

ABSTRACT
Constitutive epimutations of tumor suppressor genes are increasingly considered as cancer predisposing factors equally to sequence mutations. In light of the emerging role of the microenvironment for cancer predisposition, initiation, and progression, we aimed to characterize the consequences of a BRCA1 epimutation in cells of mesenchymal origin. We performed a comprehensive molecular and cellular comparison of primary dermal fibroblasts taken from a monozygous twin pair discordant for recurrent cancers and BRCA1 epimutation, whose exceptional clinical case we previously reported in this journal. Comparative transcriptome analysis identified differential expression of extracellular matrix-related genes and pro-tumorigenic growth factors, such as collagens and CXC chemokines. Moreover, genes known to be key markers of so called cancer-associated fibroblasts (CAFs), such as ACTA2, FAP, PDPN, and TNC, were upregulated in fibroblasts of the affected twin (BRCA1mosMe) in comparison to those of the healthy twin (BRCA1wt). Further analyses detected CAF-typical cellular features, including an elevated growth rate, enhanced migration, altered actin architecture and increased production of ketone bodies in BRCA1mosMe fibroblasts compared to BRCA1wt fibroblasts. In addition, conditioned medium of BRCA1mosMe fibroblasts was more potent than conditioned medium of BRCA1wt fibroblasts to promote cell proliferation in an epithelial and a cancer cell line. Our data demonstrate, that a CAF-like state is not an exclusive feature of tumor-associated tissue but also exists in healthy tissue with tumor suppressor deficiency. The naturally occurring phenomenon of twin fibroblasts differing in their BRCA1 methylation status revealed to be a unique powerful tool for exploring tumor suppressor deficiency-related changes in healthy tissue, reinforcing their significance for cancer predisposition.
KEYWORDS: BRCA1; cancer-associated fibroblasts, CAFs; tumor-suppressor deficiency; cancer predisposition; epimutation; mosaic; promoter methylation; primary fibroblasts
Introduction
Cancer is generally referred to as a genetic disease. However, epigenetic aberrations are also hallmarks of cancer formation, which lead to gene expression changes without affecting the DNA sequence.1-3 Cancer evolution is associated with both global DNA hypomethylation, leading to genomic instability, and a more specific hypermethylation of promoters of tumor suppressor genes, inducing their silencing.1,2,4,5 The latter not only arises in neoplastic cells but can also occur as soma-wide constitutional epimutations, increasingly considered as a first hit according to Knudson's hypothesis. As somatic mosaicism is a common facet of epimutations, it suggests that the originating events occur after fertilization during early embryo development. However, there is also increasing evidence for the existence of germline epimutations, such as epigenetic aberrations in MLH1 and MSH2 in cases of familial colorectal cancer.6-8
We previously described a 29-year-old patient with recurrent cancers that harbors a mosaic epimutation of BRCA1 in contrast to her healthy monozygous twin sister.9 The constitutive BRCA1 epimutation in one quarter of the patient's cells manifested as an elevated BRCA1 promoter methylation level in DNA from saliva and dermal fibroblasts and was identified as the most likely cause for the difference in cancer proneness.
BRCA1 is a genomic caretaker gene that when mutated is responsible for a strong cancer predisposition, in particular for the hereditary forms of early-onset breast and ovarian cancers. Hypermethylation of the BRCA1 promoter was found to be present in a subset of blood cells of mutation-negative breast and ovarian cancer patients, suggesting that epigenetic disruption of BRCA1 may be an alternative, but equivalent, mechanism to genetic alterations for cancer predisposition.10 Furthermore, in absence of sequence mutations, promoter hypermethylation of BRCA1 and its complete silencing correlate with BRCAness of breast tumors in terms of histopathological characteristics and also therapy response.11-13 The pathomechanism by which BRCA1 sequence or epimutations promote cancer formation is still not fully understood. Deficiency in DNA repair capability, which is generally considered as a causal factor for cancer proneness, could only be shown in vitro for cancer cells or cell lines but never for human primary cells harboring BRCA1 mutations.14 In light of some recent findings that cancer is not an isolated entity but stands in symbiosis with tumor microenvironment, studying the role of BRCA1 in stromal cells of mesenchymal origin, such as fibroblasts, is also important, but has been neglected in the past. However, first in vitro studies have already shown that BRCA1 deficiency leads to altered features of stromal cells potentially modulating stromal-epithelial interactions in a pro-tumorigenic manner.15
We have analyzed molecular and cellular features of primary dermal fibroblasts taken from our patient with the mosaic BRCA1 epimutation (BRCA1mosMe) compared to control fibroblasts of the healthy twin sister (BRCA1wt), giving us the unique chance to explore the consequences of BRCA1 deficiency in an genetically nearly identical system of somatic mesenchymal cells. We determined a differential gene expression profile highly consistent with that described for cancer-associated fibroblasts (CAFs) as well as CAF-typical cellular features including a significant increase in cell proliferation, migration, ketone production, and altered actin architecture. In addition, conditioned medium of the BRCA1mosMe fibroblasts similar to that of CAFs enhanced growth of tumor cells in vitro. Together, our data strengthen the hypothesis that constitutive BRCA1 epimutation modulates the phenotypic and functional characteristics of primary fibroblasts toward a state that provides a favorable environment for cancer formation.
Results
BRCA1 mRNA and protein expression is reduced in BRCA1mosMe fibroblasts in comparison to BRCA1wt fibroblasts
We analyzed BRCA1 expression at the mRNA and protein level for an initial characterization of the epigenetically mediated BRCA1 haploinsufficiency. Using quantitative reverse transcription PCR (RT-qPCR), BRCA1mosMe fibroblasts exhibited a BRCA1 mRNA expression that is 29% lower and significantly different from that of BRCA1wt fibroblasts (Fig. 1A, P = 0.017). Although immunodetection of BRCA1 in primary non-neoplastic cells is known to be challenging,16 we succeeded in detecting 220 kDa bands representing BRCA1 protein in the Western blot analysis and determined a definite difference in band intensities between the two samples (Fig. 1B). Band quantification and normalization to ACTIN control showed that the BRCA1 protein level in BRCA1mosMe fibroblasts is 32% lower compared to that in BRCA1wt fibroblasts (Fig. 1B).
Figure 1.

(A) RT-qPCR analysis of BRCA1 mRNA expression, normalized to TBP and standardized to expression of BRCA1wt fibroblasts (FIB) (n = 3,*P < 0.05). (B) Western blot analysis of BRCA1 protein in 150µg total cell extracts of BRCA1mosMe fibroblasts and BRCA1wt fibroblasts using anti-BRCA1 antibody. Immunoblotting for ACTIN serves as loading control. (C) Heatmap showing CAF-related selection of differentially expressed transcripts between BRCA1wt fibroblasts and BRCA1mosMe fibroblasts. Hierarchical clustering of DEGs was performed using the complete linkage method. (D) RT-qPCR analysis of CAF keyplayers (n = 3, * P < 0.05, **P < 0.0005, ***P < 0.00005).
