

ABSTRACT
DNA methylation is the major repression mechanism for human retrotransposons, such as the Alu family. Here, we have determined the methylation levels associated with 5238 loci belonging to 2 Alu subfamilies, AluYa5 and AluYb8, using high-throughput targeted repeat element bisulfite sequencing (HT-TREBS). The results indicate that ∼90% of loci are repressed by high methylation levels. Of the remaining loci, many of the hypomethylated elements are found near gene promoters and show high levels of DNA methylation variation. We have characterized this variation in the context of tumorigenesis and interindividual differences. Comparison of a primary breast tumor and its matched normal tissue revealed early DNA methylation changes in ∼1% of AluYb8 elements in response to tumorigenesis. Simultaneously, AluYa5/Yb8 elements proximal to promoters also showed differences in methylation of up to one order of magnitude, even between normal individuals. Overall, the current study demonstrates that early loss of methylation occurs during tumorigenesis in a subset of young Alu elements, suggesting their potential clinical relevance. However, approaches such as deep-bisulfite-sequencing of individual loci using HT-TREBS are required to distinguish clinically relevant loci from the background observed for AluYa5/Yb8 elements in general with regard to high levels of interindividual variation in DNA methylation.
KEYWORDS: DNA methylation, epigenetic biomarkers, epigenomes, retrotransposons, SINE
Abbreviations
- BRCA1
Breast cancer 1, early onset
- BRCA2
Breast cancer 2 early onset
- CEBPG
CCAAT/enhancer binding protein (C/EBP) gamma
- DHODH
Dihydroorotate dehydrogenase (quinone)
- EDAR
Ectodysplasin A receptor
- H3K27ac
Histone H3 Lysine 27 acetylation
- H3K4me1
Histone H3 Lysine 4 monomethylation
- HT-TREBS
High-Throughput Targeted Repeat Element Bisulfite Sequencing
- SINE
Short INterspersed Elements
- UBE2T
Ubiquitin-conjugating enzyme E2T
Introduction
Alu elements are one of the most successful primate retrotransposons, with over one million copies in the human genome.1 They are short interspersed elements (SINEs), derived from the 7SL RNA gene, that have a dimeric structure linked by a middle A-rich region.2 The 5′ ends of Alu elements house the A- and B-boxes, which are the internal hallmarks of a RNA polymerase III (RNAP III) promoter, and their propagation is thought to occur via transcription by RNAP III.3 The 3′ends of Alu elements have an oligo-dA-rich sequence that is crucial to their retrotransposition mechanism (target-primed reverse transcription).4-7 Currently, relatively few Alu copies are capable of retrotransposition and those elements belong largely to the young Alu (AluY) family and its derivatives.8,9 Some of these elements have undergone massive expansions, specifically in humans, thereby comprising largely human-specific subfamilies, such as AluYa5 and AluYb8.10 These expansions have been driven by both “master” Alu copies and secondary “source” elements.11 Further, some of Alu subfamilies are predicted to contain “stealth-driver” elements, which escape negative selection by mobilizing at only very low rates over long periods of time.12
DNA methylation is the major epigenetic mechanism that represses all retrotransposons in the human genome, including Alu.13 However, Alu elements may be more affected in terms of DNA methylation than other retrotransposons due to their relatively high CpG density.14 Indeed, most Alu elements tend to be heavily methylated in somatic tissues, with some locus- and tissue-specific differences.15-19 They also show a unique pattern of differential methylation in the male and female germ cells, compared to somatic cells.20 Primate oocytes and human dysgerminoma (primary germ cell tumors usually occurring in the ovary) show high levels of DNA methylation, similar to somatic cells.20 By contrast, many Alu elements, especially the young Alu (e.g., the AluYa5 subfamily), show distinct hypomethylation in sperm.16,17,21 Consequently, Alu-specific RNAs have been observed in spermatozoa, indicating their transcriptional activity.17
Hypomethylation of Alu elements is not just associated with sperm, but also with several disease states.5,22,23 It is predicted that hypomethylation may lead to Alu retrotransposition that can disrupt gene expression; it may also allow for Alu-mediated recombination, which is believed to contribute to about 0.3% of all human diseases.22 BRCA1 and BRCA2 (2 breast cancer susceptibility genes) represent 2 of the best-characterized cases of diseases caused by Alu insertions and Alu-mediated recombinations.24-26 In addition to breast cancer,27 recent reports suggest extensive hypomethylation of Alu in several other types of cancers, such as gastric carcinoma,28 multiple myeloma,29 epithelial ovarian cancer,30 and lung adenocarcinoma.31 Hypomethylation of Alu in all these types of cancers is expected to be associated with tumor progression, as a recent meta-analysis concluded that hypomethylation of multiple repetitive elements, including Alu, has significant negative effects on tumor prognosis.32
Based on the propensity for Alu hypomethylation in many cancers, efforts are underway to develop them as epigenetic cancer biomarkers.33 Thus far, these efforts have focused on Alu elements as an entire group, instead of studying the methylation levels of individual Alu loci. This has been due mainly to the lack of methods that could provide reliable methylation levels of individual retrotransposon loci in a targeted fashion and on a genome-wide scale. Hence, it has remained largely unknown which particular Alu loci, and how many, show early changes in DNA methylation specifically in response to changes in cell state, such as during tumorigenesis. In this study, we have used high-throughput targeted repeat element bisulfite sequencing (HT-TREBS)34,35 to derive the methylation levels regarding >5,000 elements belonging to 2 of the most active human Alu subfamilies, AluYa5 and AluYb8, that have been implicated in many diseases, including breast cancer.22,24 Our data show that ∼90% of AluYa5/Yb8 loci are highly methylated; however, the hypomethylated loci (∼10%) are often located close to gene promoters and show high degrees of variation in DNA methylation. We have characterized this variation in the context of tumorigenesis in the breast and with regard to interindividual differences. The results indicate that AluYa5/Yb8 loci proximal to promoter regions may respond to tumorigenic events, but this response occurs in the background of very high levels of interindividual variation in DNA methylation. In fact, genome-wide, only ∼1% of AluYb8 elements are expected to be early responders specifically to tumorigenesis, suggesting the potential use of specific Alu elements as epigenetic biomarkers for the early detection of cancer.
Results
HT-TREBS of AluYa5 and AluYb8 in human skin-derived fibroblast
Here, we have analyzed the DNA methylation of >5,000 individual Alu elements from one of the most commonly used human cell lines, skin-derived fibroblast cells, using an established protocol for HT-TREBS.34,35 We enriched for 2 human-specific subfamilies of the young Alu group, AluYa5 and AluYb8, using a primer scheme that took advantage of their subfamily-specific diagnostic mutations5 (Fig. 1A). We obtained a total of 3.9 million final library sequencing reads, after filtering for read quality. Reads were then processed and mapped to a custom database containing unidirectional, bisulfite-converted sequence for the individual loci (and flanking sequence) belonging to the AluYa5 and AluYb8 subfamilies. Given the high sequence similarity within all AluY subfamilies, we repeated the mapping process against databases for elements in the AluYc, Yd, Yf, and Yk groups and discarded reads which also mapped to those groups. In the end, ∼63% of the mapped reads belonged to the human-specific AluYa5 and Yb8 subfamilies, yielding methylation levels regarding ∼75% of all AluYa5 and Yb8 elements in the human genome (Supplemental Data 1). On average, for the 3102 AluYa5 and 2136 AluYb8 elements, ∼30 reads were used to calculate the percent methylation at each locus, with a minimum of 10X coverage.
