

Abstract
The present studies sought to determine whether the lethality of the drug combination [sorafenib + sildenafil] could be enhanced by the anti-inflammatory agent celecoxib, using ovarian cancer and other tumor cell lines as models. Also, in a dose dependent fashion celecoxib enhanced [sorafenib + sildenafil] lethality in multiple ovarian cancer cell lines. In a dose dependent fashion celecoxib enhanced the ability of [sorafenib + sildenafil] to reduce expression of multiple chaperone proteins in parallel with lower levels of the drug efflux pumps ABCB1 and ABCG2. Over-expression of GRP78 and HSP27 maintained pump expression in the presence of drugs. Cell killing by the 3 drug combination was mediated by mitochondrial / caspase 9 –dependent apoptotic signaling and by RIP-1 / caspases 2 and 4 / AIF –dependent necroptotic signaling. Pre-treatment of intrinsically resistant primary ovarian cancer cells with [celecoxib + sorafenib + sildenafil] significantly enhanced tumor cell killing by a subsequent cisplatin exposure. Similar data were obtained in some cancer cell lines, but not all, using the related platinum containing drugs, oxaliplatin and carboplatin. As our prior publications have also validated in vivo the combinations of [celecoxib + sildenafil] and [sorafenib + sildenafil] as cytotoxic to multiple tumor cell types, combined with the present findings, we would argue that the combination of celecoxib/sorafenib/sildenafil should be explored in a new phase I trial in ovarian cancer.
Abbreviations
- ERK
extracellular regulated kinase
- MEK
mitogen activated extracellular regulated kinase
- PI3K
phosphatidyl inositol 3 kinase
- ca
constitutively active
- dn
dominant negative
- ER
endoplasmic reticulum
- mTOR
mammalian target of rapamycin
- MAPK
mitogen activated protein kinase
- PTEN
phosphatase and tensin homolog on chromosome 10
- ROS
reactive oxygen species
- CMV
empty vector plasmid or virus
- si
small interfering
- SCR
scrambled
- IP
immunoprecipitation
- Ad
adenovirus
- VEH
vehicle.
Introduction
Approximately 22,000 American women are diagnosed with ovarian cancer every year and more than 14,000 die annually of the disease. It is the second most common gynecologic malignancy in the United States and is the deadliest of gynecologic cancers and the fifth leading cause of cancer death among women.1 Standard of care therapy usually involves debulking surgery followed by platinum/taxane-based chemotherapy as the first line treatment.2 Second line therapies used at recurrence depend largely on whether the tumor is still sensitive to platinum therapy (platinum-free interval ≥ 6 months) wherein platinum-based therapy is repeated or resistant to platinum therapy (platinum-free interval < 6 months) in which case non-platinum monotherapies are typically used. These salvage therapies are palliative rather than curative in nature and include liposomal doxorubicin, gemcitabine, bevacizumab and topotecan.3 As ovarian cancer often presents with dissemination throughout the abdomen and pelvis, complete surgical removal of the tumor (optimal debulking) may not be possible, and even when complete surgical removal is possible disease recurrence is frequent. Additionally, some 20% of ovarian tumors present with de novo resistance to platinum and these patients present the greatest challenge for the clinician.3 Thus, there are many cases of ovarian cancer where alternate therapeutic approaches are needed to improve upon the current 5 y survival for all forms of ovarian cancer of 45%, with stage III and stage IV having 5-year survivals of only 35% and 10%, respectively, 3 speaking to the urgent need for better therapies for this disease.
Platinum containing chemotherapeutic drugs (cisplatin; oxaliplatin; carboplatin) are also widely used in the treatment of many thoracic and GI malignancies.4,5 As with ovarian cancer, many of these tumors at initial presentation also exhibit resistance to platinum containing drugs, and previously treated tumors are very often resistant to platinum. Collectively, there clearly is an urgent need to develop new approaches that will both kill potentially platinum sensitive tumors outright, sensitize these cells to platinum, and re-establish platinum sensitivity after development of secondary resistance. Of all the drugs used to treat ovarian cancer, the platinum based drugs remain the mainstay of treatment and the ability to sensitize these tumors to initial platinum or re-establish sensitivity at the time of platinum resistant recurrence remains a major goal of cancer therapeutics.
Studies by the Dent laboratory over the previous 10 y have highlighted the usefulness of manipulating the ERK and PI3K signal transduction pathways in parallel with causing endoplasmic reticulum stress and autophagy to kill tumor cells.6-15 Most recently we have demonstrated that the multi-kinase inhibitor sorafenib (Nexavar®) or the anti-inflammatory drug celecoxib (Celebrex®) when combined with the phosphodiesterase 5 inhibitors sildenafil (Viagra®) or tadalafil (Cialis®), results in synergistic profound levels of tumor cell killing both in vitro and also in multiple animal model systems in vivo.13-15 Normal tissue toxicity was minimal and animal body mass unaltered by these 2 drug combinations; tumor cells were preferentially killed compared to non-transformed cells which may be a reflection on transformed cells expressing much higher protein levels associated with a greater expression of chaperone proteins and an elevated basal level of ER stress. Tumor cell killing with [sorafenib + sildenafil] and [celecoxib + sildenafil] occurred through activation of the death receptor CD95 and through elevated levels of toxic autophagosome formation. For [sorafenib + sildenafil] only, expression of CD95 was not absolutely essential for increased levels of death though artificial expression of the death receptor facilitated killing. As single agents, neither sorafenib nor celecoxib have any known significant anti-tumor properties in ovarian cancer patients.