Comparative transcriptome analysis shows stable expression differences between BRCA1mosMe fibroblasts and BRCA1wt control fibroblasts
To investigate molecular changes that result from the BRCA1 epimutation in our patient's fibroblasts we first conducted a comparative genome wide expression array analysis on four separately cultured samples of BRCA1mosMe fibroblasts and BRCA1wt fibroblasts using the Affymetrix U219 array. In the subsequently performed analysis of variance (ANOVA) for the identification of differentially expressed genes (DEGs), rather loose filtering criteria were used to include all relevant genes and to compensate for the fact that our sample of interest is a mixture of affected and unaffected cells. We set the fold change threshold for differentially expressed probe sets to 1.5 and the P-value to 0.05. The ANOVA analysis revealed 133 probe sets accounting for 91 genes that were significantly upregulated and 243 probe sets, representing 194 genes, that were significantly downregulated in BRCA1mosMe fibroblasts compared to BRCA1wt fibroblasts (Tables S1 and S2). Hierarchical clustering of the prefiltered probe set list resulted in two clusters of four samples that belonged to the patient and to the healthy twin sister, respectively (Fig. 1C), indicating that the expression profile is stable through fibroblast culture and specific to the fibroblast donor.
Validation of the expression differences of 10 selected genes between BRCA1mosMe fibroblasts and BRCA1wt fibroblasts by qRT-PCR showed consistency between microarray and qRT-PCR results in terms of a generally consistent trend and extent of differential expression between the two samples (Fig. S1).
BRCA1mosMe fibroblast overexpress extracellular matrix (ECM)-associated genes and pro-tumorigenic cytokines
Next we investigated the functional importance of the expression signature of BRCA1mosMe fibroblasts by gene ontology enrichment analysis with the “Database for Annotation, Visualization and Integrated Discovery” (DAVID) tool in more detail. To avoid overlapping effects, we decided to analyze the upregulated and the downregulated parts of the DEG list separately. For the upregulated genes we found that terms associated to the extracellular space and the extracellular matrix were highly enriched in the cellular component analysis (Table 1). The analysis of biological processes fittingly showed a significant enrichment of processes that take place in the surrounding of cells, such as cellular and biological adhesion, extracellular structure, and matrix organization. These term enrichments can be traced back to a large extent to genes encoding extracellular structural proteins such as collagens (COL1A1, COL5A1 COL11A1, COL8A1, and COL12A1) and filaggrin (FLG) that are highly overrepresented on the upregulated list (Fig. 1C and Table S1). Furthermore, a variety of genes encoding proteins involved in ECM remodeling, such as MGP, POSTN, or TNC, represent a huge part of the specific signature of BRCA1mosMe fibroblasts. Moreover, there is also a significant upregulation of genes encoding some cytokines and growth factors, including, for example, CXCL12, CXCL6, and IL6. These genes play important roles in processes like cell adhesion, cell motion, and cell differentiation, which were also revealed by gene ontology (GO) analysis.
Table 1.
DAVID gene ontology and pathway analysis of upregulated genes1.
| GO terms Cellular Component |
GO terms Biological Process |
||||||
|---|---|---|---|---|---|---|---|
| GO: | Term | No. | P-value | GO: | Term | No. | P-value |
| 0044421 | extracellular region part | 27 | 1.70E-11 | 0007155 | cell adhesion | 18 | 1.63E-07 |
| 0005578 | proteinaceous extracellular matrix | 17 | 4.57E-11 | 0022610 | biological adhesion | 18 | 1.65E-07 |
| 0031012 | extracellular matrix | 17 | 1.40E-10 | 0043062 | extracellular structure organization | 9 | 2.87E-06 |
| 0044420 | extracellular matrix part | 9 | 4.35E-07 | 0030198 | extracellular matrix organization | 7 | 2.17E-05 |
| 0005581 | collagen | 5 | 5.25E-05 | 0035295 | tube development | 9 | 2.58E-05 |
| 0005604 | basement membrane | 5 | 1.18E-03 | 0051094 | positive regulation of developmental | 9 | 1.34E-04 |
| 0005583 | fibrillar collagen | 3 | 2.22E-03 | 0007517 | muscle organ development | 8 | 1.51E-04 |
| 0005615 | extracellular space | 12 | 2.29E-03 | 0006928 | cell motion | 11 | 2.48E-04 |
| 0005795 | Golgi stack | 4 | 2.55E-03 | 0045597 | positive regulation of cell differentiation | 8 | 2.50E-04 |
| 0008282 |
ATP-sensitive potassium channel complex |
2 |
1.77E-02 |
0001501 |
skeletal system development |
9 |
9.51E-01 |
| GO terms Molecular Function |
KEGG pathways |
||||||
| GO: |
Term |
No. |
P-value |
hsa code |
Pathway |
No. |
P-value |
| 0005201 | extracellular matrix structural constituent | 6 | 7.93E-05 | 004512 | ECM-receptor interaction | 5 | 1.37E-03 |
| 0019838 | growth factor binding | 6 | 2.04E-04 | 004510 | Focal adhesion | 6 | 5.84E-03 |
| 0005509 | calcium ion binding | 15 | 2.05E-04 | 005410 | Hypertrophic cardiomyopathy (HCM) | 4 | 1.32E-02 |
| 0005198 | structural molecule activity | 11 | 1.43E-03 | 004350 | TGF-beta signaling pathway | 4 | 1.41E-02 |
| 0046872 | metal ion binding | 33 | 4.75E-03 | 004060 | Cytokine-cytokine receptor interaction | 5 | 6.58E-02 |
| 0043169 | cation binding | 33 | 5.56E-03 | ||||
| 0043167 | ion binding | 33 | 7.10E-03 | ||||
| 0005539 | glycosaminoglycan binding | 4 | 3.56E-02 | ||||
| 0030247 | polysaccharide binding | 4 | 4.52E-02 | ||||
| 0000187 | pattern binding | 4 | 4.52E-02 | ||||
1genes that are more than 1.5-fold upregulated with a P-value smaller than 0.05
The expression profile of BRCA1mosMe fibroblasts shows a downregulation of HOX genes and suggests an inhibition of BRCA1-related DNA repair
A significant portion of downregulated genes in the BRCA1mosMe fibroblast expression profile belongs to the HOX gene family and, consistently, the GO biological processes analysis showed a strong enrichment of associated terms. HOX gene expression profiles in dermal fibroblasts display a positional memory and are known to be variable depending on minimal differences in the localization from which the biopsy was taken.17 To exclude overestimation of such effects and avoid possible masking of other important results by the very strong expression changes of HOX genes, we have repeated the GO analysis with the downregulated gene list after exclusion of HOX genes (Table 2). There was still an enrichment of terms associated with the ECM. However, in contrast to the GO analysis of upregulated genes, the ECM term enrichment could be attributed to the downregulation of secreted growth factors such as FGF13 and FGF6, rather than structural proteins or ECM-modulating enzymes. Interestingly, some of the accumulated biological processes and molecular function analysis terms were linked to DNA damage and repair, such as nucleotide kinase activity or double strand break repair. Many of these enriched terms included BRCA1, which we have shown to be downregulated by its epimutation in BRCA1mosMe fibroblasts (Fig. 1A and 1B). Consistent with this, genes encoding binding partners of BRCA1, such as MLH2, CHEK2, and RAD51AP1, were also downregulated in the BRCA1mosMe fibroblasts (Fig. 1C) and responsible for the enrichment of repair-associated terms in the GO analysis.