Figure 1.
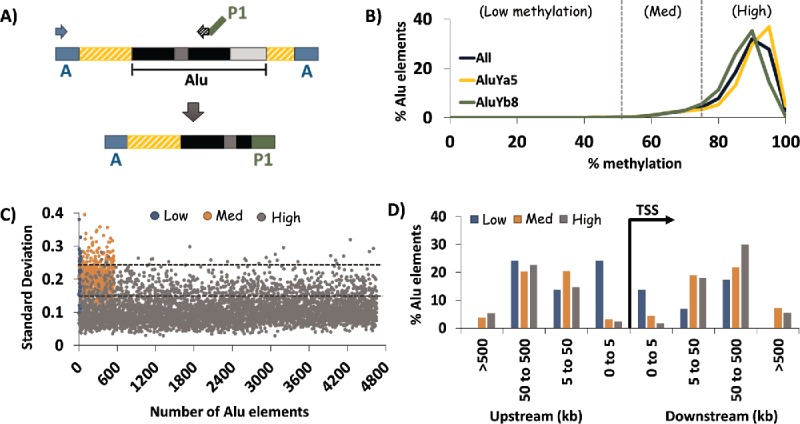
HT-TREBS of AluYa5 and AluYb8 in human fibroblast cells. (A) Primer scheme depicting the enrichment of AluYa5 and AluYb8 sequences using HT-TREBS. Briefly, methylated Ion Torrent “A” adaptors were ligated to sheared and size-selected genomic DNA, and the sample was bisulfite-treated. Ion “A” primers and subfamily-specific primers (with Ion “P1” adaptor on their 5′ ends) amplified fragments containing part of the Alu element (the first monomer and middle A-rich region) along with some flanking genomic sequence. (B) Methylation profile for AluYa5 and AluYb8 elements, binned in increments of 5%, for the 5238 loci sequenced. Loci were subdivided into 3 broad groups roughly corresponding to the inflection points in the graph: Low (0–50%), Medium (50–75%) and High methylation (75-100%). (C) Characterization of loci in terms of variation in methylation level (expressed in standard deviation, SD) for the 3 methylation groups. Dashed horizontal lines represent the thresholds used in the analysis (SD 0.15 and 0.25). Variation in DNA methylation was generally observed to be lower for highly methylated elements compared with those in the medium and low methylation groups. (D) Distance of AluYa5 andYb8 elements to the nearest transcription start site (TSS) based on methylation level. Alu elements with low levels of methylation appeared more likely to be present close to gene promoters compared with those methylated at >50% levels.
The results indicated that both AluYa5 and Yb8 elements tend to be highly methylated in the human epigenome (Fig. 1B). For these analyses, the entire data set of 5238 loci was divided into 3 groups: Low (0–50% methylation); Medium (50–75% methylation) and; High (75–100% methylation). For human skin-derived fibroblast cells, high methylation levels were observed for nearly 90% of these human-specific Alu elements (2788 in Ya5; 1867 in Yb8). By contrast, there were only 30 loci (∼0.5%) in the “Low” category and 533 loci (∼10%) in the “Medium” category. The distribution of Ya5:Yb8 loci within each methylation category ranged from ∼60:40 to ∼55:45.
For these elements within the fibroblast cell line, we also assessed the level of variability in DNA methylation as calculated by the standard deviation in methylation levels for all reads mapped to each specific locus (Fig. 1C). Variation appeared to be inversely correlated with the methylation categories, with 84% of the elements (3903) in the “High” category having the lowest levels of standard deviation (SD 0-0.15) while 40% of the elements (12 out of 30) in the “Low” category had SD values in the highest range (≥0.25). Loci in the “Medium” category showed intermediate levels of variation, with 75% (413 elements) having SD values between 0.15 and 0.25.
To test whether the methylation level of the AluYa5 and Yb8 elements correlated with any positional bias, we mapped their distance to the nearest transcription start site (TSS) using the genomic regions enrichment of annotations tool (GREAT 36) (Fig. 1D). For AluYa5 and Yb8 elements methylated at high or medium levels, most were found 5–500 kb from the nearest TSS. In contrast, those methylated at low levels were often (though not exclusively) found within 5 kb upstream of the TSS. Thus, most AluYa5 and AluYb8 elements are not present near gene-rich regions; however, those with low levels of methylation may be more frequently associated with proximity to gene promoters.
Characteristics of AluYa5 and AluYb8 elements located near gene promoters
A search for the nearest gene with a TSS within 1 kb of the AluYa5 and Yb8 elements in our data set detected 18 genes, with equal numbers associated with AluYa5 vs. AluYb8 elements. With respect to their association with Alu loci methylation categories, the 18 genes were distributed as follows: “Low”, 7 genes (∼40%); “Medium”, 4 genes (∼20%) and; “High”, 7 genes (∼40%). This highlights the proximity bias of low-methylation AluYa5/Yb8 elements; over 20% of the 30 elements with low levels of methylation were located within 1 kb of the nearest transcription start site, whereas only 0.1% of loci in the high-methylation (7 out of 4655) and 0.7% of those in the medium-methylation group (4 out of 553) were present so close to a gene (Fig. 1D, Table 1).
Table 1.
Characteristics of AluYa5/Yb8 elements located within 1-kb of the nearest transcription start site (TSS) and their associated gene. Loci were subdivided into three broad groups: Low (0-50%), Medium (50-75%) and High methylation (75-100%). Negative sign (-) indicates that the gene is upstream of the transcription start site (TSS).
| Gene | Alu Subfamily | Methylation Level (%) | Group | Distance to nearest TSS (bp) | H3K27ac shore | Hypo-methylated Tissue(s) |
|---|---|---|---|---|---|---|
| OR10Q1 | Ya5 | 82 | High | -160 | — | Sperm |
| OSBPL10 | Ya5 | 17 | Low | -160 | Yes | Various |
| TMSB4Y | Ya5 | 24 | Low | -200 | — | Various |
| †UBE2T | †Ya5 | 8 | Low | 529 | Yes | Various |
| †CEBPG | †Ya5 | 83 | High | -578 | Yes | Various |
| HIGD1B | Yb8 | 66 | Medium | -603 | — | — |
| †DHODH | †Yb8 | 21 | Low | 634 | Yes | Various |
| RABEPK | Yb8 | 44 | Low | -660 | Yes | Various |
| UGGT2 | Ya5 | 17 | Low | -698 | Yes | Various |
| HENMT1 | Ya5 | 66 | Medium | 729 | — | Various |
| INTS5 | Yb8 | 97 | High | -779 | Yes | Sperm |
| HTR3E | Yb8 | 83 | High | -783 | Yes | Sperm |
| †MAP3K7 (TAK1) | †Yb8 | 20 | Low | 798 | Yes | Various |
| AASDH | Yb8 | 66 | Medium | 830 | Yes | Various |
| ADAD1 | Ya5 | 88 | High | -854 | — | Sperm |
| IDNK | Yb8 | 84 | High | -931 | — | Sperm |
| †EDAR | †Yb8 | 61 | Medium | -936 | — | — |
| EIF2B1 | Ya5 | 89 | High | 952 | — | Sperm |
† Loci further characterized in this study
The data in Table 1 also suggest that most AluYa5 and Yb8 elements found proximal to gene promoters often bear active epigenetic signatures in a tissue-specific manner. For instance, in several tissues, nearly 60% (10/18) of these Alu elements were found along the shores of regions bearing histone marks indicative of enhancer regions,37-39 with especially strong acetylation on lysine 27 of histone H3 (H3K27ac) signals, and somewhat weaker signals for monomethylation on lysine 4 of histone H3 (H3K4me1) for some of these elements (data not shown). Next, consistent with the results of our study, nearly all elements from the “Low” and “Medium” methylation groups were also found to be hypomethylated in various other normal and cancer cell lines.40-45 Finally, nearly all AluYa5/Yb8 elements in the high methylation category in fibroblast cells (as well as most loci in the low and medium categories) were nonetheless hypomethylated in sperm.21
Gene-associated AluYa5/Yb8 elements show variable DNA methylation
Most of the genes located within 1 kb of the 18 Alu elements have previously been found to be misregulated in various different cancers (Supplemental Fig. 1). Hypothesizing that such changes could affect nearby Alu elements, we assessed the methylation levels of Alu elements located near 5 genes (CEBPG, UBE2T, EDAR, DHODH, and MAP3K7/TAK1) as well as their endogenous promoters. Two genes, CEBPG and UBE2T, were associated with elements from the AluYa5 subfamily; the other 3 genes were associated with elements from the AluYb8 subfamily. Using two independent approaches, combined bisulfite restriction assay (COBRA46) and NGS-based bisulfite sequencing of the amplicons, we tested the methylation level of these 5 Alu loci in 5 samples: matched breast normal, matched breast tumor, breast cancer, lung normal, and lung cancer.