The present studies were performed to determine whether [sorafenib + sildenafil] or [celecoxib + sildenafil] killed ovarian cancer cells in vitro and in vivo. Compared to many other tumor cell types previously tested, such as breast and brain tumor cells, ovarian tumor cells were generally more resistant to being killed by both 2-drug combinations. In a dose-dependent fashion, low concentrations of celecoxib significantly increased the killing efficacy of [sorafenib + sildenafil] against established and primary ovarian tumor cells. In addition, 3 drug combination exposure sensitized ovarian tumor cells to cisplatin, and the 3 drug combination platinum sensitized primary ovarian tumor cells that were de novo highly resistant in the patient to cisplatin therapy. Collectively, our findings strongly argue that the combination of clinically relevant levels of sorafenib, celecoxib and sildenafil has the potential to be a new approach for ovarian cancer treatment.
Materials and Methods
Materials
Phospho-/total- antibodies were purchased from Cell Signaling Technologies (Danvers, MA) and Santa Cruz Biotech. (Santa Cruz, CA). All drugs were purchased from Selleckchem (Houston, TX). Commercially available validated short hairpin RNA molecules to knock down RNA / protein levels were from Qiagen (Valencia, CA). At least 2 different validated siRNA molecules were independently used to confirm the effects observed were not due to non-specific effects. The plasmid to express GRP78/BiP/HSPA5 was kindly provided to the Dent laboratory by Dr. A.S. Lee (University of Southern California, Los Angeles, CA). All other plasmids were purchased from Addgene under material transfer agreements. Antibody reagents, other kinase inhibitors, caspase inhibitors cell culture reagents, and non-commercial recombinant adenoviruses have been previously described. Established cell lines were purchased from the ATCC and were not further validated in the Dent lab.8-17 De novo carboplatin / paclitaxel resistant “Spiky” ovarian cancer cells as well as CTG-1677 and CTG-1703 CDDP resistant cells, PDX models, were kindly provided by Dr. Karen Paz (Champions Oncology, NJ). GBM cells were obtained from the Mayo Clinic repository (Rochester, MN).
Methods
Culture and in vitro exposure of cells to drugs
All cell lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short term cell killing assays, immunoblotting / IF studies, cells were plated at a density of 3 × 103 per cm2 (∼2 × 105 cells per well of a 12 well plate) and 24 h after plating treated with various drugs, as indicated. In vitro drug treatments were from 100 mM stock solutions of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
Cell death measurements by live / dead assay
Cells were grown in 96 well plates with each well containing ∼10,000 cells in 200 ml of media. Cells were treated with the indicated concentrations of drugs for the indicated amounts of time in each panel. Plates were then centrifuged (500 rpm, 5 min) to re-adhere floating dead cells to the base of each well. The media was removed and live / dead assay reagent added and cells incubated for 10 min before the reagent was removed. Cells were imaged in a Hermes WiScan instrument under 10× magnification. Green cells = viable; yellow/red cells = dying / dead. The numbers of viable and dead cells were counted manually from several images taken from one well together with images from another 2 wells.
Transfection of cells with siRNA or with plasmids
For plasmids
Cells were plated as described above and 24 h after plating, transfected. For mouse embryonic fibroblasts (2–5 μg) or other cell types (0.5 μg) plasmids expressing a specific mRNA (or siRNA) or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium (1 portion for each sample). Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well / dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl, and the cells were incubated for 4 h at 37°C. An equal volume of 2× medium was then added to each well. Cells were incubated for 48 h, then treated with drugs.
Transfection for siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used. Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37°C for 2h. One ml of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37°C for 48h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with drugs (0–24 h).
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a 2 tailed Student's t-test. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Results
Initial studies were performed in 2 well studied established ovarian tumor cell lines, OVCAR and SKOV-3. Treatment of tumor cells with [sorafenib + sildenafil] for 24 h resulted in a reduction in total cell numbers and a modest albeit significant ∼10% increase in the percentage of dead cells over control cell death values (Fig. 1A, p < 0.05). In OVCAR cells, at concentrations of celecoxib as low as 1–2 μM, the lethality of [sorafenib + sildenafil] treatment was profoundly enhanced; SKOV-3 cells were more resistant requiring >3 μM celecoxib for an obvious clear increase in [sorafenib + sildenafil] lethality (both, p < 0.05). In multiple other ovarian cancer cell lines as well as in primary / PDX ovarian tumor isolates (Spiky, MCVH OP1, CTG-1677, CTG-1703) 3 drug combined [celecoxib + sorafenib + sildenafil] treatment caused significant amount of tumor cell death (Figs. 1B–1E, p < 0.05). Very similar data to that in ovarian cancer cells was also obtained examining primary PDX human glioblastoma cells (Fig. 1F, p < 0.05).
Figure 1.

Celecoxib enhances the lethality of [sorafenib + sildenafil] in ovarian cancer cells. (A) OVCAR and SKOV3 cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM)], and with increasing concentrations of celecoxib (0–4 μM). Twenty four h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (B) CAOV-3, CAOV-4 and PA-1 established ovarian tumor cells and MCVH OP1 fresh primary ovarian cancer cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)], as indicated in the panel. Twenty four h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (C) CRL-1572 established ovarian tumor cells and Spiky primary ovarian cancer cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)], as indicated in the panel. Twenty four h and 48h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (D and E) CTG-1677 and CTG-1703 PDX primary ovarian tumor cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)], as indicated in the panel. Twenty four h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (F) GBM5/6/12/14 primary glioblastoma cancer cells were treated with vehicle or [sorafenib (0.5, 2.0 μM) + sildenafil (2 μM) + celecoxib (5 μM)], as indicated in the panel. Twenty four h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
It has been well known for many years that cancer cells isolated from recurrent ovarian cancer patients are variably resistant to prior standard of care therapies, particularly cisplatin and carboplatin. One well characterized mechanism of chemotherapy resistance is by increased expression of the plasma membrane drug efflux pumps ABCB1 and ABCG2. Treatment of ovarian cancer cells with [sorafenib + sildenafil] reduced expression of ABCB1, an effect that was enhanced by simultaneous exposure to increasing concentrations of celecoxib (Figs. 2A and 2B). Very similar data was obtained when the expression of ABCG2 was examined after drug exposure (Figs. 2C and 2D). In addition to altering the expression of drug efflux pumps, treatment of cells with [celecoxib + sorafenib + sildenafil] for 6 h consistently resulted in reduced expression of the chaperone proteins GRP78, HSP70 and HSP27, and the HSP70 chaperone modulator protein BAG3 (Figs. 2E–2G). Overexpression of the chaperone proteins HSP27 and GRP78 prevented [celecoxib + sorafenib + sildenafil] from reducing the expression of ABCB1 and ABCG2 (Fig. 2H).