Table 2.
DAVID gene ontology and pathway analysis of downregulated genes1.
| GO terms Biological Process (incl. HOX genes) |
GO terms Biological Process (excl. HOX genes) |
||||||
|---|---|---|---|---|---|---|---|
| GO: | Term | No. | P-value | GO: | Term | No. | P-value |
| 0009952 | anterior/posterior pattern formation | 9 | 8.33E-05 | 0009153 | purine deoxyribonucleotide biosynthesis | 2 | 1.90E-02 |
| 0003002 | regionalization | 9 | 8.43E-04 | 0010033 | response to organic substance | 14 | 2.01E-02 |
| 0048706 | embryonic skeletal system development | 6 | 1.06E-03 | 0006302 | double-strand break repair | 4 | 2.13E-02 |
| 0007389 | pattern specification process | 10 | 1.50E-03 | 0009719 | response to endogenous stimulus | 9 | 4.00E-02 |
| 0048568 | embryonic organ development | 8 | 1.76E-03 | 0009265 | 2'-deoxyribonucleotide biosynthesis | 2 | 4.68E-02 |
| 0043009 | chordate embryonic development | 11 | 1.87E-03 | 0006072 | glycerol-3-phosphate metabolism | 2 | 5.59E-02 |
| 0009792 | embryonic development ending in birth | 11 | 2.00E-03 | 0016053 | organic acid biosynthesis | 5 | 6.09E-02 |
| 0048704 | embryonic skeletal system morphogenesis | 5 | 2.56E-03 | 0046394 | carboxylic acid biosynthesis | 5 | 6.09E-02 |
| 0001501 | skeletal system development | 10 | 4.92E-03 | 0018065 | protein-cofactor linkage | 2 | 7.38E-02 |
| 0048705 |
skeletal system morphogenesis |
6 |
5.42E-03 |
0009263 |
deoxyribonucleotide biosynthesis |
2 |
7.38E-02 |
| GO terms Cellular Component (excl. HOX genes) |
GO terms Molecular Function (excl. HOX genes) |
||||||
| GO: |
Term |
No. |
P-value |
GO: |
Term |
No. |
P-value |
| 0043005 | neuron projection | 8 | 2.82E-02 | 0042802 | identical protein binding | 13 | 193E-02 |
| 0044421 | extracellular region part | 14 | 6.85E-02 | 0003747 | translation release factor activity | 2 | 5.60E-02 |
| 0042995 | cell projection | 11 | 7.71E-02 | 0008079 | translation termination factor activity | 2 | 5.60E-02 |
| 0005739 | mitochondrion | 15 | 8.28E-02 | 0019205 | nucleobase, nucleoside, nucleotide kinase act. | 3 | 6.34E-02 |
| 0031233 | intrinsic to external side of plasma membrane | 2 | 8.28E-02 | 0003697 | single-stranded DNA binding | 3 | 9.69E-02 |
| 0005576 |
extracellular region |
24 |
8.89E-02 |
0004896 | cytokine receptor activity | 3 | 9.69E-02 |
| KEGG pathways (excl. HOX genes) |
|||||||
| hsa code |
Pathway |
No. |
p-value |
|
|
|
|
| 00270 | Cysteine and methionine metabolism | 3 | 5.17E-02 | ||||
1genes that are more than 1.5-fold downregulated with
a P-value smaller than 0.05
BRCA1mosMe fibroblasts reflect BRCA1 haploinsufficiency with an expression signature highly similar to that of CAFs
To test if the fibroblasts contain expression changes in addition to those of the DNA-repair-associated genes that can be traced back to BRCA1 haploinsufficiency, we compared our obtained signature to published data that had been produced on lymphocytes to identify BRCA1 mutation carriers based on their expression profiles.18 When using our original filtering criteria, we found an overlap of about 10%. Considering the fact that our sample represents a BRCA1 haploinsufficiency present in a ˜25% mosaic, we also included genes whose expression differed significantly but only minimally to an extent of 25% and thereby found an overlap of 25% between our BRCA1mosMe fibroblast profile and the signature predicting BRCA1 mutation carrier status. Shared genes were, for example, PXDN, CSRP2, ENPP2, and FOXP1 (Fig. 1C). However, the by far larger portion of deregulated genes in BRCA1mosMe fibroblasts and, in particular, the overexpression of ECM-associated genes, cytokines, and growth factors could not be directly explained and connected to BRCA1 disruption. For this reason, we searched in the literature if these expression changes had already been described for primary fibroblasts in any conditions. We found a massive concordance of our BRCA1mosMe fibroblast profile with the expression signature of CAFs. This overlap mainly involved the genes that constitute the core signature of CAFs, being equally deregulated in CAFs isolated from a variety of different tumor types, in contrast to normal fibroblasts from the same tissue.
CAF keyplayer genes are deregulated in BRCA1mosMe fibroblasts
To further verify the hypothesis that our BRCA1mosMe fibroblasts share transcriptomic characteristics with CAFs, we specifically re-analyzed the expression of known CAF key genes with qRT-PCR. All analyzed genes showed the expected expression differences, based on the assumption that BRCA1mosMe fibroblasts in contrast to BRCA1wt fibroblasts have a CAF-like transcription profile (Fig. 1D). The fold changes of ACTA2 and FAP, the most established CAF markers, were as high as 7.29 and 3.86, respectively. We also observed a strong, even though not significant, decrease (40%) of CAV1 mRNA, which is a common feature of many types of CAFs. In addition, there were significant expression differences ranging from 1.8 to 9.5 in cytokines and ECM-related genes known to determinably trigger CAF-specific phenotypes and functions, such as CXCL12, FN1, IL8, MGP, PDPN, TGFB1, and TNC. Taken together, these results let us assume that the BRCA1mosMe fibroblasts show transcriptional characteristics that demarcate them from the BRCA1wt fibroblasts, in the same way CAFs differ from normal fibroblasts.