The COBRA results showed that 4 out of the 5 loci showed significant variation in at least one of the 3 cancer samples (Fig. 2A-F). CEBPG-Alu was the most affected, showing significant hypermethylation by 20% on average in the matched breast tumor, and hypomethylation by 16% on average in the unmatched breast cancer sample. This particular locus was also affected especially severely in the lung cancer sample, showing near-complete demethylation (Fig. 2A). UBE2T-Alu and EDAR-Alu also seemed affected in the context of breast tumorigenesis (Fig. 2B, C), with UBE2T-Alu showing almost no methylation in both the matched breast tumor as well as the unmatched breast cancer sample, and EDAR-Alu showing 25% less methylation only in the unmatched breast cancer. In addition to CEBPG-Alu, lung cancer samples were hypomethylated by 10% for EDAR-Alu and by 12% for DHODH-Alu (Fig. 2C, D). The fifth locus, MAP3K7-Alu, showed no significant difference between any of the normal and cancer samples (Fig. 2E). Fig. 2F summarizes the COBRA-derived variation in methylation in the cancer samples relative to their normal controls.
Figure 2.
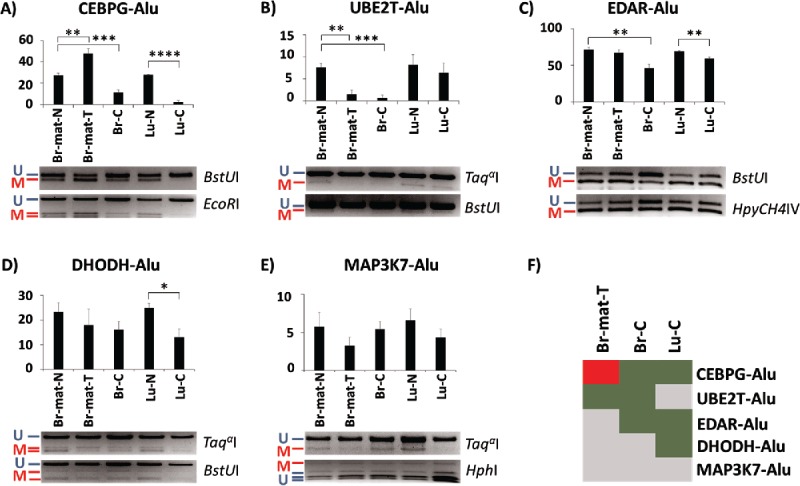
Methylation variation of AluYa5 and Yb8 loci closely associated with a gene in normal and cancer samples. Alu elements are named after their associated gene. (A-E) Methylation levels are quantified through COBRA, followed by densitometry, in 5 tissues: matched breast normal (Br-mat-N); matched breast primary tumor (Br-mat-T); breast cancer (Br-C); normal lung (Lu-N); and lung cancer (Lu-C). Bands are labeled as either methylated (red “M”) or unmethylated (blue “U”), along with the appropriate restriction enzyme. * P < 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001. Error bars indicate standard error of mean (SEM). (F) Relative to the appropriate control, CEBPG-Alu, UBE2T-Alu, EDAR-Alu and DHODH-Alu showed significant difference in methylation in at least one test sample. Heatmap summarizes the results as hypermethylation (red), hypomethylation (green), and no significant change (gray).
The methylation patterns of 3 out of the 5 loci, CEBPG-Alu, EDAR-Alu and DHODH-Alu, were further tested with next-generation-sequencing on the bisulfite-PCR products. Overall, the bisulfite-NGS confirmed the COBRA results, demonstrating hypermethylation of CEBPG-Alu in the matched breast tumor and hypomethylation in the lung cancer samples (Fig. 3), as well as being concordant for EDAR-Alu and DHODH-Alu (Supplemental Figs. 2–3). Detailed analysis of the sequencing pattern also showed that the methylation of the CpG sites associated with the A- and B-boxes of the elements showed no unique patterns and was similar to nearby CpG sites which did not correspond to any functional element (Fig. 3B). Additionally, variation in Alu methylation was unrelated to the methylation status of the endogenous gene promoters, which were completely unmethylated in all samples, aside from some hypermethylation at the CEBPG promoter in lung cancer (Supplemental Figs. 4–5). In sum, based on the COBRA and bisulfite-NGS data, we conclude that locus-specific differences in methylation exist for AluYa5 and AluYb8 elements proximal to 4 cancer-associated genes in breast and lung cancer samples. The changes observed in the breast tumor relative to the matched normal can be attributed to tumorigenesis, suggesting that methylation levels of UBE2T-Alu and CEBPG-Alu are likely to respond to changes in cell state.
Figure 3.
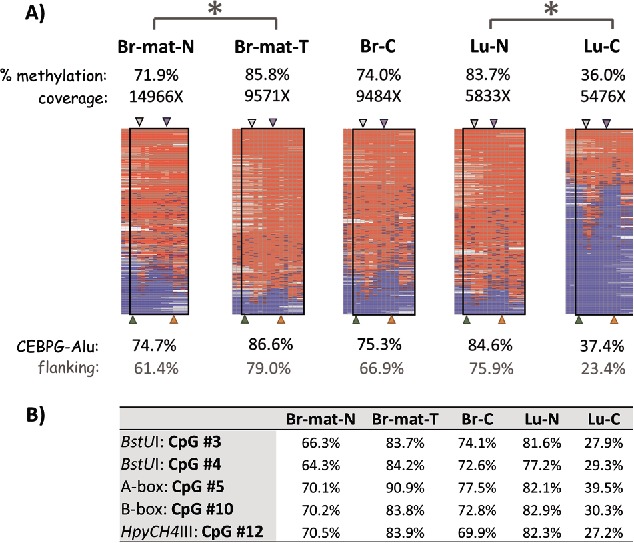
Methylation variation of CEBPG-Alu in normal and cancer samples. (A) Heatmaps show the methylation patterns of the CEBPG-Alu locus (after nested PCR) in the 5 samples described in Fig. 2. Red and blue in the heatmap indicate methylated and unmethylated CpG sites, whereas white marks CpG sites with unknown methylation status. CpG sites within actual Alu elements are enclosed by black rectangles. The % methylation is shown for the entire amplicon (top) as well as for CpG sites within vs. flanking the AluYa5 element (bottom). Significant differences (P < 0.0001) are shown with a large asterisk. Arrowheads indicate the A-box (gray) and B-box (purple) in the Alu element [top], and CpG sites analyzed by COBRA (BstUI, green; HpyCH4III, orange) [bottom]. (B) Methylation levels of a few pertinent CpG sites within the locus (marked by the arrowheads) in the 5 samples.