Figure 2.

Celecoxib / Sorafenib / Sildenafil treatment decreases the expression of the plasma membrane drug efflux pumps ABCB1 and ABCG2. (A–D) OVCAR and SKOV3 cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM)], and with increasing concentrations of celecoxib (0–4 μM). Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of ABCB1 and ABCG2 at 10X using a Hermes WiScan wide field microscope. (E and F) Spiky primary ovarian cancer cells and OVCAR established ovarian cancer cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of: BAG1, BAG2, BAG3, HSP70, HSP90, GRP78, GRP75, mitochondrial HSP70 and HSP60 using a Hermes WiScan wide field microscope. (G) Spiky primary ovarian cancer cells and OVCAR, CAOV3 and PA-1 established ovarian cancer cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of HSP27 using a Hermes WiScan wide field microscope. (H) Spiky and OVCAR cells were transfected with empty vector plasmid (CMV); a plasmid to express GRP78; a plasmid to express HSP27; or to express GRP78 and HSP27. Twenty four h after transfection cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of ABCB1 and ABCG2 using a Hermes WiScan wide field microscope. (I) OVCAR and SKOV3 cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)] in the presence or absence of elacridar (Pi, 80 nM). Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
The drug elacridar (in the Figure as pump inhibitor, “Pi”) was developed as a high specificity potent inhibitor of ABCB1 and ABCG2 that failed in the clinic due to poor PK/PD in patients with a C max of approximately 100 nM, where a sustained required therapeutic level of the drug, ≥ 200 nM, could not be achieved. As treatment of tumor cells with [sorafenib + sildenafil + celecoxib] caused a reduction in total ABCB1 and ABCG2 levels, and these pumps can cause sorafenib efflux from the cell, we investigated the impact of elacridar on [sorafenib + sildenafil + celecoxib] lethality. Twelve h after drug treatment there was no significant difference in the lethality of [sorafenib + sildenafil + celecoxib] regardless of elacridar (at 80 nM) addition (∼50% of the OVCAR cells dead at 12 h) (Fig. 2I). In the much more chemotherapy resistant Apaf-1 mutant cell line SKOV-3, elacridar had no effect on [sorafenib + sildenafil + celecoxib] toxicity, even though [sorafenib + sildenafil + celecoxib] treatment reduced ABCB1 and ABCG2 expression. Collectively these data suggests that modulation of pump expression / function is unlikely to fully explain how [sorafenib + sildenafil + celecoxib] can kill ovarian tumor cells.
We next determined the molecular mechanisms by which [celecoxib + sorafenib + sildenafil] interacted to kill ovarian tumor cells. Treatment of control transfected cells with the 3 drug [celecoxib + sorafenib + sildenafil] combination increased the true percentage of cell death by 27% over basal levels, an effect that was significantly reduced by knock down of the AMP-dependent protein kinase α and β subunits (AMPK) (Fig. 3A). Knock down of PKR-like endoplasmic reticulum kinase (PERK); and its downstream effectors ATF4 and the transcription factor CHOP also significantly reduced drug combination lethality (in all manipulations, p < 0.05). As noted in Figure 2, the expression of GRP78 is reduced by drug combination treatment and in agreement with reduced levels of this chaperone, this de-repression of PERK activity results in elevated levels of eIF2α Serine 51 phosphorylation being detected (Figs. 3B–3F). Expression of a dominant negative eIF2α S51A protein or over-expression of GRP78 protected cells from [celecoxib + sorafenib + sildenafil] treatment whereas knock down of GRP78 enhanced tumor cell killing (Fig. 3G).
Figure 3.

Part 1: Multiple apoptotic and necroptotic mechanisms facilitate ovarian tumor cell killing by [sorafenib + sildenafil + celecoxib]. (A) Upper: OVCAR cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM); or all 3 drug together for 12h. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. Lower: OVCAR cells were transfected with scrambled siRNA (siSCR); an siRNA to knock down AMPKα; to knock down PERK, ATF4 and CHOP. Twenty four h after transfection cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)] for 12h. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (B) Spiky cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM)]; or all 3 drug together for 6h. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of GRP78 and the Serine 51 phosphorylation of eIF2α using a Hermes WiScan wide field microscope. (C–D) Spiky cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM)]; or all 3 drug together for 12h. Twelve h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of GRP78 and the Serine 51 phosphorylation of eIF2α using a Hermes WiScan wide field microscope. (E–F) OVCAR cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM)]; or all 3 drug together for 12h. Twelve h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of GRP78 and the Serine 51 phosphorylation of eIF2α using a Hermes WiScan wide field microscope. (G) Spiky and OVCAR cells were transfected with, as indicated: an empty vector plasmid (CMV); a scrambled siRNA (siSCR); an siRNA to knock down GRP78 expression; a plasmid to express a dominant negative eIF2α S51A protein. Twenty four h after transfection cells were treated with vehicle or with [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
In addition to the studies in Figure 3, we also explored whether [celecoxib + sildenafil + sildenafil] treatment utilized apoptotic, necrotic or autophagy pathways in the cell killing process. Treatment of cells with [celecoxib + sorafenib + sildenafil] reduced the expression of MCL-1, BCL-XL and c-FLIP-s in an eIF2α-dependent mechanism, which was associated with pro-caspase 3 cleavage (Fig. 4A). Of note, in the de novo highly chemotherapy resistant PDX ovarian cancer model, Spiky; the basal levels of MCL-1, BCL-XL and c-FLIP-s were all very much higher than in chemotherapy sensitive OVCAR cells (anywhere from 2–10 –fold increased, all p < 0.05).