BRCA1mosMe fibroblasts show accelerated proliferation and migration
Next, we investigated if BRCA1mosMe fibroblasts also exhibit functional features of CAFs. We analyzed the growth rates of BRCA1mosMe fibroblasts compared to BRCA1wt fibroblasts, as an increased proliferative index is a common feature of CAFs. We established growth curves over 10 days for different culture passages testing every day for the amount of living cells by a luminescence assay. The increased growth rates of BRCA1mosMe fibroblasts already manifested itself significantly at day 3 for passage 6 cells and even already at day 2 for passage 11 cells, and became more prominent in the course of the experiment (Fig. 2A). At day 10, BRCA1mosMe fibroblasts demonstrated an increase of normalized Relative Light Unit (RLU) signals of 157 and 185 percentage points, respectively, compared to BRCA1wt fibroblasts. Consistently, immunostaining against Ki-67 showed a 3-fold higher amount (P < 0.05) of Ki-67-positive cells in the BRCA1mosMe fibroblast culture than in the control culture (Fig. 2B). Moreover, there was a small but significant increase in migratory capability of BRCA1mosMe fibroblasts as determined by means of IBIDI chambers (Fig. 2C).
Figure 2.

(A) Growth curves of BRCA1wt and BRCA1mosMe fibroblasts (FIB) as measured by Cell titer glo assay (n = 6, *P < 0.005, **P < 0.0005, ***P < 0.0005); RLU=Relative Light Units (B) Representative microphotographs of Ki-67 immunostaining of BRCA1wt and BRCA1mosMe fibroblasts and calculation of Ki-67 index (n = 3, *P < 0.05). (C) Migration assay of BRCA1wt and BRCA1mosMe fibroblasts and quantification (n=3, *P < 0.05).
BRCA1mosMe fibroblasts show changes in collagen abundance and actin architecture
As CAFs usually also strongly produce collagen, we immunostained BRCA1mosMe fibroblasts for type I collagen to analyze this feature not only on the RNA but also on the protein level. All BRCA1mosMe fibroblasts were positive for cell-associated collagen I in contrast to BRCA1wt fibroblasts that displayed almost no collagen I staining (Fig. 3B).
Figure 3.
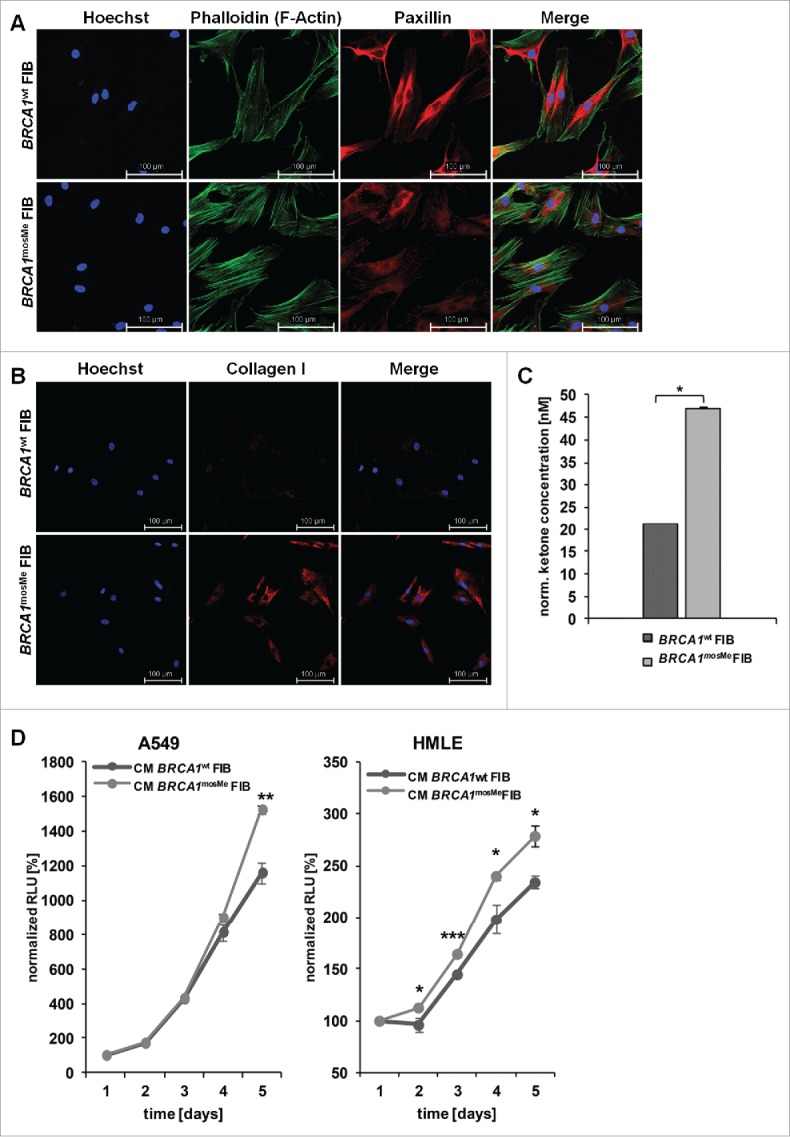
(A) Phalloidin staining of BRCA1wt and BRCA1mosMe fibroblasts (FIB). (B) Collagen I immunostaining of BRCA1wt and BRCA1mosMe fibroblasts. (C) Ketone concentrations of cell culture medium of BRCA1wt and BRCA1mosMe fibroblasts as determined by B-hydroxybutyrate assay (n = 6, *P < 0.05). Values were normalized to protein mass per well. (D) Growth curves of a cancer cell line and a non-cancerous epithelial cell line cultured in conditioned medium of BRCA1wt and BRCA1mosMe fibroblasts as determined by Cell titer glo assay. (n=5, *P < 0.005, **P < 0.0005, ***P < 0.0005).
Next, we visualized the cytoskeletal organization of BRCA1mosMe fibroblasts using phalloidin staining. As predicted, BRCA1mosMe fibroblasts showed a more organized actin skeleton characterized by a large number of thick and parallel F-actin-based stress fibers extending across the cell (Fig. 3A). In contrast, the staining in BRCA1wt fibroblasts was weaker, and the actin skeleton looked more relaxed with the strongest staining along the membranes. Paxillin staining was much weaker in BRCA1mosMe fibroblasts than in BRCA1wt fibroblasts. However, in BRCA1mosMe fibroblasts paxillin tended to show some punctual staining at the ends of stress fibers in contrast to BRCA1wt fibroblasts, where the staining was completely diffuse and disorganized in particular around the nucleus. This observation can indicate that focal adhesion signaling is more active in BRCA1mosMe fibroblasts than in the control fibroblasts.
Increased ketone body generation in BRCA1mosMe fibroblasts
Metabolic reprogramming is a hallmark of CAFs, and fueling cancer cells with energy-rich metabolites is one way by which CAFs promote proliferation of surrounding cancer cells. To test if BRCA1mosMe fibroblasts also show hallmarks for such a mitochondrial dysfunction, we compared the concentration of ketone bodies as energy-rich molecules between conditioned medium of BRCA1mosMe fibroblasts and that of BRCA1wt fibroblasts. We found the concentration of β-hydroxybutyrate to be reproducibly and significantly increased by more than two-fold in BRCA1mosMe fibroblasts in comparison to BRCA1wt fibroblasts (Fig. 3C).