Interindividual variation in AluYa5 and AluYb8 elements
Various molecular events during normal development, independent of those related to tumorigenesis, may potentially lead to interindividual variation in Alu DNA methylation. To assess the background level of variation in DNA methylation at these Alu loci, we tested the DNA methylation levels in 8 normal breast tissues by COBRA and bisulfite-NGS for CEBPG-Alu, DHODH-Alu, EDAR-Alu and UBE2T-Alu. Methylation levels were high enough for accurate quantitation in only 2 of the 4 loci, with the range in normal tissue being ∼9–85% for CEBPG-Alu and ∼9–80% for DHODH-Alu (Fig. 4A). Further, NGS-based bisulfite sequencing supported the COBRA data by showing various different methylation patterns between the 8 normal and 8 tumor samples (Supplemental Fig. 6A-C). Overall, the data suggest that AluYa5 and Yb8 loci close to gene promoters are likely to show high levels of interindividual variation in DNA methylation.
Figure 4.
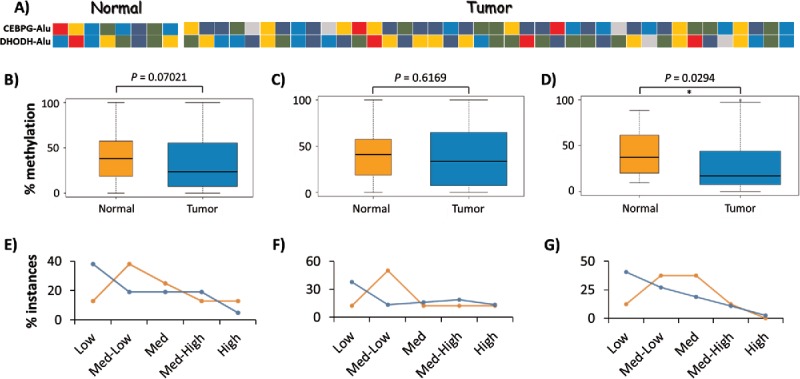
Interindividual variation in DNA methylation of AluYa5 and Yb8. (A) Heatmap showing the average methylation level of CEBPG-Alu and DHODH-Alu in 8 normal and 40 breast tumor samples as determined by the COBRA assay: dark blue, 0–20% methylation (Low); blue, 20–40% (Medium-Low); green, 40–60% (Medium); gold, 60–80% (Medium-High); and red, 80–100% (High). (B-G) Variation in methylation levels for CEBPG-Alu in normal vs. tumor samples for CpG sites recognized by: both BstUI and HpyCH4III (B, E); BstUI only (C, F); and, HpyCH4III only (D, G). Panels B-D represent statistical analyses of data from all samples as pooled into normal vs. tumor, with significant hypomethylation only at the HpyCH4III site; Panels E-G depict the relative numbers of samples in each group as pooled into 5 levels of variation in methylation, with more tumor samples methylated at Low (0–20%) levels at all tested CpG sites.
To determine whether some of the variation could be tumor-related, we then compared the COBRA methylation data obtained from the normal breasts to those from 40 breast tumor samples. For these analyses, we grouped all individual methylation counts into normal vs. tumor samples. With respect to overall methylation state, the pooled site data (BstUI and HpyCH4III) for CEBPG-Alu showed no significant differences between normal and tumor samples (Wilcoxon-Mann-Whitney test, Fig. 4B). However, analyzed separately, significant hypomethylation in the tumor was observed at the HpyCH4III site (Fig. 4C-D). We also quantified the percentage of normal and tumor samples in 5 methylation-ranges: Low (0–20%), Medium-Low (20–40%), Medium (40–60%), Medium-High (60–80%) and High (80–100%). Here, regardless of whether the COBRA sites for CEBPG-Alu were analyzed separately or together, ∼25% more tumor samples were methylated in the low range compared to normal (Fig. 4E-G). Altogether, this analysis indicated the presence of interindividual variations at AluYa5 and AluYb8 loci located close to gene promoters, with the methylation level at one CpG site in CEBPG-Alu being significantly lower in the breast tumor sample set.
Aberrant DNA methylation of AluYa5 and AluYb8 in response to tumorigenesis
Despite the large interindividual variation in DNA methylation documented through the 8 normal samples, initial analysis of the matched breast pair suggested that at least 2 loci, CEBPG-Alu and UBE2T-Alu, responded specifically to tumorigenesis (Fig. 2A-B). To determine how many AluYa5 and Yb8 elements are likely to respond similarly genome-wide, we performed another HT-TREBS on the same matched breast normal and primary tumor. Reads for normal (6.3 million) and tumor (6.8 million) samples were processed as described for the fibroblast cells, yielding methylation values regarding 104 AluYa5 and 1776 AluYb8 elements with a minimum of 10X coverage in both the normal and tumor samples (Supplemental Data 2).
Initial analysis of the HT-TREBS results revealed a subtle trend toward hypomethylation of AluYa5/Yb8 elements in the breast tumor compared to normal (Fig. 5A). For instance, in terms of their percentages of Alu elements, there was a disparity between the tumor and normal samples within the 85–100% methylation level, with ∼10% of the tumor elements being shifted into the 65–85% methylation range. In fact, 36% of all loci with methylation levels of <85 % were unique to the tumor sample, while 55% were common to both the normal and the tumor (inset in Fig. 5A). Next, to determine the pattern of change for individual elements, the methylation level of each AluYa5/Yb8 locus in the tumor sample was plotted against the normal (Fig. 5B). Most loci clustered between 80–100% methylation, with a minority of loci ranging between 20–80% methylation. Loci falling on the diagonal y = x line represent Alu elements showing no change in DNA methylation during tumorigenesis; deviations from the line represent loci showing differential methylation between the 2 samples. Loci were considered to be responding to tumorigenesis if they deviated by at least 20% in in their methylation between normal and tumor samples; at this threshold, as few as 10 reads were sufficient to establish statistical significance. Twenty-two loci (all AluYb8) were differentially methylated at >20% levels, with 19 loci (∼86%) showing hypomethylation in the tumor (Supplemental Data 2). Overall, the HT-TREBS data on the matched breast pair revealed that the primary breast tumor had at least 4X more uniquely hypomethylated loci than its matched normal sample. Furthermore, about 1% of AluYb8 loci are expected to show at least 20% difference in methylation in response to tumorigenesis.
Figure 5.