Figure 4.

Part 2: Multiple apoptotic and necroptotic mechanisms facilitate ovarian tumor cell killing by [sorafenib + sildenafil + celecoxib]. (A) Spiky and OVCAR cells were transfected with an empty vector plasmid (CMV) or a plasmid to express a dominant negative eIF2α S51A protein. Twenty four h after transfection cells were treated with vehicle or with [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Twelve h after drug treatment, cells were processed were fixed in place and immuo-staining performed to detect the expression of: MCL-1, BCL-XL, c-FLIP-s and cleaved caspase 3, using a Hermes WiScan wide field microscope. (B) OVCAR cells were transfected with, as indicated: an empty vector plasmid (CMV); a scrambled siRNA (siSCR); an siRNA to knock down RIP-1 expression; and plasmids to express c-FLIP-s, BCL-XL and dominant negative caspase 9 proteins. Twenty four h after transfection cells were treated with vehicle or with [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (C) OVCAR cells were transfected with, as indicated: a scrambled siRNA (siSCR); and siRNA molecules to knock down ATG5 or Beclin1 expression. Twenty four h after transfection cells were treated with vehicle or with [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (D) OVCAR and Spiky cells were transfected with, as indicated: a scrambled siRNA (siSCR); and siRNA molecules to knock down BID or [caspase 2 + caspase 4] expression. Twenty four h after transfection cells were treated with vehicle or with [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. Twelve h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
We next delved into mechanisms of tumor cell killing. Treatment of control transfected cells with the 3 drug combination increased the true percentage cell death by 38% over basal levels, an effect that was significantly reduced by overexpression of the caspase 8/10 inhibitor c-FLIP-s, although of note, expression of c-FLIP-s did not modify the obvious decrease in cell numbers caused by drug treatment (Fig. 4B, p < 0.05). Over-expression of either BCL-XL or dominant negative caspase 9 also significantly reduced drug combination lethality and largely abolished the anti-proliferative effects of drug treatment (both for BCL-XL and dom. neg. caspase 9, p < 0.05). Knock down of RIP-1, an upstream regulator of necroptosis, also significantly reduced drug lethality and the anti-proliferative effects of drug exposure (p < 0.05). The relative contributions of death receptor – caspase 8 and death receptor – RIP-1 signaling at causing ovarian tumor cell death will require studies beyond the scope of this manuscript.
In our prior studies in other tumor types we had demonstrated that [sorafenib + sildenafil] or [celecoxib + sildenafil] as 2 drug treatments increased the levels of autophagosomes in cells which was associated with increased tumor cell killing. In ovarian cancer cells knock down of the autophagy regulatory proteins Beclin1 or ATG5 unexpectedly and modestly increased [celecoxib + sorafenib + sildenafil] killing (Fig. 4C). Downstream of RIP-1 signaling in the necroptotic pathway to cell death are caspases 2 and 4, and the toxic BH3 domain protein BID. Knock down of caspase 2 and caspase 4 together, or knock down of BID, was significantly protective against [celecoxib + sorafenib + sildenafil] toxicity (Fig. 4D, p < 0.05).
We next determined the impact of [celecoxib + sorafenib + sildenafil] treatment on well-known signal transduction pathways whose activities have been linked to tumor growth and invasion, as well as resistance to chemotherapy. In the primary “Spiky” ovarian cancer isolate, [celecoxib + sildenafil] increased the phosphorylation of JNK1/2 (Fig. 5A). Both [celecoxib + sildenafil] and [sorafenib + sildenafil] reduced STAT5 regulatory tyrosine phosphorylation but only the 3 drug combination reduced STAT3 tyrosine phosphorylation. Treatment with the 3 drug combination was required to suppress the phosphorylation of AKT (T308); p65 NFκB; and ERK1/2. Based on these data, we transfected cells to express an activated form of MEK1; an activated form of AKT; an activated for of STAT3; and a dominant negative super-repressor of p65, IκB S32A S36A. A separate set of control transfected cells were also treated with a molecular tool to inhibit JNK signaling; the JNK inhibitory peptide. Inhibition of JNK signaling was modestly protective in OVCAR cells and strongly protective in Spiky cells (Fig. 5B). Expression of dominant negative IκB strongly enhanced drug combination killing in Spiky cells, but surprisingly not OVCAR cells. Consistently in both cell lines, expression of an activated form of AKT strongly protected cells from drug toxicity (p < 0.05).
Figure 5.

Ovarian tumor cell killing requires activation of the JNK pathway and inactivation of [STAT3 + AKT + NFκB]. (A) Spiky ovarian tumor cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM)]; or all 3 drugs together for 6h. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the phosphorylation of JNK1/2; STAT3; STAT5; AKT T308; p65 NFκB; and ERK1/2, using a Hermes WiScan wide field microscope. (B) OVCAR and Spiky cells were transfected with empty vector plasmid (CMV) or plasmids to express activated MEK1; activated AKT; activated STAT3; and the dominant negative super-repressor IκB S32A S36A. A portion of CMV transfected cells was treated with the JNK inhibitory peptide (10 μM). Twenty four h after transfection, cells were treated with vehicle; [sorafenib (2 μM) + sildenafil (2 μM)]; [celecoxib (2 μM) + sildenafil (2 μM)]; or all 3 drugs together for 24h. Twenty four h after drug treatment, cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
A significant problem in the treatment of recurrent ovarian cancer is that the cells have become resistant to the most efficacious primary standard of care therapy, platinum.16 Some unfortunate patients present with tumors that are already de novo resistant to cisplatin and also taxane therapy, as was the case for the patient whose cells in these studies have been termed “Spiky” based on their morphology. This patient did not respond to carboplatin or paclitaxel at presentation with only a brief subsequent response to gemcitabine therapy and died approximately 4.5 months after initial diagnosis.