Conditioned medium of BRCA1mosMe fibroblasts enhances proliferation of cancer cells and non-neoplastic epithelial cells
Next, we investigated whether the potentially pro-tumorigenic features that we found in BRCA1mosMe fibroblast actually have a similar influence on tumorous or non-tumorous epithelial cells as CAFs. We performed proliferation assays with the tumorous human lung adenocarcinoma epithelial cell line A549 and non-tumorous human mammary epithelial cell line (HLME) cells in the presence of conditioned medium of the fibroblast cultures. Growth of A549 cells fed with conditioned medium of BRCA1mosMe fibroblasts started to differ from that of A549 cells cultured in conditioned medium of the control fibroblasts after four days of culture with a significant difference occurring only after five days of culture (Fig. 3D). The proliferation-enhancing effect of conditioned medium of BRCA1mosMe fibroblasts on HLME cells was much stronger and already apparent on the second day of culture with a significant difference present throughout the whole experiment. These results suggest that BRCA1mosMe fibroblasts not only show phenotypic features of CAFs but also harbor the ability to promote cell growth of cancer and epithelial cells.
Discussion
CAFs are the major part of the tumor stroma and play a pivotal role in carcinogenesis and tumor progression.19 A huge variety of cell types such as epithelial cells, mesenchymal stem cells, resident fibroblasts or endothelial cells have been reported to be able to transdifferentiate into CAFs through epithelial–mesenchymal transition (EMT), mesothelial-to-mesenchymal transition (MMT), or endothelial-mesenchymal transition (EndMT).20-24 All these models for CAF development assume exogenous stimuli that initiate transformation and, in most cases, are thought to be sent from adjacent cancer cells in form of growth factors, such as TGF-ß and CXCL12.23
There is experimental evidence that the CAF phenotype can also be initiated by intrinsic factors. In particular depletion of tumor suppressor genes was reported to convert normal fibroblasts to CAFs.15,25,26 As a result the question arises, if constitutional disruption of tumor suppressors may also provoke a CAF-like state in vivo in healthy tissue of persons epigenetically predisposed to hereditary cancers.
In the current study, we systematically compared for the first time primary fibroblasts affected by epigenetically mediated haploinsufficiency of the tumor suppressor BRCA1 to CAFs and found remarkable similarities on the molecular, cellular, and functional level. We showed that key transcriptional features of CAFs, such as increased expression of pro-tumorigenic genes and ECM-associated genes, can also be detected in our BRCA1mosMe fibroblasts, although taken from healthy non-neoplastic tissue. Ingenuity identified TWIST1, TGF-ß, CXCL12, IL6, TNF, and ethanol as possible upstream regulators, i.e., effectors of these expression changes (Fig. S2). As all these factors are important regulators or inducers of the CAF phenotype,27-30 it stands to reason that the transcriptional program of BRCA1mosMe fibroblasts actually resembles that of CAFs. To our knowledge, a CAF-like expression signature was only detected once before in healthy skin of individuals predisposed to basal cell carcinoma by mutations in the tumor suppressor gene PTCH1.31 As BRCA1 was reported to regulate PTCH1 and other genes in Hedgehog pathways,32 a convergent mechanism in induction of the CAF phenotype may be suspected.
We demonstrated that BRCA1mosMe fibroblasts display an accelerated proliferation and migration compared to control fibroblasts. This finding is comparable to previous studies that described skin fibroblasts from individuals with hereditary breast cancer and from their relatives to have abnormal cellular phenotypes, including altered migratory behavior and proliferation.33,34 From today's perspective, the similarities between these anomalies and those of CAFs are eye-catching, as a high proliferative index and increased motility are common features of CAFs from all different sources. Moreover, in vitro disruption of BRCA1 by knockdown in fibroblasts, as well as epithelial cells and cancer cell lines, was also shown to accelerate cell proliferation.15,35,36
Cytoskeletal changes observed in BRCA1mosMe fibroblasts, like bundling of actin stress fibers and also upregulation of α-smooth muscle actin (α-SMA) on mRNA level, are a known key feature of activated fibroblasts, such as myofibroblasts and also CAFs. The formation of stress fibers is required for matrix organization and remodeling,37 which is a major function of CAFs in building a favorable environment for cancer growth and metastasis. In dermal fibroblasts of individuals predisposed to retinoblastoma, polyposis coli, and basal cell carcinoma, an increased actin content and an accelerated actin reorganization were found,38 indicating that this kind of change seems to be common to cancer-predisposed fibroblasts independent of the tumor suppressor gene causally affected.
CAFs are known to fuel growth of neighboring cancer cells by supplying energy-rich metabolites such as lactate and ketones, which is named “reverse Warburg effect” or “two-compartment tumor metabolism”.39,40 In several studies, CAFs from different sources were shown to produce more ketone bodies than paired normal fibroblasts. Ketone elevation was also detected for BRCA1mosMe fibroblasts in contrast to twin control fibroblasts in the current study, although cells were neither taken from malignant tissue nor co-cultured with cancer cells in vitro. It was previously hypothesized that the first step of metabolic reprogramming of CAFs is production of oxidative stress by cancer cells that subsequently leads to DNA damage in CAFs, which in turn triggers autophagy and mitochondrial degradation, causing shifts of the metabolism to aerobic glycolysis.40-42 This process is crucially regulated by elevation of HIF-1α, which, in turn, is depending on BRCA1.43,44 As BRCA1 seems to be an emerging regulator of mitochondrial integrity, we hypothesize that its disruption in primary fibroblasts of persons with germline or somatic alterations also may induce CAF-like metabolic changes in the absence of cancer cells. This is supported by experimental data from a stable knockdown of BRCA1 in immortalized fibroblasts that results in upregulation of HIF-1α, autophagy, and ketone body production.15 In addition, BRCA1-knockdown fibroblasts were shown to enhance cancer cell line proliferation when co-injected into xenografts. This effect is known from CAFs taken from tumor tissue.