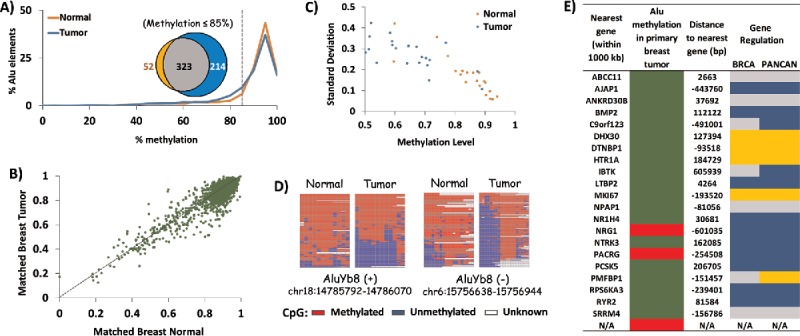
Tumorigenesis-related variation in DNA methylation of AluYb8. HT-TREBS analysis of matched breast normal and primary tumor tissues showing tumorigenesis-related variations in DNA methylation of AluYb8 elements. (A) Methylation profile of the normal (orange) and tumor (blue) samples, based on the percent Alu elements belonging to methylation bins in increments of 5%. The venn-diagram (inset) shows the 4X higher number of hypomethylated loci (<85% methylation; dotted line) in the tumor (blue) compared to the normal tissue (orange). (B) Methylation level of individual Alu loci in the matched breast tumor vs. normal, with each dot representing one Alu locus. Most loci follow the y = x dotted line, indicating no major difference in methylation between the 2 tissues. (C) Variation in methylation (standard deviation) vs. methylation level of the 22 differentially methylated AluYb8 loci, indicating lower methylation and higher levels of variation in the tumor tissue (blue) compared to the normal tissue (orange). (D) Heatmaps showing methylation levels of 2 AluYb8 loci in normal and tumor tissues. (E) Summary of information with respect to the nearest gene associated with the 22 differentially methylated AluYb8 loci (green for hypomethylation and red for hypermethylation). Negative sign (−) indicates that the gene is upstream of the Alu element; “N/A” indicates no gene within 1000 kb of the Alu element. Gene regulation information is from the invasive breast cancer (BRCA) and Pan-Cancer (PANCAN) data sets from TCGA; using Student t-test (P <0.05), genes were classified as downregulated (blue) or upregulated (gold). Gray indicates no significant change.
Close examination of the 22 AluYb8 loci showed that, along with a decrease in DNA methylation, these loci also displayed an increase in the variability of their methylation state (Fig. 5C). As seen in the individual heatmaps, this change is the result of read-specific hypomethylation, either in the entire span or in only the 5′ end of the Alu element (Fig. 5D). Assuming that each individual methylation read is derived from the DNA from different cells, this pattern indicates the accumulation of cells in the primary tumor which are losing DNA methylation at these loci. To test whether these early changes in DNA methylation correlate with events in later stages of breast cancer, we searched The Cancer Genome Atlas (TCGA) for the gene nearest to the Alu elements and determined whether that gene showed any misexpression in the invasive breast cancer (BRCA) or Pan-Cancer (PANCAN47) data set from TCGA (Fig. 5E). Of the 22 loci responding to tumorigenesis, 14 had their nearest gene misexpressed in both the invasive breast cancer as well as in the PANCAN set. Considering the 2 data sets combined, the AluYb8 elements appeared to be associated with a significantly greater number of downregulated genes than upregulated genes (P = 0.02, chi-square test). This gave rise to a slight correlation between hypomethylated loci in breast tumor and downregulated genes in the invasive breast cancer stage. With regard to the BRCA data set, 8 out of 19 (42%) loci hypomethylated in the primary breast tumor were associated with a gene downregulated in breast cancer, and 4 (21%) loci were associated with an upregulated gene (Fig. 5E). Unfortunately, however, the significance of this correlation could not be established due to the small sample number. Overall, detailed analysis of the 1% AluYb8 elements responding to breast tumorigenesis indicates that a fraction of the tumor cells may show complete unmethylation of the Alu locus, either in part or in its entirety. Further, such changes in DNA methylation of Alu elements may serve as early indicators of downstream aberrations in gene expression, characteristic in the later stages of breast cancer.
Discussion
Here, we have determined DNA methylation levels regarding 5238 individual Alu loci belonging to the AluYa5 and AluYb8 subfamilies. Consistent with previous results, we find that ∼90% of these elements are highly (>75%) methylated (Fig. 1B), presumably as a means of repressing expression of retrotransposons in the human genome. Nevertheless, ∼10% of the elements are hypomethylated and tend to be located in regions of active chromatin (Fig. 1D, Table 1), potentially allowing them to escape this repression mechanism. Interestingly, these hypomethylated loci exhibit high variation in their methylation status from cell to cell in a tissue sample (Fig. 1C). Loci near gene promoters also tend to show high levels of interindividual variation in DNA methylation (Fig. 4A, Supplemental Fig. 6).
The existence of Alu elements with variable levels of retrotransposition activity, secondary and “stealth” drivers, have been proposed in previous studies;11,12 yet, the molecular mechanisms underlying these models remain unclear. Given that DNA methylation is one of the major epigenetic mechanisms repressing Alu retrotransposition, we speculate that interindividual variations in DNA methylation, as reported here for AluYa5 and AluYb8 elements near gene promoters (Fig. 4, Supplemental Fig. 6), might play an important role. After all, for “stealth” drivers and secondary elements to be successful, they must be repressed in most individuals in order to maintain low retrotranspositional activity within the overall population. Differential repression of Alu elements between individuals is likely to be at the epigenetic level because the epigenome is highly plastic and easily influenced by various environmental factors. Further, because only a few individuals in a population are expected to have very low levels of DNA methylation at an Alu locus, very few copies should be active at the population level, with others being methylated (and perhaps active) at various different levels. Thus, differential methylation of the human-specific Alu elements, as found in this study, may decouple them from selection pressures and allow their continued propagation according to the Stealth Model.12
With respect to disease, hypomethylation at Alu elements has been associated with various cancer types, and it may have a bearing on tumor prognosis;32 however, there is a low penetrance of cancer incidences which can be directly attributed to Alu activity. The high interindividual variation in methylation of Alu elements proximal to promoter regions may be a contributing factor toward the low penetrance phenotype. Only a small fraction of the population is expected to harbor severely hypomethylated Alu loci, potentially increasing their chances of developing cancer. The underlying mechanism could be either Alu retrotransposition or its activity in cis. As discussed below, cis activity is especially interesting with regard to Alu elements proximal to cancer-associated genes, such as CEBPG-Alu (Fig. 4, Supplemental Fig. 6). In addition to the evolutionary perspective, further investigations regarding interindividual variation in Alu methylation may prove fruitful in relation to elucidating disease susceptibility factors.
A long standing question regarding the role of Alu retrotransposons in cancer has been whether they can be drivers of tumorigenesis or if they are simply “passengers.” Answering this question involves understanding how many Alu elements are likely to affect nearby gene promoters in cis. Here, focusing on only 2 subfamilies of human-specific Alu elements, we report characteristics for 18 AluYa5 and Yb8 elements located within 1 kb of a gene promoter (Table 1). Many of these loci are both hypomethylated and are located near enhancer regions in various tissues, with some bearing active histone marks themselves. Moreover, nearly all of these loci are associated with genes that are frequently misregulated in cancer (Supplemental Fig. 1). Two loci, UBE2T-Alu (chr1:202310424-202310734) and CEBPG-Alu (chr19:33863506-33863809) are particularly noteworthy for the following reasons:
-
1.
Early changes in DNA methylation during tumorigenesis were displayed by the AluYa5 elements associated with both of these genes, as seen in significant changes in a primary breast tumor relative to the matched normal. CEBPG-Alu was hypermethylated; by contrast, UBE2T-Alu was hypomethylated (Figs. 2–3).
-
2.
Both elements are closely associated with tumor-suppressor/oncogenes and are located within the active promoter regions of their respective genes. In addition, CEBPG-Alu shows some enhancer-related histone marks in some cell lines (e.g., HMEC, normal human mammary epithelial cells),38,39 suggesting potential functional exaptation of the locus.