Ovarian tumor cell lines, as well as Spiky, were treated with vehicle control or with [celecoxib + sorafenib + sildenafil] for 12 h, at which time cells were additionally then treated with vehicle control or cisplatin, and then cell viability assessed at 24 h. As previously noted, after 24 h of [celecoxib + sorafenib + sildenafil] treatment, SKOV-3 and Spiky cells exhibited modest increases in cell killing compared to control cells, whereas PA-1 and CAOV-3 cells exhibited strong levels of cell death (Fig. 6A, p < 0.05). Other than in CAOV-3 cells where a modest response was observed, cisplatin did not alter tumor cell viability at this time point. In all cell lines tested except SKOV-3, [celecoxib + sorafenib + sildenafil] treatment profoundly enhanced cisplatin toxicity 12 h after the start of cisplatin exposure. Thus SKOV-3 cells are more resistant to platinum-induced cell killing than Spiky cells which are derived from a patient whose tumor was de novo platinum resistant.
Figure 6.

Treatment of tumor cells with [sorafenib + sildenafil + celecoxib] sensitizes them to chemotherapy drugs containing platinum. (A) Spiky, PA-1, SKOV3 and CAOV-3 cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, cisplatin (CDDP, 50 nM) was added to the culture media. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (B) CTG-1677 and CTG-1703 PDX primary ovarian tumor cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, cisplatin (CDDP, 50 nM) was added to the culture media. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (C) Spiky cells as separated single cells were plated in 6 well plates (250–1,500 cells per well). Twelve h after plating cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, cisplatin (CDDP, 50 nM) was added to the culture media. At the 24 h time point the drug containing media was removed, the cells washed once with warm drug free media, and the cells cultured for another 7–10 d in drug free media. Cells were then fixed and stained with crystal violet and colonies of > 50 cells counted (n = 6 ++− SEM). (D) Non-small cell lung cancer cells: Left: were treated for 12 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 6h after the initiation of the experiment, cisplatin (CDDP, 50 nM) was added to the culture media. At the 12 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. Right: Non-small cell lung cancer cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (E) Testicular carcinoma cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, cisplatin (CDDP, 50 nM) was added to the culture media. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (F) OVCAR cells were treated with vehicle control or with [sorafenib (2 μM) / celecoxib (2 μM) + sildenafil (2 μM)] for 6h after which the drug containing media was removed, the cells washed once with warm drug free media, and the cells cultured for another 18h (24 h assay total time) in media treated with vehicle control or cisplatin (CDDP, 50 nM). At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (G) Spiky and OVCAR ovarian cancer cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, oxaliplatin (OX, 50 nM) or carboplatin (CARBO, 50 nM) was added to the culture media. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (H and I) PA-1 and CAOV-3, and CGT-1677 and CGT-1703 ovarian cancer cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, oxaliplatin (OX, 50 nM) or carboplatin (CARBO, 50 nM) or cisplatin (CDDP, 50 nM) was added to the culture media. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system. (J) Tumor cells from various tissues were analyzed: NSCLC (ADOR, H1299); stomach (AGS); colorectal (HCT116); pancreatic (Mia Paca 2, PANC1); hepatoma (HuH7, HEP3B). Cells were treated for 24 h with either vehicle control or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)]. In portions of cells, 12 h after the initiation of the experiment, oxaliplatin (OX, 50 nM) or carboplatin (CARBO, 50 nM) was added to the culture media. Nota bene, hepatoma cells were treated for only 12 h. At the 24 h time point cells were processed and subjected to live/dead assays in a Hermes Wiscan system.
In colony formation assays where Spiky cells were treated for only 24 h with drugs as per the short-term death assays in Figure 6A, the outgrowth of cisplatin treated cells into colonies of 50 or greater cells was significantly reduced at clinically relevant cisplatin concentrations ≤ 50 nM by [celecoxib + sorafenib + sildenafil] (Fig. 6B, p < 0.05). Other tumor types are also treated with platinum containing drugs, notably non-small cell lung cancer and testicular cancer. Lung cancer cells were in general more efficiently killed by [celecoxib + sorafenib + sildenafil] treatment than were ovarian cancer cells with all lines tested showing significant increases in cell death after 24 h of exposure (Fig. 6C, p < 0.05). Studies with cisplatin were performed with cisplatin addition at 6 h after the 3 drug combination, and cell death then assessed at 12 h. In all lines tested [celecoxib + sorafenib + sildenafil] treatment enhanced the lethality of cisplatin. Similar killing and platinum sensitization data were obtained in 2 testicular cancer cell lines (Fig. 6D). Of note, the use of recurrent testicular tumor cells, previously exposed to cisplatin and carboplatin therapies, would a priori be predicted to generate isolates that are cisplatin and carboplatin resistant, and our data shows that such cells are not sensitized to cisplatin or carboplatin by [celecoxib + sorafenib + sildenafil]; yet oxaliplatin, that uses a different mechanism of killing and would not normally be considered as a testicular cancer therapeutic, was very effective in combination with [celecoxib + sorafenib + sildenafil] treatment in killing.
We next attempted to approximately determine how much time [celecoxib + sorafenib + sildenafil] treatment of ovarian tumor cells was required to facilitate a cisplatin sensitization effect. As we observed profound changes in efflux pump expression; chaperone function; and signaling pathway alterations after 6 h of treatment we chose this time point. OVCAR cells were treated for 6 h with vehicle control or with [celecoxib + sildenafil] / [sorafenib + sildenafil] or the 3 drug combination and the drugs removed and fresh media added; media containing vehicle control or cisplatin (50 nM). In viability assays performed 18 h after cisplatin addition we noted that a 6 h pre-treatment of OVCAR cells with [celecoxib + sorafenib + sildenafil] significantly increased the true percentage cell death caused by cisplatin over basal levels at 24 h by 39% (Fig. 6E, p < 0.05).