Moreover, it was demonstrated that conditioned medium of CAFs is enough to enhance cancer cell proliferation in in vitro experiments,45,46 probably due to secretion of tumorigenic growth factors and energy-rich metabolites. In the current study, we observed a comparable effect when culturing A549 lung carcinoma cells and non-tumorous HMLE mammary epithelial cells in the presence of conditioned medium of BRCA1mosMe fibroblasts. This strongly supports our hypothesis that BRCA1 epimutation in our proband's fibroblasts induces phenotypic, molecular, and functional changes resembling features of CAFs. Considering the data of Salem et al.15 that show comparable changes in a shBRCA1 fibroblast cell line, it stands to reason that the observed phenotype in BRCA1mosMe fibroblasts is causally linked to the downregulation of BRCA1. Nevertheless, the copy number variations of the genes RSPO3 and NREP (C5orf13), which we previously reported to be present in a 50% mosaic in the analyzed cells,9 could also play a contributory role that was not specifically analyzed in this study. RSPO3 acts as a proto-oncogene whose gain-of-expression promotes tumorigenic processes. In contrast, a reduction of RSPO3 expression, which is the consequence of the mosaic RSPO3 deletion in our proband's cells,9 was already linked to the opposite of our findings, namely a reduction in cell proliferation and migration, in RSPO3 knockdown cancer cells.47,48 The expression of NREP in BRCA1mosMe was unaffected by the deletion9 and no connection of this gene to cancerogenesis is known to date. It is beyond the scope of our present study, but remains to be determined, whether the deletions of RSPO3 and NREP co-exist with the BRCA1 epimutation in the same cells of the BRCA1mosMe fibroblasts.
Based on our findings, we speculate that epigenetic BRCA1 deficiency in predisposed persons may lead to a CAF-like state of stromal cells prior to cancer onset and perhaps even from birth on. This CAF-like state may promote tumorigenic transformation of neighboring epithelial cells by altered stromal-epithelial interactions and thereby explain the predisposition of the mutation carriers to carcinogenic conditions.
If this concept is proven to be generally applicable to mesenchymal cells also with BRCA1 sequence mutations, it may open completely new perspectives on hereditary BRCA1-related carcinogenesis and provide impetus for the development of new preventive strategies. According to this idea, therapeutic approaches for cancer treatment targeting the tumor microenvironment are already in clinical trials, and can possibly be transferred to the prevention of tumor development in hereditary cancers by interfering either with the maintenance of the CAF-like state of fibroblasts or with the secretion of protumorigenic signals.49 Another potential strategy for chemoprevention is to compensate for oxidative stress that leads to mitochondrial dysfunction and autophagy and shifting of the metabolism in BRCA1 deficient cells.15 Interestingly, antioxidant treatment just like genetic complementation of BRCA1 in BRCA1 null breast cancer cells reverts functional markers of oxidative stress, namely CAV1 loss and MCT4 induction in co-cultured fibroblasts.50
In conclusion, we have shown an example of transcriptional and functional CAF-like state existing in fibroblasts of healthy tissue, which was probably induced by epigenetically mediated BRCA1 haploinsufficiency. Thus, we suggest building a favorable environment for cancer development and growth by CAF-like fibroblasts to be one of many mechanisms by which cancer proneness is mediated in individuals with BRCA1 epimutation. This hypothesis brings many new potential targets for cancer prevention in genetically predisposed individuals into play.
Materials and methods
Case presentation
The biological material that was analyzed in this study was taken from a proven monozygous twin pair with discordance for recurrent cancers. First cancer occurred in the affected individual at an age of 4 years and was characterized as precursor B-cell lymphoblastic leukemia. Thyroid carcinoma was diagnosed at an age of 25 years. The affected twin was the only individual being suffering from cancer in the family pedigree including four generations. The clinical case has been previously described in more detail in Galetzka et al.9
Isolation and culture of primary fibroblasts
Genetic counseling was offered and informed written consent was obtained from both probands. This study was approved by the Ethics Committee of the Medical Association of Rhineland-Palatinate (No. 837.440.03[4102]). Skin biopsies were taken from the probands at an age of 29 years from the left upper arm, and fibroblasts cultures were established. Cells were cultured in their original primary state without immortalization or any other genetic manipulation. The fibroblasts were cultured in Dulbecco's modified eagles medium (Thermo Fisher Scientific, #11960) supplemented with 10% fetal calf serum (Thermo Fisher Scientific, #10270) and penicillin-streptomycin (Thermo Fisher Scientific, # 15070063). The cells were passaged and harvested at subconfluency.
Gene expression microarray analysis
The array experiment was performed by Affymetrix using the GeneAtlas™ Personal Microarray System (Affymetrix). RNA samples extracted from primary fibroblasts using TRIzol reagent (Thermo Fisher Scientific, #15596) were hybridized on a HG-U219 array strip (Affymetrix). Microarray data have been deposited at the Gene Expression Omnibus (GEO) database with the accession number GSE71078. Data normalization by Robust Multi-array Average (RMA) approach and analysis of differentially expressed probe sets by Tukey's Bi-weight average algorithm and Analysis of Variance (ANOVA) were done with Affymetrix Expression Console Software and Affymetrix Transcriptome Analysis Console respectively. Functional enrichment, network analysis and upstream regulator analysis were performed using Database of Annotation, Visualization and Integrated Discovery (DAVID)51 and Ingenuity Pathways Analysis tool (Qiagen).
Quantitative real time PCR
RNA was extracted using TRIzol® reagent (Thermo Fisher Scientific) and RNA quality and purity were checked photometrically. Reverse transcription reaction was performed by means of SuperScript® III RT kit (Thermo Fisher Scientific) using a combination of random and oligo (dT) primers. Primers for real time PCR were designed with Primer3 version 4.0.0 as detailed in Table S3 and commercially available primers for TBP served as endogenous control (Qiagen QuantiTect #QT00000721).52 Data acquisition was performed with the StepOnePlus Real-Time PCR System with following PCR cycling conditions: 40 cycles of 30 sec at 95°C, 30 sec at 60°C and 40 sec at 72°C. To avoid and check for genomic contamination, primers were designed exon spanning, water and minus-reverse-transcriptase controls were included and melting curve analysis was done. Comparative 2−ΔΔCt method was used for relative quantification of gene expression with error bars indicating ±RQmin/max = 2[ΔΔCt±T*SD(ΔCt)] as T = confidence level and SD = standard deviation. Statistical analysis was performed using the student's t-test and P-values ≤ 0.05 were considered statistically significant.
Immunoblot analysis
Protein extraction was performed according to Bräutigam et al.53 Different amounts of total protein were separated on a 6% acrylamide gel for BRCA1 blotting and on a 10% acrylamide gel for later ACTIN blotting. Western Blot analyses were performed with anti-BRCA1 (Merck Millipore, MS110, #MABC 199) and anti-ACTIN (Sigma Aldrich, #A2228) respectively, as primary antibodies, and HRP-coupled anti-rabbit and anti-mouse (Dianova, #111–036, 111–035), respectively, as secondary antibodies. Signal detection was achieved by ChemiDoc XRS digital imager (Biorad). The software ChemiDoc Image Lab (Biorad) was used for densitometric analyses of BRCA1 bands and normalization to ACTIN band intensities.
