a constant amount of cynomolgus monkey IL-17 (60 ng/mL), in the presence of either ixekizumab or control human IgG4 at the indicated concentrations for 48 hours, and GROα in the culture media measured by ELISA. Results in (A) and (B) are shown as the mean of triplicate treatments ± SD and are representative of seven independent experiments. Results in (C) are shown as the mean of triplicate treatments ± SD and are representative of two independent experiments. Human and cynomolgus monkey IL-17A were tested for neutralization in separate experiments.
Abbreviations: cyno, cynomolgus; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; SD, standard deviation; GRO, human growth-regulated oncogene.
These results demonstrate that ixekizumab is able to neutralize human and cynomolgus monkey IL-17A in vitro, indicating that ixekizumab could be tested in human and cynomolgus monkey in vivo.
Epitope identification for ixekizumab
To understand the interaction between ixekizumab and human IL-17A, studies were undertaken to determine the epitope targeted by this antibody. Using hydrogen–deuterium exchange mass spectrometry, the predominant epitope potentially contributing to the antibody-binding site for ixekizumab was determined to include amino acids 80–87 and amino acids 24–54 (Supplementary materials). Consistent with the hydrogen–deuterium exchange mass spectrometry data, ixekizumab was unable to recognize IL-17A in a reduced, denatured state, suggesting that the epitope was conformational and nonlinear (data not shown).
To further define the binding epitope, mutational analysis was performed with human IL-17A. Ixekizumab does not bind rodent IL-17A, and interestingly, the alignment of the amino acid sequences for human, cynomolgus monkey, mouse, and rat IL-17A revealed the differences between primates and the two rodent species within the putative epitope regions. Multiple mutants of human IL-17A were designed (Figure S2A) and tested for the ability to induce the secretion of KC (mouse equivalent of human GROα) from mouse 4T1 cells because human IL-17A can bind to the mouse IL-17 receptor. These data provided evidence that the mutations did not substantially affect either the overall protein structure or its activity (Figure S2B, isotype control data). Ixekizumab was tested for the ability to block KC secretion induced by these IL-17A mutants. Mutant h1 that converted the amino acids located within 81–87 (DGNVDYH) to those found in mouse IL-17A (D80E/N82K/V83L/Y85H) was still able to induce KC secretion in the presence of ixekizumab in the 4T1 cell-based assay (Figure 4A), indicating that these mutations sufficiently altered the epitope such that it no longer bound ixekizumab. Conversely, five other mutants located in the region from amino acids 27 to 52 still bound ixekizumab (Figure S2B), indicating that either the major contact points between the antibody and antigen are outside these regions or these changes to mouse amino acids do not perturb contact points with ixekizumab sufficiently to affect binding and neutralization. The mutants were also tested in HT-29 cells. Results were very similar to those in 4T1 cells (data not shown).
Figure 4.

Ixekizumab epitope.
Notes: (A) 4T1 cells were treated with a constant amount of human IL-17A wild type, mutant h1, or vector control supernatant in the presence of increasing amounts of ixekizumab (closed symbols) or isotope control antibody (open symbols). After 48 hours, KC in the supernatant was measured by ELISA. Results are shown as the mean of triplicate treatments ± SD and are representative of four independent experiments. (B) Structure of the IL-17A:IL-17RA complex (4hsa) with key amino acid residues in the epitope for ixekizumab highlighted. IL-17RA is colored in magenta, and the IL-17A dimer subunits are colored in cyan and green. The key epitope region (DGNVDYH) in IL-17A for ixekizumab is highlighted in yellow or brown in each subunit of the cytokine. This figure was generated using the PyMOL Molecular Graphics System (Version 1.7.0.3; Schrödinger, LLC).
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IL, interleukin; SD, standard deviation; KC, keratinocyte chemoattractant.
The structure of the IL-17A:IL-17RA complex (4hsa)14 with the key amino acids comprising the epitope for ixekizumab is highlighted in Figure 4B. The IL-17A dimer is shown in cyan and green, and IL-17RA is colored in magenta. The key epitope region (DGNVDYH) in IL-17A for ixekizumab is highlighted in yellow or brown on each subunit of the cytokine. Although it is possible that mutations made in mut_h1 can disrupt the interaction with ixekizumab either directly or indirectly through an effect caused by global structural alteration, our data indicate that the mutations likely disrupt the interaction with ixekizumab directly. The mut_h1 protein can still interact with the receptor and cause KC production by the 4T1 cells, indicating that it is biologically active (Figure 4A, isotype control data). The epitope is on the edge of the interface of IL-17A and IL-17 receptor, and ixekizumab binding efficiently competes with the IL-17 receptor for binding to human IL-17A, explaining the ability of ixekizumab to block the binding of IL-17RA and neutralize the IL-17A activity.