The molecular mechanisms by which cisplatin and carboplatin the related platinum containing drug oxaliplatin kill tumor cells are not identical, and cells that are cisplatin resistant are very often still sensitive to oxaliplatin. Platinum drugs are used in the treatment of multiple thoracic and GI malignancies including lung cancer and colorectal cancer. In a similar manner to our studies in Figure 6A, we determined whether [celecoxib + sorafenib + sildenafil] treatment could enhance oxaliplatin / carboplatin toxicity in ovarian tumor cells as well as in multiple thoracic and GI tumor cell types. Treatment of Spiky and OVCAR tumor cells with [celecoxib + sorafenib + sildenafil] significantly enhanced the levels of cell death caused by oxaliplatin and by carboplatin in some tumor cell lines e.g. OVCAR, but did not enhance sensitivity in others e.g., Spiky (Fig. 6F, p < 0.05). Similar sensitization data were also obtained in the established ovarian cancer lines PAI-1 and CAOV3; as well as in the CDDP-resistant PDX ovarian cancer models CTG-1677 and CTG-1703 cells, with cells being sensitized to CDDP and to carboplatin, but not oxaliplatin (Figs. 6G–6I).
Several proteins have been linked with the resistance of tumor cells to cisplatin, most recently the proteins GRP78, GST-π, WASF3 and ATAD3A.17-19 In particular, GRP78 is believed to act as an essential chaperone for the WASF3 and ATAD3A. Spiky ovarian tumor cells, which were found to be inherently resistant to cisplatin in the patient and in vitro exhibited the highest levels of GRP78, GST-π, WASF3 and ATAD3A expression when compared to platinum sensitive OVCAR, CAOV3 and PA-I cells (Fig. 7, upper images). Combined overexpression of HSP27 and GRP78 prevented [celecoxib + sorafenib + sildenafil] treatment from reducing the expression of GST-π, WASF3 and ATAD3A within 6 h (Fig. 7, lower images).
Figure 7.
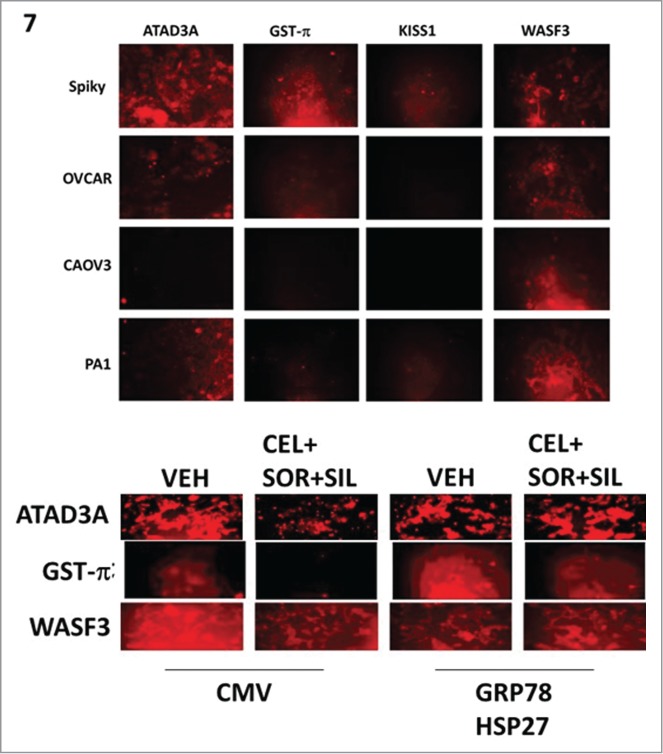
Treatment of ovarian carcinoma cells with [celecoxib + sorafenib + sildenafil] reduces expression of ATAD3A, WASF3 and GST-p that is prevented by over-expression of GRP78 and HSP27. Upper: The basal expression of ATAD3A, GST-Pi, KISS1 and WASF3 was measured in Spiky, OVCAR, CAOV3 and PA-1 ovarian carcinoma cells by immuno-fluorescence in the Hermes Wiscan system at 10X magnification. Lower: Spiky tumor cells were transfected with an empty vector plasmid or plasmids to express HSP27 and GRP78 together. Twenty four h after transfection, cells were treated with vehicle or [sorafenib (2 μM) + sildenafil (2 μM) + celecoxib (2 μM)] for 6h. Six h after drug treatment, cells were fixed in place and immuo-staining performed to detect the expression of ATAD3A; GST-Pi; KISS1; and WASF3, using a Hermes WiScan wide field microscope at 10X.
Discussion
The present studies were performed with the intention of developing a novel therapeutic drug combination for ovarian cancer, based on the foundation of recent prior published studies from the Dent laboratory. Factoring into this research process was also the realization that many of the most novel drugs / targeted therapies are very expensive to purchase and will not be paid for by health insurance in the United States for malignancies in which they are not FDA approved. Such cost factors would thus very likely preclude clinical translation in the USA, and hence probably in all other developed nations. Using three well-established orally available drugs, all soon to be losing patent protection i.e. much cheaper generics will become available by 2017, we have demonstrated a greater than additive level of tumor cell killing with their combination, and furthermore noted that this drug combination also sensitizes / re-sensitizes tumor cells to platinum therapy.