Immunofluorescence microscopy
Equal amounts of BRCA1mosMe and BRCA1wt fibroblasts were seeded in chamber well slides and grown over night. Cells were fixed in 4% paraformaldehyde (PFA), blocked in 3% BSA in PBST and permeabilized in 0.2% saponin (Carl Roth, #6857). The slides were sequentially probed with primary and secondary antibodies or stained with Fluor-488-Phalloidin (Thermo Fisher Scientific, #A12379). Nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific, #62249). The primary antibodies used were mouse anti-human paxillin (R&D Systems, #AF4259), rabbit anti-human Ki67 (Thermo Fisher Scientific, #RM-9106) and mouse anti-human collagen I (Abcam, #ab88147) the secondary antibodies used alexa-flour-488 mouse anti-rabbit, alexa-flour-488 goat anti-mouse and alexa-flour-546 goat anti rabbit (Thermo Fisher Scientific, #11059, #11001, #11035). The slides were imaged by Zeiss confocal laser scan microscope CLSM 710-NLO.
Proliferation assays of fibroblasts and conditioned medium proliferation assays of HMLE and A549 cells
The proliferation assays were conducted with the CellTiter-Glo Luminescent Cell Viability Assay (Promega, #G7570) according to the manufacturer's protocol. In brief, 2,000 fibroblasts were seeded per well in a 96 well plate. On the next day and then every 24 hours cell viability was measured and values were normalized to the first measurement.
For conditioned medium proliferation assays 2000 HMLE cells or 500 A549 cells were seeded in a 96 well plate, respectively. On the next day the medium was changed to either conditioned medium of BRCA1mosMe fibroblasts or BRCA1wt fibroblasts. The conditioned medium had been prepared by cultivation of 500,000 exponentially growing fibroblasts in 3 ml of DMEM for 72 hours. Cell debris was separated by centrifugation at 4000 rpm for 10 minutes, and the conditioned medium was stored at −80°C.
Fibroblast migration assays
Fibroblasts (12,000) were seeded in 70 µl medium into each chamber of a culture insert (IBIDI, #80206). After 24 hours the silicone inserts were removed and the gap between the cell fields was photographed every 6 hours in the same optical field. The pictures were analyzed using the Scratch Assay Analyzer plugin from the extension package MiToBo in ImageJ.
Quantification of ketone bodies
Fibroblasts (100,000/per well) were plated in a 24-well plate in complete media. The next day the cells were washed twice with dPBS and the medium was changed to optiMEM (Thermo Fisher Scientific, #31985062) containing 2% FBS. After 48 hours of incubation the cell culture supernatant was collected and separated from cell debris by centrifugation. The concentration of ketone bodies was determined using the β-hydroxybutyrate assay (Sigma Aldrich, #MAK041).
Statistical analysis
If not indicated differently, the values are given as mean ± standard error of the mean (SEM). The Student t-test was performed for statistical analysis. P-values ≤ 0.05 were considered to be statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by the FAZIT-STIFTUNG Gemeinnützige Verlagsgesellschaft mbH. The authors wish to thank the probands for consent to share their case with the scientific community and Benjamin Irmscher, Jennifer Winter, Markus Eich, Katharina Baus, Henning Janssen, Daniela Gottfried and Ronald Unger for technical support.
References
- 1.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400-13; PMID:12154403; http://dx.doi.org/ 10.1038/sj.onc.1205651 [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27-36; PMID:19752007; http://dx.doi.org/ 10.1093/carcin/bgp220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:245-54; PMID:12610534; http://dx.doi.org/ 10.1038/ng1089 [DOI] [PubMed] [Google Scholar]
- 4.De Smet C, Loriot A. DNA hypomethylation in cancer: epigenetic scars of a neoplastic journey. Epigenetics 2010; 5:206-13; PMID:20305381; http://dx.doi.org/ 10.4161/epi.5.3.11447 [DOI] [PubMed] [Google Scholar]
- 5.Zilberman D. The human promoter methylome. Nat Genet 2007; 39:442-3; PMID:17392803; http://dx.doi.org/ 10.1038/ng0407-442 [DOI] [PubMed] [Google Scholar]
- 6.Hitchins MP, Wong JJL, Suthers G, Suter CM, Martin DIK, Hawkins NJ, Ward RL. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 2007; 356:697-705; PMID:17301300; http://dx.doi.org/ 10.1056/NEJMoa064522 [DOI] [PubMed] [Google Scholar]
- 7.Chan TL, Yuen ST, Kong CK, Chan YW, Chan ASY, Ng WF, Tsui WY, Lo MWS, Tam WY, Li VSW, et al.. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet 2006; 38:1178-83; PMID:16951683; http://dx.doi.org/ 10.1038/ng1866 [DOI] [PubMed] [Google Scholar]
- 8.Crucianelli F, Tricarico R, Turchetti D, Gorelli G, Gensini F, Sestini R, Giunti L, Pedroni M, Ponz de Leon M, Civitelli S, et al.. MLH1 constitutional and somatic methylation in patients with MLH1 negative tumors fulfilling the revised Bethesda criteria. Epigenetics 2014; 9:1431-8; PMID:25437057; http://dx.doi.org/ 10.4161/15592294.2014.970080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galetzka D, Hansmann T, El Hajj N, Weis E, Irmscher B, Ludwig M, Schneider-Rätzke B, Kohlschmidt N, Beyer V, Bartsch O, et al.. Monozygotic twins discordant for constitutive BRCA1 promoter methylation, childhood cancer and secondary cancer. Epigenetics 2012; 7:47-54; PMID:22207351; http://dx.doi.org/ 10.4161/epi.7.1.18814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, Preisler-Adams S, Wieacker P, Haaf T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet 2012; 21:4669-79; PMID:22843497; http://dx.doi.org/ 10.1093/hmg/dds308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tapia T, Smalley S V, Kohen P, Muñoz A, Solis LM, Corvalan A, Faundez P, Devoto L, Camus M, Alvarez M, et al.. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics 2008; 3:157-63; PMID:18567944; http://dx.doi.org/ 10.4161/epi.3.3.6387 [DOI] [PubMed] [Google Scholar]
- 12.Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer 2004; 4:814-9; PMID:15510162; http://dx.doi.org/ 10.1038/nrc1457 [DOI] [PubMed] [Google Scholar]
- 13.Stefansson OA, Villanueva A, Vidal A, Martí L, Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics 2012; 7:1225-9; PMID:23069641; http://dx.doi.org/ 10.4161/epi.22561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel W, Surowy H. Reduced DNA repair in BRCA1 mutation carriers undetectable before onset of breast cancer? Br J Cancer 2007; 97:1184-6; author reply 1187; PMID:17848944; http://dx.doi.org/ 10.1038/sj.bjc.6603977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1α, autophagy and ketone body production. Cell Cycle 2012; 11:4167-73; PMID:23047605; http://dx.doi.org/ 10.4161/cc.22316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milner R, Wombwell H, Eckersley S, Barnes D, Warwicker J, Van Dorp E, Rowlinson R, Dearden S, Hughes G, Harbron C, et al.. Validation of the BRCA1 antibody MS110 and the utility of BRCA1 as a patient selection biomarker in immunohistochemical analysis of breast and ovarian tumors. Virchows Arch 2013; 462:269-79; PMID:23354597; http://dx.doi.org/ 10.1007/s00428-012-1368-y [DOI] [PubMed] [Google Scholar]