PK and pharmacodynamic studies
Serum PK of ixekizumab was determined after IV and SC administrations in male cynomolgus monkeys. Following IV administration of 1 mg/kg, ixekizumab was eliminated with a mean half-life of 6.5 days (Figure S3). After SC administration of 1 mg/kg, ixekizumab reached an average maximal plasma concentration of 11.1 µg/mL ∼72 hours postdose. The mean elimination half-life following the SC injection was 10.3 days (Figure S3). The results indicate that ixekizumab has very high bioavailability from the SC site. The cynomolgus monkey PK profile and clearance values for ixekizumab (Figure S3) were typical for an antibody whose elimination is not mediated by target. These results suggested no formation of significant antidrug antibody during the course of the experimental period. As a result, we did not directly measure for antidrug antibody in the preclinical studies.
Following the administration of ixekizumab to normal cynomolgus monkeys, no pharmacodynamic changes were observed. As demonstrated earlier, using in vitro assays, human IL-17A will bind and activate mouse IL-17 receptor. In order to develop an in vivo assay to evaluate ixekizumab neutralization, human IL-17A was injected into mice, and changes in cytokine levels were evaluated. The administration of human IL-17A to C57BL/6 mice increased the mouse KC levels in plasma in a time- and dose-dependent fashion. Ixekizumab was able to decrease human IL-17A-induced KC secretion in the plasma of the C57BL/6 mice in a dose-dependent manner (Figure 5). There was a statistically significant reduction in plasma KC when the mice were given 20 µg of ixekizumab compared to isotype control treatment. These results demonstrate that ixekizumab was able to neutralize human IL-17A in vivo.
Figure 5.
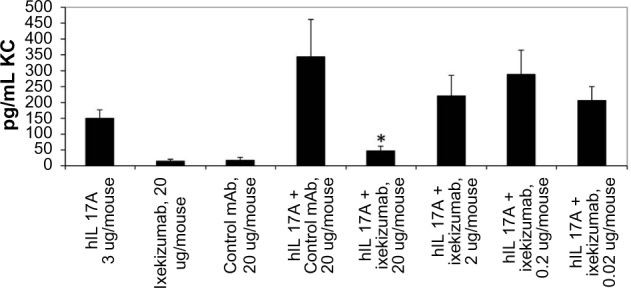
Ixekizumab inhibits human IL-17A-induced KC secretion in a mouse PD model.
Notes: Human IL-17A (3 µg) was administered to mice SC 1 hour after IV injection of 1 mg/kg, 0.1 mg/kg, 0.01 mg/kg, or 0.001 mg/kg ixekizumab (corresponding to 20 µg, 2 µg, 0.2 µg, and 0.02 µg per mouse, respectively). KC levels were determined by ELISA 2 hours posthuman IL-17A injection. n=5 mice per group. One-way ANOVA was used for the pairwise comparison of KC chemokine levels between treatments. *The unadjusted P-value is 0.0012 comparing the IL-17A +20 µg control mAb group and the IL-17A +20 µg ixekizumab group. The figure represents one experiment of two separate studies.
Abbreviations: SC, subcutaneous; ANOVA, analysis of variance; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; IV, intravenous; KC, keratinocyte chemoattractant; mAb, monoclonal antibody; PD, pharmacodynamic.
Discussion
This report describes the generation and characterization of ixekizumab, a new humanized anti-IL-17A mAb. The humanization process used to engineer ixekizumab included steps for optimizing its affinity for IL-17A,11 resulting in a molecule with a very high binding affinity (KD <3 pM). Ixekizumab binds to human and cynomolgus monkey IL-17A but does not bind to mouse or rat IL-17A. Ixekizumab directly blocks IL-17A binding to IL-17RA but does not bind to other IL-17 family members.