In prior studies with the 2 drug combinations of [celecoxib + sildenafil] and [sorafenib + sildenafil] we demonstrated that cell killing either required (celecoxib) or was facilitated by (sorafenib) expression of the death receptor CD95. The present studies argued that the 3 drug combination also utilized caspase 8/10 signaling in the killing process but inhibition of these apical caspases did not suppress the drug-induced decline in proliferation and viability. Knock down of CD95 expression modestly, though significantly, suppressed 3 drug combination-induced killing (unpublished results). The role, if any, of TNFR1 in killing was not determined. Downstream of death receptors also exists a necroptotic signaling pathway through RIP-1 / caspases 2 and 4, and molecular knock down of these proteins resulted in significantly lower level of cell killing as well as a reduced ability of the drug combination to suppress growth. Also in contrast to data with c-FLIP-s, protection of the mitochondria and endoplasmic reticulum by over-expression of BCL-XL or by expression of dominant negative caspase 9 both suppressed killing and growth arrest. Thus [celecoxib + sorafenib + sildenafil] treatment causes both endoplasmic reticulum stress and mitochondrial –mediated cell death.
Plasma membrane drug efflux pumps such as ABCB1 and ABCG2 are known to transport from the cytosol to the extracellular environment many established DNA damaging agents as well as more recently novel “targeted” drugs i.e., cisplatin, sorafenib, and celecoxib. Thus, developing inhibitors of plasma membrane drug efflux pumps has been a “holy-grail” for cancer focused drug companies over the past 25 years, though no inhibitor has yet received FDA approval in any malignancy. Elacridar (GF120918) was originally developed by Glaxo Smith-Kline and although highly promising data from in vitro work and mouse studies were obtained, in rats and some larger animals and in human subjects the drug had dose-limiting PK/PD issues with non-linear uptake which prevented a therapeutic dose of ∼200 nM being achieved.20 Elacridar is now owned by Izumi Biosciences Inc.. who hope to reformulate and increase drug bio-availability. Our data using elacridar demonstrated, for [sorafenib + sildenafil + celecoxib], which itself reduces ABCB1 and ABCG2 expression, that elacridar did not significantly alter the lethality of [sorafenib + sildenafil + celecoxib] when measured after 12 h. However, in SKOV-3 cells no effect of elacridar was observed under any condition arguing that the inherent resistance of SKOV-3 cells to chemotherapeutic drugs is not due to efflux pump expression. And in studies with cisplatin, SKOV-3 cells were even more resistant to drug-induced toxicity than the patient validated primary isolate, Spiky, which was de novo platinum resistant. It is known in ovarian cancer cells, but particularly in SKOV-3, that a functional apoptosome downstream of cytochrome c and Apaf-1 does not form with reduced levels of complexed pro-caspase 9, which would in theory act to suppress drug lethality.
Treatment of ovarian cancer cells with the 3 drug combination consistently reduced expression of the chaperone proteins GRP78, HSP70 and BAG3 in both Spiky and OVCAR cells. Reduced expression of GRP78 results in elevated levels of unfolded proteins within the endoplasmic reticulum in parallel with activation of the stress-sensing kinase PERK, which through eIF2α Serine 51 phosphorylation causes a general overall reduction in cellular transcription. HSP70 activity will be reduced by lower levels of BAG3 due to a lack of ATP for ADP exchange chaperone loading, and the total expression of HSP70 itself was also shown to be reduced. In tumor cells HSP70 is known to be over-expressed and the induction of its expression following various types of cellular stress has been linked to multiple mechanisms related to maintaining cell viability. Notably, HSP70 overexpression has been linked to inhibition of the pro-apoptotic functions of BAX and of AIF; hence reduced HSP70 levels will facilitate mitochondrial dysfunction and a greater ability of AIF to promote DNA fragmentation and tumor cell death.
Platinum containing drugs are used to treat a wide variety of thoracic, GI and GU and gynecologic malignancies including tumors of the lung, colon, ovary and testes. We discovered that treatment of a wide variety of different tumor cell types with [celecoxib + sorafenib + sildenafil] enhanced the ability of cisplatin to cause tumor cell death. Of particular note, the Spiky PDX isolate was found to be inherently cisplatin resistant in the donor patient but was sensitized to both cisplatin and carboplatin in our in vitro studies. In NSCLC cells, treatment with [celecoxib + sorafenib + sildenafil] caused cell death and sensitized tumor cells to cisplatin but not to either carboplatin or oxaliplatin. Prior studies, in vitro and in vivo, have demonstrated that [sorafenib + sildenafil] and [celecoxib + sildenafil] can kill a variety of cancer cell types and in the present studies we found the combination of [celecoxib + sorafenib + sildenafil] strongly enhanced the lethality of oxaliplatin. Based on our prior data with celecoxib, sorafenib and sildenafil, coupled to our present findings with this 3 drug combination's ability to enhance platinum drug lethality, we argue a logical next step for our studies is to propose a phase I trial of this combination targeting lung, colorectal and ovarian cancer patients to determine safe doses of the 3 drug combination to be followed by phase II trials in the same tumors in conjunction with platinum in patients who have displayed platinum resistance to confirm the ability of this combination to re-sensitize tumors to platinum.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Support for the present study was funded from PHS grants from the National Institutes of Health [R01-CA141704, R01-CA150214, R01-DK52825, R01-CA192613]. Services in support of the research project were provided by the VCU Massey Cancer Center Tissue and Data Acquisition and Analysis Core, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. Thanks to the Betts family fund for support in the purchase of the Hermes Wiscan instrument. PD is the holder of the Universal Inc.. Chair in Signal Transduction Research.