-
3.
Both elements have potential function as cis elements, being associated with genes that are directly involved in tumorigenesis. UBE2T is frequently overexpressed in breast cancer and is thought to aid in breast carcinogenesis by downregulating BRCA1.48 CEBPG, by contrast, is mainly a lung cancer gene, with interindividual differences in expression level of CEBPG being associated with the risk of lung cancer.49,50 In this study, CEBPG-Alu showed interindividual differences in methylation in the breast tissue (Fig. 4); assuming similar interindividual variation exists at the Alu locus in the lung, it may correlate with interindividual variation in CEBPG expression.
In summary, based on their methylation profiles in normal and cancer samples, we have identified several Alu elements for further research into their potential roles as drivers of tumorigenesis.
Regardless of their role as “drivers” or “passengers,” Alu elements have the potential to serve as epigenetic cancer biomarkers. We performed HT-TREBS on a primary breast tumor and its matched normal sample, and found 22 elements (∼1%) with signatures of being early responders to breast tumorigenesis. These elements all belong to the AluYb8 subfamily and they are all located far from promoter regions (Fig. 5). Alu hypomethylation has been considered to be a later event in breast carcinogenesis, given that significant changes in overall Alu methylation were not seen until the later more invasive stages.27 Thus, the percentage of ‘early responders’ may vary among tumor types; for example, we expect it to be much higher in lung cancer where Alu hypomethylation is considered to be an early event.31 Here, even in an early stage of breast tumor and using very stringent criteria, deep-sequencing of 1800 individual AluYa5/Yb8 loci revealed that the primary tumor had 4 times more uniquely hypomethylated loci (with ≤ 85% methylation) than the normal tissue (Fig. 5A), and that 19 of these loci showed significant hypomethylation in a read-specific manner only in the tumor (Fig. 5D). The greater number of reads showing complete unmethylation at the 5′ end of the Alu loci (which house the regulatory elements for Alu propagation) may indicate an accumulation of cells within the tumor sample which are completely losing DNA methylation at this locus.
These results demonstrate that HT-TREBS is capable of detecting when very few cells within a potentially cancerous tissue show loss of methylation at a locus. In fact, as few as 10 reads were deemed sufficient to establish statistical significance when the difference in methylation was at least 20%. Further, according to TCGA, many of the genes found within 1000 kb of these 22 AluYb8 elements showed a significant change in their expression level in invasive breast cancer as well as in the PANCAN data set (Fig. 5E).47 This suggests that Alu elements located far from genes, which show an early change in DNA methylation during tumorigenesis, may be good biomarkers for the early detection and risk-assessment of cancer. However, given their distance from the associated genes, it seems likely that these Alu elements are merely neutral bystanders (a.k.a., “passengers”) in the genome, and that their methylation levels are only indicative of a certain cell state. Taken together, these factors render HT-TREBS a potentially useful technique in detecting epigenetic biomarkers for the early detection of cancer through the deep-bisulfite-sequencing of individual loci of the targeted retrotransposon.
Materials and methods
HT-TREBS analysis of AluYa5 and AluYb8
The established protocol for HT-TREBS34,35 was used for this study, with some modifications. Purified DNA was sonicated to a mode of ∼700-bp fragments, end-repaired and ligated to custom-designed Ion Torrent “A” adaptors with methylated cytosines (Integrated DNA Technologies). Excess adaptors and DNA fragments <300-bp were then removed using Agencourt AMPure XP beads (Beckman Coulter). The final product was subjected to a bisulfite conversion reaction using EZ DNA Methylation™ kit (Zymo Research) according to manufacturer's protocol. The bisulfite-converted DNA was then amplified with a forward primer complementary to the Ion Torrent “A” adaptor and reverse primers specifically designed to select for AluYa5 (5′- CCACTACGCCTCCGCTTTCCT CTCTATGGGCAGTCGGTGAT^CAAATAACTAAAACTACAAACRCCCRCCACT -3′) and AluYb8 (5′- CACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGAT^CTCTATCRCCCAAACCRAACTACTA -3′) using their diagnostic mutations.5 The sequence before the caret (^) belongs to the Ion Torrent “P1” adaptor which was included as a 5′-extension on the reverse primers; ‘R’ stands for either ‘A’ or ‘G’ (IUPAC DNA nomenclature). The PCR products from the separate “AluYa5” and “AluYb8” reactions were then size-selected to a range of 450–500 bp in length using the E-gel Precast Agarose Electrophoresis System (Life Technologies), purified by Select-A-Size DNA columns (Zymo Research), quantified on the Bioanalyzer DNA-HS chip (Agilent Technologies) and combined in equimolar concentrations. The combined library was templated on the Ion™ OneTouch 2 (using the Ion PGM Hi-Q™ OT2 Kit) and then sequenced on the Ion PGM (using the Ion PGM HiQ Sequencing Kit and a 318-v2 chip). Sequencing reads were first filtered for quality and size by the Ion Torrent Suite software (v-4.4.3). All remaining reads >100-bp in length were then used for mapping using Bowtie2.51 Reference genomes were custom-prepared for the mapping, which consisted of ∼20,000 Alu sequences from 6 AluY subfamilies (a, b, c, d, f, and k) plus 700-bp flanking regions, from Human Genome Build hg19, in which all non-CpG cytosines were converted to thymines and all CpG cytosines to ‘Y’ (IUPAC ambiguous base for C/T). The mapped reads were then processed using various Perl scripts to yield only those sequences which contained the Alu and at least 10-bp of flanking genomic sequence. The filtered reads were then processed through BiQAnalyzerHT52 to derive the individual methylation levels of each Alu locus as defined by the following equation: [(# methylated CpG sites from all reads)/(# all CpG sites from all reads)]*100. Due to the nature of the HT-TREBS protocol, these values applied to ∼60% of each AluYa5 element and ∼85% of each AluYb8 element; the remaining portions of each element were not part of the amplification products and thus were not sequenced. All HT-TREBS datasets have been added to the NCBI's Gene Expression Omnibus53 data repository and can be viewed under the accession number GSE74420 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74420).
COBRA (Combined Bisulfite Restriction Analysis)
Purified DNA (∼500 ng) was treated with sodium bisulfite according to the EZ DNA Methylation™ kit protocol (Zymo Research). The bisulfite-treated DNA (∼1 µL; ∼20 ng) was then amplified by PCR using primers (Supplemental Data 3) lacking any CpG-dinucleotides and in which all cytosines were converted to thymines. PCR products were digested using a restriction enzyme that recognizes at least one CpG site within the Alu sequence and none in the flanking sequence (New England BioLabs). The gel band densities of the restriction products were analyzed using the Quantity One software (BioRad) to determine the percent methylation of the Alu elements. Data from at least 2 restriction enzyme sites were used for all AluYa5/Yb8 tested in this manner. All densitometric analyses were repeated using an independent software, ImageJ, to ensure consistency. Error bars in Fig. 2 were derived from repeating the entire process at least 3 times, starting with bisulfite-conversion and ending with the densitometry. By contrast, for the data in Fig. 4 & Supplemental Fig. 6, the bisulfite-conversion process could not be repeated due to insufficient DNA stocks; thus, the error bars in Fig. 4 were derived from repeating the process from PCR to COBRA-densitometry 2–3 times for each locus. The boxplots in these figures were generated by combining all the data from all the independent trials of all restriction enzyme sites tested for the 8 normal and 40 tumor samples.