Ixekizumab is a human IgG4 variant antibody with a specific serine-to-proline change engineered into the hinge region connecting the CH1 and CH2 of the heavy chain. This specific mutation makes the hinge more similar to that of the human IgG1 subclass and has been shown to prevent half-mer formation.12 Immune activation is not a part of the intended mechanism of action for ixekizumab; therefore, it was constructed as an IgG4 isotype, known to have low binding ability to Fcγ receptors or components of the complement system. Using a binding assay for Fc receptors and C1q, ixekizumab does not bind to any of the Fcγ receptors. Therefore, it is highly unlikely that ixekizumab will be able to induce Fc-mediated biological effects in vivo, reducing the potential for effector function.
Ixekizumab was humanized to reduce the potential for immunogenicity during chronic administration to humans. Often investigators view human antibodies in a more favorable light compared to humanized not realizing that those labels simply connote the means by which the antibody was generated and have nothing to do with the potential for immunogenicity in human.15 All antibodies, regardless of their origin, have the same overall molecular modules such that the variable region is composed of framework regions (FR) and three CDRs.11 It is the CDRs that bind antigen, and they are structurally supported by the FR. All FR in ixekizumab are of human germline origin, meaning that they are native antibody sequences for all humans. As expected, the 12 amino acids that are not of germline origin are in the CDRs, since this is the most diverse region of an antibody due to the direct interaction with a specific antigen, in this case, human IL-17A. Both human and humanized therapeutic antibodies can offer patients excellent therapeutic options, yet each has the potential for immunogenicity due to unique CDRs.16
While IL-17A is an important cytokine involved in host defense against extracellular bacteria and fungi, several lines of evidence suggest that IL-17A plays a critical role in the pathophysiology of autoimmune diseases, such as psoriasis, psoriatic arthritis, ankylosing spondylitis, and rheumatoid arthritis.8 Ixekizumab has completed Phase III clinical trials in moderate-to-severe plaque psoriasis showing rapid, significant, dose-dependent improvements in clinical endpoints, quality of life scores, and itching severity.9,17 Skin biopsies of psoriatic lesions taken pre- and post-ixekizumab treatment showed that there were significant dose-dependent reductions from baseline in keratinocyte proliferation, hyperplasia, epidermal thickness, infiltration into the dermis and epidermis by T-cells and dendritic cells, and keratinocyte expression of innate defense peptides at 2 weeks.18 Clearly, for most patients with psoriasis, neutralization of a single cytokine, IL-17A, is necessary and sufficient for rapid and clinically meaningful benefit.9 Ixekizumab has also demonstrated efficacy in rheumatoid arthritis19 and is currently in clinical development for psoriatic arthritis.
Other antibodies targeting the IL-17 pathway are currently in clinical development. Brodalumab is a human IgG2 that binds the receptor subunit IL-17RA shared by IL-17A, IL-17C, IL-17E, IL-17F, and IL-17A/F heterodimer and has been evaluated in Phase II studies in psoriasis and psoriatic arthritis.8 The roles of IL-17C and IL-17E in psoriasis and other autoimmune conditions have not been established. Secukinumab is a human IgG1 mAb that neutralizes IL-17A and was recently approved for the treatment of moderate- to-severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy20 and has been tested in rheumatoid arthritis21 and ankylosing spondylitis.22
Conclusion
Ixekizumab (LY2439821) is a humanized IgG4 mAb with high affinity and specificity for IL-17A. Its high binding affinity to IL-17A translates to high potency in blocking the IL-17A-mediated biological activities in vitro and in vivo. Ixekizumab has shown a high-level clinical efficacy in patients with moderate-to-severe plaque psoriasis.
Supplementary materials
Humanization and optimization of ixekizumab
The goal of the engineering process was to generate a stable, high-affinity antibody with a potent interleukin (IL)-17A neutralization containing completely human germline frameworks to minimize the potential immunogenicity for chronic administration. Beginning with a murine anti-human IL-17A monoclonal antibody (clone 2321) with high affinity and specificity for human IL-17A, human heavy- and light-chain frameworks were selected for the synthesis of humanization libraries based on the V-region analysis and canonical structure homology. Libraries were constructed in a bacteriophage M13 expression vector containing human constant regions and frameworks fused with completely randomized CDRH3 and CDRL3 variant sequences based on clone 2321.1–3 In addition, a deamidation site (NG motif) present in CDRL1 of clone 2321 was simultaneously randomized while excluding the parental asparine residue. The diversity and quality of the library was determined by the sequence analysis of random clones. This library was initially screened by capture lift using 1 nM biotinylated human IL-17A to identify humanized Fab variants retaining the binding activity. Variants of interest were sequenced, and unique clones were further characterized by titration on immobilized antigen in a dissociation-driven enzyme-linked immunosorbent assay-based format.