References
- 1.Howlander N, Noone AN, Krapcho M, Garshall J, Miller D, Altekruse SF, Kpsary G, Yu M, Ruhl J, Tatalovich Z et al,, editors. SEER Cancer Statistics Review, 1975-2011. Natl Cancer Institute; 2014. (www.seer.cancer.gov) [Google Scholar]
- 2.McGuire WP, Ozols RF. Chemotherapy of advanced ovarian cancer. Semin Oncol 1998. 25:340-348; PMID:9633846 [PubMed] [Google Scholar]
- 3.Tummala MK, McGuire WP. Recurrent ovarian cancer. Clin Adv Hematol Oncol 2005. 3:723-736; PMID:16224447 [PubMed] [Google Scholar]
- 4.Stintzing S. Management of colorectal cancer. F1000Prime Rep 2014. 6:108; PMID:25580262; http://dx.doi.org/ 10.12703/P6-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 2014. 740:364-78; PMID:25058905; http://dx.doi.org/ 10.1016/j.ejphar.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts JL, Tavallai M, Nourbakhsh A, Fidanza A, Cruz-Luna T, Smith E, Siembida P, Plamondon P, Cycon KA, Doern CD, et al.. GRP78/Dna K Is a Target for Nexavar/Stivarga/Votrient in the Treatment of Human Malignancies, Viral Infections and Bacterial Diseases. J Cell Physiol 2015. 230:2552-78; PMID:25858032; http://dx.doi.org/ 10.1002/jcp.25014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamed HA, Tavallai S, Grant S, Poklepovic A, Dent P. Sorafenib/regorafenib and lapatinib interact to kill CNS tumor cells. J Cell Physiol 2015. 230:131-9; PMID:24911215; http://dx.doi.org/ 10.1002/jcp.24689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavallai M, Hamed HA, Roberts JL, Cruickshanks N, Chuckalovcak J, Poklepovic A, Booth L, Dent P. Nexavar/Stivarga and viagra interact to kill tumor cells. J Cell Physiol 2015. 230:2281-98; PMID:25704960; http://dx.doi.org/ 10.1002/jcp.24961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booth L, Roberts JL, Cash DR, Tavallai S, Jean S, Fidanza A, Cruz-Luna T, Siembiba P, Cycon KA, Cornelissen CN, et al.. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. J Cell Physiol 2015. 230:1661-76; PMID:25546329; http://dx.doi.org/ 10.1002/jcp.24919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts JL, Booth L, Conley A, Cruickshanks N, Malkin M, Kukreja RC, Grant S, Poklepovic A, Dent P. PDE5 inhibitors enhance the lethality of standard of care chemotherapy in pediatric CNS tumor cells. Cancer Biol Ther 2014. 15:758-67; PMID:24651037; http://dx.doi.org/ 10.4161/cbt.28553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth L, Roberts JL, Cruickshanks N, Conley A, Durrant DE, Das A, Fisher PB, Kukreja RC, Grant S, Poklepovic A, Dent P. Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol Pharmacol 2014. 85:408-19; PMID:24353313; http://dx.doi.org/ 10.1124/mol.113.090043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sajithlal GB, Hamed HA, Cruickshanks N, Booth L, Tavallai S, Syed J, Grant S, Poklepovic A, Dent P. Sorafenib/regorafenib and phosphatidyl inositol 3 kinase/thymoma viral proto-oncogene inhibition interact to kill tumor cells. Mol Pharmacol 2013. 84:562-71; PMID:23877009; http://dx.doi.org/ 10.1124/mol.113.088005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Booth L, Roberts JL, Tavallai M, Nourbakhsh A, Chuckalovcak J, Carter J, Poklepovic A, Dent P. OSU-03012 and Viagra Treatment Inhibits the Activity of Multiple Chaperone Proteins and Disrupts the Blood-Brain Barrier: Implications for Anti-Cancer Therapies. J Cell Physiol 2015. 230:1982-98; PMID:25736380; http://dx.doi.org/ 10.1002/jcp.24977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Booth L, Roberts JL, Cruickshanks N, Grant S, Poklepovic A, Dent P. Regulation of OSU-03012 toxicity by ER stress proteins and ER stress-inducing drugs. Mol Cancer Ther 2014. 13:2384-98; PMID:25103559; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Booth L, Roberts JL, Cruickshanks N, Tavallai S, Webb T, Samuel P, Conley A, Binion B, Young HF, Poklepovic A, et al.. PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J Cell Physiol 2015. 230:1115-27; PMID:25303541; http://dx.doi.org/ 10.1002/jcp.24843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bogliolo S, Cassani C, Gardella B, Musacchi V, Babilonti L, Venturini PL, Ferrero S, Spinillo A. Oxaliplatin for the treatment of ovarian cancer. Expert Opin Investig Drugs 2015. 24:1275-86; PMID:26125301 [DOI] [PubMed] [Google Scholar]
- 17.Teng Y, Ren X, Li H, Shull A, Kim J, Cowell JK. Mitochondrial ATAD3A combines with GRP78 to regulate theWASF3 metastasis-promoting protein. Oncogene 2015 Mar 30. doi: 10.1038/onc.2015.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang HY, Chang CL, Hsu SH, Huang CY, Chiang SF, Chiou SH, Huang CH, Hsiao YT, Lin TY, Chiang IP, et al.. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J Cell Sci 2010. 123: 1171-80; PMID:20332122; http://dx.doi.org/ 10.1242/jcs.062034 [DOI] [PubMed] [Google Scholar]
- 19.Yacoub A, Liu R, Park MA, Hamed HA, Dash R, Schramm DN, Sarkar D, Dimitriev IP, Bell JK, Grant S, et al.. Cisplatin enhances protein kinase R-like endoplasmic reticulum kinase- and CD95-dependent melanoma differentiation-associated gene-7/interleukin-24-induced killing in ovarian carcinoma cells. Mol Pharmacol 2010. 77:298-310; PMID:19910452; http://dx.doi.org/ 10.1124/mol.109.061820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, de Gooijer MC, Buil LC, Beijnen JH, Li G, van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors. Int J Cancer 2015. 137:2007-18; PMID:25868794; http://dx.doi.org/ 10.1002/ijc.29566 [DOI] [PubMed] [Google Scholar]