Bisulfite-NGS
One set of PCR products used for the COBRA analyses was used for NGS-based bisulfite sequencing. This was accomplished as follows. Multiple PCR products of the same sample were combined into one barcoded library, with different barcodes for the different samples. These libraries were then end-repaired and ligated to Ion Torrent “A” and “P1” adaptors lacking 5′-phosphate, using a novel scheme that uses T4 ligase and Bst 2.0 WarmStart® DNA Polymerase (New England BioLabs). Unligated adaptors were then removed by agarose gel extraction and further purification by Select-A-Size DNA columns (Zymo Research), and the libraries were quantified on the Bioanalyzer (Agilent Technologies). Next, each library, with its unique barcode, was combined for multiplexed next-generation-sequencing using the Ion Torrent 318-v2 chip as described above for HT-TREBS. After processing for quality and size (Torrent Suite 4.4.3), reads were filtered first by barcode sequence and then by the primer sequence for each locus from each sample. To derive individual methylation heatmaps and levels, the processed reads were analyzed using BiQ Analyzer54 against the unconverted sequence for each amplification product. Percent methylation was calculated using: [(# methylated CpG sites from all reads)/(# all CpG sites from all reads)]*100. Separate methylation levels for only the Alu locus were calculated by applying the formula above to only the CpG sites that mapped within the Alu sequence.
Statistical analyses
COBRA-densitometry data in Fig. 2–3 were analyzed by the Student's t-test (P < 0.05), given that all analyzed cohorts were normally distributed (mean = median). For HT-TREBS and bisulfite-NGS data, at least one group in each case was not normally distributed (mean ≠ median); thus, these data were analyzed by the non-parametric Wilcoxon-Mann-Whitney test in RStudio. For data derived from next-generation-sequencing, significance was set at P < 0.001 due to the high depth of coverage at each locus. The boxplots in Fig. 4 were also subjected to the Wilcoxon-Mann-Whitney test; however, as these data were derived from COBRA analyses, P < 0.05 was deemed statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Muhammad Ekram for initial help in library construction and bioinformatics. We also thank Jerilyn Walker, Dr. Hana Kim, B. Pinithi Perera, Corey Bretz and Terri Cain for their careful reading and discussion of the manuscript. This research was supported by the National Institutes of Health R01 GM066225 (J.K.), R01 GM097074 (J.K.) and R01 GM59290 (M.A.B.).
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al.. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921; PMID:11237011; http://dx.doi.org/ 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- 2.Deininger PL, Moran JV, Batzer MA, Kazazian HH Jr.. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 2003; 13:651-8; PMID:14638329; http://dx.doi.org/ 10.1016/j.gde.2003.10.013 [DOI] [PubMed] [Google Scholar]
- 3.Chu WM, Liu WM, Schmid CW. RNA polymerase III promoter and terminator elements affect Alu RNA expression. Nucleic Acids Res 1995; 23:1750-7; PMID:7540287; http://dx.doi.org/ 10.1093/nar/23.10.1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy-Engel AM, Salem AH, Oyeniran OO, Deininger L, Hedges DJ, Kilroy GE, Batzer MA, Deininger PL. Active Alu element “A-tails”: size does matter. Genome Res 2002; 12:1333-44; PMID:12213770; http://dx.doi.org/ 10.1101/gr.384802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet 2002; 3:370-9; PMID:11988762; http://dx.doi.org/ 10.1038/nrg798 [DOI] [PubMed] [Google Scholar]
- 6.Dewannieux M, Heidmann T. Role of poly(A) tail length in Alu retrotransposition. Genomics 2005; 86:378-81; PMID:15993034; http://dx.doi.org/ 10.1016/j.ygeno.2005.05.009 [DOI] [PubMed] [Google Scholar]
- 7.Deininger P.Alu elements: know the SINEs. Genome Biol 2011; 12:236; PMID:22204421; http://dx.doi.org/ 10.1186/gb-2011-12-12-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen MR, Batzer MA, Deininger PL. Evolution of the master Alu gene(s). J Mol Evol 1991; 33:311-20; PMID:1774786; http://dx.doi.org/ 10.1007/BF02102862 [DOI] [PubMed] [Google Scholar]
- 9.Bennett EA, Keller H, Mills RE, Schmidt S, Moran JV, Weichenrieder O, Devine SE. Active Alu retrotransposons in the human genome. Genome Res 2008; 18:1875-83; PMID:18836035; http://dx.doi.org/ 10.1101/gr.081737.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll ML, Roy-Engel AM, Nguyen SV, Salem AH, Vogel E, Vincent B, Myers J, Ahmad Z, Nguyen L, Sammarco M, et al.. Large-scale analysis of the Alu Ya5 and Yb8 subfamilies and their contribution to human genomic diversity. J Mol Biol 2001; 311:17-40; PMID:11469855; http://dx.doi.org/ 10.1006/jmbi.2001.4847 [DOI] [PubMed] [Google Scholar]
- 11.Cordaux R, Hedges DJ, Batzer MA. Retrotransposition of Alu elements: how many sources? Trends Genet 2004; 20:464-7; PMID:15363897; http://dx.doi.org/ 10.1016/j.tig.2004.07.012 [DOI] [PubMed] [Google Scholar]
- 12.Han K, Xing J, Wang H, Hedges DJ, Garber RK, Cordaux R, Batzer MA. Under the genomic radar: the stealth model of Alu amplification. Genome Res 2005; 15:655-64; PMID:15867427; http://dx.doi.org/ 10.1101/gr.3492605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu WM, Maraia RJ, Rubin CM, Schmid CW. Alu transcripts: cytoplasmic localisation and regulation by DNA methylation. Nucleic Acids Res 1994; 22:1087-95; PMID:7512262; http://dx.doi.org/ 10.1093/nar/22.6.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xing J, Hedges DJ, Han K, Wang H, Cordaux R, Batzer MA. Alu element mutation spectra: molecular clocks and the effect of DNA methylation. J Mol Biol 2004; 344:675-82; PMID:15533437; http://dx.doi.org/ 10.1016/j.jmb.2004.09.058 [DOI] [PubMed] [Google Scholar]
- 15.Schmid CW. Human Alu subfamilies and their methylation revealed by blot hybridization. Nucleic Acids Res 1991; 19:5613-7; PMID:1945838; http://dx.doi.org/ 10.1093/nar/19.20.5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hellmann-Blumberg U, Hintz MF, Gatewood JM, Schmid CW. Developmental differences in methylation of human Alu repeats. Mol Cell Biol 1993; 13:4523-30; PMID:8336699; http://dx.doi.org/ 10.1128/MCB.13.8.4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochanek S, Renz D, Doerfler W. DNA methylation in the Alu sequences of diploid and haploid primary human cells. EMBO J 1993; 12:1141-51; PMID:8384552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez J, Vives L, Jorda M, Morales C, Munoz M, Vendrell E, Peinado MA. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res 2008; 36:770-84; PMID:18084025; http://dx.doi.org/ 10.1093/nar/gkm1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie H, Wang M, Bonaldo M, de F, Smith C, Rajaram V, Goldman S, Tomita T, Soares MB. High-throughput sequence-based epigenomic analysis of Alu repeats in human cerebellum. Nucleic Acids Res 2009; 37:4331-40; PMID:19458156; http://dx.doi.org/ 10.1093/nar/gkp393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubin CM, VandeVoort CA, Teplitz RL, Schmid CW. Alu repeated DNAs are differentially methylated in primate germ cells. Nucleic Acids Res 1994; 22:5121-7; PMID:7800508; http://dx.doi.org/ 10.1093/nar/22.23.5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011; 146:1029-41; PMID:21925323; http://dx.doi.org/ 10.1016/j.cell.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab 1999; 67:183-93; PMID:10381326; http://dx.doi.org/ 10.1006/mgme.1999.2864 [DOI] [PubMed] [Google Scholar]