Based on both the relative binding affinity and the chemical nature of the selected mutations, an initial humanized Fab clone (B3) was chosen for an additional round of affinity optimization. The process consisted of the following: 1) single residue complete randomization and screening of all CDR amino acids, 2) sequence analysis of beneficial mutations, 3) enzyme-linked immunosorbent assay titration of unique clones, and 4) combination of beneficial mutations in a combinatorial library.1–3 The combinatorial library was screened using the reduced (200 pM) antigen and included a prolonged wash step to enhance the resolution of clones with slower dissociation rates, yielding a panel of high-affinity humanized Fabs. Six Fabs with diverse sequence were converted into full-length human IgG4 variant with an improved stability and expressed and purified to homogeneity for further characterization.
Binding epitope analysis
The epitope of ixekizumab was mapped by hydrogen–deuterium exchange mass spectrometry (H/DXMS). Hydrogen–deuterium exchange reaction of human IL-17A was carried out with or without ixekizumab. IL-17A regions that gained less deuterium in the presence of ixekizumab than in its absence were identified as the epitope(s) for the antibody. Site-directed mutagenesis was then performed to verify the key region of epitope identified by H/DXMS.
Exchange experiments
Antigen/antibody complex was prepared by mixing 10 µg aliquot of IL-17A-Flis (human IL-17A FLAG/His tagged) and 100 µg of ixekizumab with a molar ratio of IL-17A/mAb of 1:2 in a Microcon centrifugal filters (EMD Millipore, Billerica, MA, USA), in duplicate. A third Microcon centrifugal filter contained 20 µg of IL-17A-Flis solution; however, no antibody was added. Phosphate buffered saline (PBS) was added to bring the total volume to ∼180 µL for each Microcon centrifugal filter. Each Microcon filter was centrifuged at 14,000× g for 14 minutes followed by the addition of 150 µL of PBS and then centrifuged for a second time at 14,000× g for 14 minutes. The protein portion was collected, and the final volume was adjusted to 50 µL for the complex or 80 µL for IL-17A-Flis only by the addition of PBS. Six microliters of IL-17A-Flis or the complex of IL-17A-Flis with ixekizumab were mixed with 14 µL of 100% D2O (Cambridge Isotope Laboratories, Inc., Andover, MA, USA) (70% D2O solution). The solution was incubated at ambient temperature for 10 minutes.
Peptic digest
The exchange was immediately quenched with 20 µL of 1% formic acid solution and then digested with 2 µL of 2 mg/mL pepsin solution (Sigma-Aldrich Co., St Louis, MO, USA) at ambient temperature for 30 seconds or at 0°C for 10 minutes. The digest was immediately injected onto the liquid chromatography (LC) column manually.
LC/mass spectrometry
Waters 2795 high-performance LC (HPLC) (Waters, Milford, MA, USA) and Micromass LCT Premier (Waters) were used for all analyses. HPLC stream from HPLC pump was connected to a metal tube (∼1 mL), to a manual injector, to a Zorbax C18 column (2.1 mm ×50 mm) (Agilent Technologies, Santa Clara, CA, USA), and then to mass spectrometry (MS) The metal tube, injector loop, and column were submerged in ice water during experiments. A gradient with the mobile phases consisted of 0.15% formic acid in H2O and 0.12% formic acid in acetonitrile was applied over 23 minutes. The MS was performed using a Micromass LCT Premier Mass Spectrometer with a positive spray and the following settings: a cone voltage of 80 V, a mass scan range of 300–2,000 mass to charge ratio, a desolvation temperature of 300°C, and a desolvation gas flow of 500 L/h.
Data processing
Mass spectrum of each peptic peptide of IL-17A was obtained after hydrogen–deuterium exchange with or without ixekizumab. For small peptides, average mass of each peptide was calculated based on its isotopic ions and intensities. For larger peptides, their average masses were obtained from deconvoluted mass spectra after internal calibration.
Major mass difference of IL-17A-Flis peptide between the complex and free IL-17A-Flis protein was found for the peptic peptides 24–87+117–133 and 66–87+117–134. The key epitope was further narrowed down to amino acid residues DGNVDYH based on the mutagenesis analysis.
Site-directed mutagenesis
To further narrow down the key amino acids that ixekizumab binds to, multiple mutants of human IL-17A were designed that incorporated key amino acid residues from mouse IL-17A in these regions (Figure S2A). Supernatants generated from transiently transfected HEK293 cells containing each of the IL-17A mutants were analyzed for interaction with ixekizumab. Since ixekizumab is known to neutralize human IL-17A, if ixekizumab binds an IL-17A mutant, this would result in a dramatic decrease in the amount of KC produced by the 4T1 cells. Of these six mutants, only mutant h1 was still able to induce KC secretion in the presence of ixekizumab, suggesting that these mutations sufficiently altered the epitope such that it no longer bound the antibody (Figure S2B). This mutant contained four amino acid changes over the span of six amino acids and is within the major region identified through H/DXMS. Ixekizumab was able to bind and neutralize the other five mutant IL-17A proteins. This indicates that either the major contact points between the antibody and antigen are outside these regions or these changes to the equivalent mouse amino acids do not perturb the contact points with ixekizumab sufficiently to affect binding and neutralization. The same mutants were also tested in human HT-29 cells (data not shown), and the results were consistent that the amino acids in the region of mutant h1 (DGNVDYH) provide the major contact points for ixekizumab.
Ixekizumab humanization.
Notes: (A) Variable region sequence alignment of selected human germline genes, VH1–69 and VκA18, parental mouse antibody 2321, and ixekizumab. Germline homologous residues of 2321 are indicated by a dash. CDR residues are underlined, and Kabat antibody numbering system used. The CDR residues that differ from VH1–69 or VκA18 germline sequence are shown in green (different from VH1–69 or VκA18 but present in other human germline sequences) and red (not present in any human germline sequences). (B) Calculation of homology to human germline. ‡Excluding HCDR3 and *including CDR residues present at the homologous location in other human germline genes.
Abbreviations: CDR, complementarity-determining region; mAb, monoclonal antibody; VH, variable heavy; Vκ, variable kappa; N/A, not applicable.
Ixekizumab epitope mapping.
Notes: (A) Mutagenesis of the six regions of the potential binding sites. (B) 4T1 cells were treated with a constant amount of human IL-17A wild type, mutant, or vector control supernatant in the presence of increasing amounts of ixekizumab (closed symbols) or isotope control antibody (open symbols). After 48 hours, KC in the supernatant was measured by ELISA. Results are shown as the mean of triplicate treatments ± standard deviation and are representative of four independent experiments.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IL, interleukin; KC, keratinocyte chemoattractant.
Serum pharmacokinetic profile of ixekizumab in male cynomolgus monkeys.
Notes: Following IV administration of 1 mg/kg, ixekizumab was eliminated with a mean half-life of 6.5 days. After SC administration of 1 mg/kg, it reached an average maximal plasma concentration of 11.1 µg/mL ∼72 hours postdose. The mean elimination half-life following subcutaneous injection was 10.3 days. The figure illustrates mean curve and data from individual animals (n=2).
Abbreviations: IV, intravenous; SC, subcutaneous.
References
- 1.Huse WD, Sastry L, Iverson SA, et al. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989;246(4935):1275–1281. doi: 10.1126/science.2531466. [DOI] [PubMed] [Google Scholar]
- 2.Wu H, Beuerlein G, Nie Y, et al. Stepwise in vitro affinity maturation of Vitaxin, an alphav beta3-specific humanized mAb. Proc Natl Acad Sci U S A. 1998;95(11):6037–6042. doi: 10.1073/pnas.95.11.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu H, Nie Y, Huse WD, Watkins JD. Humanization of a murine monoclonal antibody by simultaneous optimization of framework and CDR residues. J Mol Biol. 1999;294(1):151–162. doi: 10.1006/jmbi.1999.3141. [DOI] [PubMed] [Google Scholar]
Acknowledgments
The authors thank Angela Okragly, Ningjie Nancy Hu, Nicole New, Robert Benschop, Stuart Bright, and Yong Wang for their technical assistance. The authors also thank Yun-fei Chen for the statistical analysis.
Footnotes
Funding
This study was funded by Eli Lilly and Company.
Disclosure
All authors are the employees of Eli Lilly and Company. The authors report no other conflicts of interest in this work.