- 23.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet 2009; 10:691-703; PMID:19763152; http://dx.doi.org/ 10.1038/nrg2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miki Y, Katagiri T, Kasumi F, Yoshimoto T, Nakamura Y. Mutation analysis in the BRCA2 gene in primary breast cancers. Nat Genet 1996; 13:245-7; PMID:8640237; http://dx.doi.org/ 10.1038/ng0696-245 [DOI] [PubMed] [Google Scholar]
- 25.Teugels E, De Brakeleer S, Goelen G, Lissens W, Sermijn E, De Greve J. De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum Mut 2005; 26:284; PMID:16088935; http://dx.doi.org/ 10.1002/humu.9366 [DOI] [PubMed] [Google Scholar]
- 26.Peixoto A, Pinheiro M, Massena L, Santos C, Pinto P, Rocha P, Pinto C, Teixeira MR. Genomic characterization of two large Alu-mediated rearrangements of the BRCA1 gene. J Hum Genet 2013; 58:78-83; PMID:23223007; http://dx.doi.org/ 10.1038/jhg.2012.137 [DOI] [PubMed] [Google Scholar]
- 27.Park SY, Seo AN, Jung HY, Gwak JM, Jung N, Cho NY, Kang GH. Alu and LINE-1 hypomethylation is associated with HER2 enriched subtype of breast cancer. PLoS One 2014; 9:e100429; PMID:24971511; http://dx.doi.org/ 10.1371/journal.pone.0100429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae JM, Shin SH, Kwon HJ, Park SY, Kook MC, Kim YW, Cho NY, Kim N, Kim TY, Kim D, et al.. ALU and LINE-1 hypomethylations in multistep gastric carcinogenesis and their prognostic implications. Int J Cancer 2012; 131:1323-31; PMID:22120154; http://dx.doi.org/ 10.1002/ijc.27369 [DOI] [PubMed] [Google Scholar]
- 29.Bollati V, Fabris S, Pegoraro V, Ronchetti D, Mosca L, Deliliers GL, Motta V, Bertazzi PA, Baccarelli A, Neri A. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009; 30:1330-5; PMID:19531770; http://dx.doi.org/ 10.1093/carcin/bgp149 [DOI] [PubMed] [Google Scholar]
- 30.Akers SN, Moysich K, Zhang W, Collamat Lai G, Miller A, Lele S, Odunsi K, Karpf AR. LINE1 and Alu repetitive element DNA methylation in tumors and white blood cells from epithelial ovarian cancer patients. Gynecol Oncol 2014; 132:462-7; PMID:24374023; http://dx.doi.org/ 10.1016/j.ygyno.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhee YY, Lee TH, Song YS, Wen X, Kim H, Jheon S, Lee CT, Kim J, Cho NY, Chung JH, et al.. Prognostic significance of promoter CpG island hypermethylation and repetitive DNA hypomethylation in stage I lung adenocarcinoma. Virchows Arch 2015; 466:675-83; PMID:25772390; http://dx.doi.org/ 10.1007/s00428-015-1749-0 [DOI] [PubMed] [Google Scholar]
- 32.Li J, Huang Q, Zeng F, Li W, He Z, Chen W, Zhu W, Zhang B. The prognostic value of global DNA hypomethylation in cancer: a meta-analysis. PLoS One 2014; 9:e106290; PMID:25184628; http://dx.doi.org/ 10.1371/journal.pone.0106290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khakpour G, Pooladi A, Izadi P, Noruzinia M, Tavakkoly Bazzaz J. DNA methylation as a promising landscape: A simple blood test for breast cancer prediction. Tumour Biol 2015; 36:4905-12; PMID:26076810; http://dx.doi.org/ 10.1007/s13277-015-3567-z [DOI] [PubMed] [Google Scholar]
- 34.Ekram MB, Kim J. High-throughput targeted repeat element bisulfite sequencing (HT-TREBS): genome-wide DNA methylation analysis of IAP LTR retrotransposon. PLoS One 2014; 9:e101683; PMID:25003790; http://dx.doi.org/ 10.1371/journal.pone.0101683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakshi A, Ekram MB, Kim J. Locus-specific DNA methylation analysis of retrotransposons in ES, somatic and cancer cells using High-throughput targeted repeat element bisulfite sequencing. Genom Data 2015; 3:87-9; PMID:25554740; http://dx.doi.org/ 10.1016/j.gdata.2014.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010; 28:495-501; PMID:20436461; http://dx.doi.org/ 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Sci Acad 2010; 107:21931-6; PMID:21106759; http://dx.doi.org/20657582 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 2010; 28:817-25; PMID:20657582; http://dx.doi.org/ 10.1038/nbt.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; http://dx.doi.org/ 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al.. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315-22; PMID:19829295; http://dx.doi.org/ 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al.. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2012; 44:40-6; PMID:22120008; http://dx.doi.org/21706001 10.1038/ng.969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al.. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43:768-75; PMID:21706001; http://dx.doi.org/ 10.1038/ng.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al.. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 2011; 44:17-28; PMID:21924933; http://dx.doi.org/ 10.1016/j.molcel.2011.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, et al.. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 2012; 22:246-58; PMID:22156296; http://dx.doi.org/ 10.1101/gr.125872.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al.. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500:477-81; PMID:23925113; http://dx.doi.org/ 10.1038/nature12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 1997; 25:2532-4; PMID:9171110; http://dx.doi.org/ 10.1093/nar/25.12.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein JN, Collisson EA, Mills GB, Shaw KM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat Genet 2013; 45:1113-20; PMID:24071849; http://dx.doi.org/ 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ueki T, Park JH, Nishidate T, Kijima K, Hirata K, Nakamura Y, Katagiri T. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res 2009; 69:8752-60; PMID:19887602; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1809 [DOI] [PubMed] [Google Scholar]
- 49.Blomquist TM, Brown RD, Crawford EL, de la Serna I, Williams K, Yoon Y, Hernandez DA, Willey JC. CEBPG exhibits allele-specific expression in human bronchial epithelial cells. Gene Regul Syst Biol 2013; 7:125-38; http://dx.doi.org/ 10.4137/GRSB.S11879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford EL, Blomquist T, Mullins DN, Yoon Y, Hernandez DR, Al-Bagdhadi M, Ruiz J, Hammersley J, Willey JC. CEBPG regulates ERCC5/XPG expression in human bronchial epithelial cells and this regulation is modified by E2F1/YY1 interactions. Carcinogenesis 2007; 28:2552-9; PMID:17893230; http://dx.doi.org/ 10.1093/carcin/bgm214 [DOI] [PubMed] [Google Scholar]
- 51.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357-9; PMID:22388286; http://dx.doi.org/ 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lutsik P, Feuerbach L, Arand J, Lengauer T, Walter J, Bock C. BiQ Analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res 2011; 39:W551-6; PMID:21565797; http://dx.doi.org/ 10.1093/nar/gkr312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30:207-10; PMID:11752295; http://dx.doi.org/ 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005; 21:4067-8; PMID:16141249; http://dx.doi.org/ 10.1093/bioinformatics/bti652 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.