References
- 1.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hot A, Zrioual S, Toh M-L, Lenief V, Miossec P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis. 2011;70(2):341–348. doi: 10.1136/ard.2010.132233. [DOI] [PubMed] [Google Scholar]
- 3.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22(3):285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 5.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 6.Cypowyj S, Picard C, Marodi L, Casanova JL, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol. 2012;42(9):2246–2254. doi: 10.1002/eji.201242605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe. 2012;11(5):425–435. doi: 10.1016/j.chom.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths CEM, Reich K, Lebwohl M, et al. UNCOVER-2 and UNCOVER-3 Investigators Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386(9993):541–551. doi: 10.1016/S0140-6736(15)60125-8. [DOI] [PubMed] [Google Scholar]
- 10.Langley RG, Elewski BE, Lebwohl M, et al. ERASURE Study Group. FIXTURE Study Group Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med. 2014;371(4):326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 11.Vasserot AP, Dickinson CD, Tang Y, Huse WD, Manchester KS, Watkins JD. Optimization of protein therapeutics by directed evolution. Drug Discov Today. 2003;8(3):118–126. doi: 10.1016/s1359-6446(02)02590-4. [DOI] [PubMed] [Google Scholar]
- 12.Labrijn AF, Buijsse AO, van den Bremer ET, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27(8):767–771. doi: 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- 13.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997;6(2):407–415. doi: 10.1002/pro.5560060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Song X, Chrunyk BA, et al. Crystal structures of interleukin 17A and its complex with IL-17 receptor A. Nat Commun. 2013;4:1888. doi: 10.1038/ncomms2880. [DOI] [PubMed] [Google Scholar]
- 15.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301–316. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 16.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2(3):256–265. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366(13):1190–1199. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 18.Krueger JG, Fretzin S, Suarez-Farinas M, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145.e–154.e. doi: 10.1016/j.jaci.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese MC, Greenwald M, Cho CS, et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014;66(7):1693–1704. doi: 10.1002/art.38617. [DOI] [PubMed] [Google Scholar]
- 20.Langley RG, Rich P, Menter A, et al. Improvement of scalp and nail lesions with ixekizumab in a phase 2 trial in patients with chronic plaque psoriasis. J Eur Acad Dermatol Venereol. 2015;29(9):1763–1770. doi: 10.1111/jdv.12996. [DOI] [PubMed] [Google Scholar]
- 21.Genovese MC, Durez P, Richards HB, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis. 2013;72(6):863–869. doi: 10.1136/annrheumdis-2012-201601. [DOI] [PubMed] [Google Scholar]
- 22.Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9906):1705–1713. doi: 10.1016/S0140-6736(13)61134-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ixekizumab humanization.
Notes: (A) Variable region sequence alignment of selected human germline genes, VH1–69 and VκA18, parental mouse antibody 2321, and ixekizumab. Germline homologous residues of 2321 are indicated by a dash. CDR residues are underlined, and Kabat antibody numbering system used. The CDR residues that differ from VH1–69 or VκA18 germline sequence are shown in green (different from VH1–69 or VκA18 but present in other human germline sequences) and red (not present in any human germline sequences). (B) Calculation of homology to human germline. ‡Excluding HCDR3 and *including CDR residues present at the homologous location in other human germline genes.
Abbreviations: CDR, complementarity-determining region; mAb, monoclonal antibody; VH, variable heavy; Vκ, variable kappa; N/A, not applicable.
Ixekizumab epitope mapping.
Notes: (A) Mutagenesis of the six regions of the potential binding sites. (B) 4T1 cells were treated with a constant amount of human IL-17A wild type, mutant, or vector control supernatant in the presence of increasing amounts of ixekizumab (closed symbols) or isotope control antibody (open symbols). After 48 hours, KC in the supernatant was measured by ELISA. Results are shown as the mean of triplicate treatments ± standard deviation and are representative of four independent experiments.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IL, interleukin; KC, keratinocyte chemoattractant.
Serum pharmacokinetic profile of ixekizumab in male cynomolgus monkeys.
Notes: Following IV administration of 1 mg/kg, ixekizumab was eliminated with a mean half-life of 6.5 days. After SC administration of 1 mg/kg, it reached an average maximal plasma concentration of 11.1 µg/mL ∼72 hours postdose. The mean elimination half-life following subcutaneous injection was 10.3 days. The figure illustrates mean curve and data from individual animals (n=2).
Abbreviations: IV, intravenous; SC, subcutaneous.