- 17.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet 2006; 2:e119; PMID:16895450; http://dx.doi.org/ 10.1371/journal.pgen.0020119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuillaume ML, Uhrhammer N, Vidal V, Vidal VS, Chabaud V, Jesson B, Kwiatkowski F, Bignon Y-J. Use of gene expression profiles of peripheral blood lymphocytes to distinguish BRCA1 mutation carriers in high risk breast cancer families. Cancer Inform 2009; 7:41-56; PMID:19352458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432:332-7; PMID:15549095; http://dx.doi.org/ 10.1038/nature03096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007; 67:10123-8; PMID:17974953; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-3127 [DOI] [PubMed] [Google Scholar]
- 21.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392-401; PMID:16572188; http://dx.doi.org/ 10.1038/nrc1877 [DOI] [PubMed] [Google Scholar]
- 22.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110:341-50; PMID:12163453; http://dx.doi.org/ 10.1172/JCI0215518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, et al.. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 2010; 107:20009-14; PMID:21041659; http://dx.doi.org/ 10.1073/pnas.1013805107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010; 17:135-47; PMID:20138012; http://dx.doi.org/ 10.1016/j.ccr.2009.12.041 [DOI] [PubMed] [Google Scholar]
- 25.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al.. Pten in stromal fibroblasts suppresses mammary epithelial tumors. Nature 2009; 461:1084-91; PMID:19847259; http://dx.doi.org/ 10.1038/nature08486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pickard A, Cichon AC, Barry A, Kieran D, Patel D, Hamilton P, Salto-Tellez M, James J, McCance DJ. Inactivation of Rb in stromal fibroblasts promotes epithelial cell invasion. EMBO J 2012; 31:3092-103; PMID:22643222; http://dx.doi.org/ 10.1038/emboj.2012.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KW, Yeo SY, Sung CO, Kim SH. Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Res 2015; 75:73-85; PMID:25368021; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0350 [DOI] [PubMed] [Google Scholar]
- 28.Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, And∫ S, Martinez-Outschoorn U, et al.. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012; 11:3019-35; PMID:22874531; http://dx.doi.org/ 10.4161/cc.21384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res 2013; 73:5016-28; PMID:23824740; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, Sotgia F, Rubin E, Lisanti MP. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle 2013; 12:289-301; PMID:23257780; http://dx.doi.org/ 10.4161/cc.23109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valin A, Barnay-Verdier S, Robert T, Ripoche H, Brellier F, Chevallier-Lagente O, Avril M-F, Magnaldo T. PTCH1 +/− dermal fibroblasts isolated from healthy skin of Gorlin syndrome patients exhibit features of carcinoma associated fibroblasts. PLoS One 2009; 4:e4818; PMID:19287498; http://dx.doi.org/ 10.1371/journal.pone.0004818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Luca P, Moiola CP, Zalazar F, Gardner K, Vazquez ES, De Siervi A. BRCA1 and p53 regulate critical prostate cancer pathways. Prostate Cancer Prostatic Dis 2013; 16:233-8; PMID:23670255; http://dx.doi.org/ 10.1038/pcan.2013.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durning P, Schor SL, Sellwood RA. Fibroblasts from patients with breast cancer show abnormal migratory behaviour in vitro. Lancet 1984; 2:890-2; PMID:6148619; http://dx.doi.org/ 10.1016/S0140-6736(84)90653-6 [DOI] [PubMed] [Google Scholar]
- 34.Haggie JA, Sellwood RA, Howell A, Birch JM, Schor SL. Fibroblasts from relatives of patients with hereditary breast cancer show fetal-like behaviour in vitro. Lancet 1987; 1:1455-7; PMID:2885452; http://dx.doi.org/ 10.1016/S0140-6736(87)92206-9 [DOI] [PubMed] [Google Scholar]
- 35.Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A 2005; 102:9176-81; PMID:15967981; http://dx.doi.org/ 10.1073/pnas.0503793102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet 1995; 9:444-50; PMID:7795653; http://dx.doi.org/ 10.1038/ng0495-444 [DOI] [PubMed] [Google Scholar]
- 37.Mar PK, Roy P, Yin HL, Cavanagh HD, Jester JV. Stress fiber formation is required for matrix reorganization in a corneal myofibroblast cell line. Exp Eye Res 2001; 72:455-66; PMID:11273673; http://dx.doi.org/ 10.1006/exer.2000.0967 [DOI] [PubMed] [Google Scholar]
- 38.Antecol MH, Darveau A, Sonenberg N, Mukherjee BB. Altered biochemical properties of actin in normal skin fibroblasts from individuals predisposed to dominantly inherited cancers. Cancer Res 1986; 46:1867-73; PMID:3948169 [PubMed] [Google Scholar]
- 39.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, et al.. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506-14; PMID:20818174; http://dx.doi.org/ 10.4161/cc.9.17.12731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012; 11:3956-63; PMID:23082721; http://dx.doi.org/ 10.4161/cc.22136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al.. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984-4001; PMID:19923890; http://dx.doi.org/ 10.4161/cc.8.23.10238 [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al.. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256-76; PMID:20814239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyo JK, Hee JK, Rih J-KK, Mattson TL, Kyu WK, Cho C-HH, Isaacs JS, Bae I, Kang HJ, Kim HJ, et al.. BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J Biol Chem 2006; 281:13047-56; PMID:16543242; http://dx.doi.org/ 10.1074/jbc.M513033200 [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, et al.. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515-33; PMID:20855962; http://dx.doi.org/ 10.4161/cc.9.17.12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu LN, Xu BN, Cai J, Yang JB, Lin N. Tumor-associated fibroblast-conditioned medium promotes tumor cell proliferation and angiogenesis. Genet Mol Res 2013; 12:5863-71; PMID:24301956; http://dx.doi.org/ 10.4238/2013.November.22.14 [DOI] [PubMed] [Google Scholar]
- 46.Subramaniam KS, Tham ST, Mohamed Z, Woo YL, Mat Adenan NA, Chung I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS One 2013; 8:e68923; PMID:23922669; http://dx.doi.org/ 10.1371/journal.pone.0068923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong X, Yi J, Carmon KS, Crumbley CA, Xiong W, Thomas A, Fan X, Guo S, An Z, Chang JT, et al.. Aberrant RSPO3-LGR4 signaling in Keap1-deficient lung adenocarcinomas promotes tumor aggressiveness. Oncogene 2015; 34:4692-701; PMID:25531322; http://dx.doi.org/ 10.1038/onc.2014.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carmon KS, Gong X, Yi J, Thomas A, Liu Q. RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc Natl Acad Sci U S A 2014; 111:E1221-9; PMID:24639526; http://dx.doi.org/ 10.1073/pnas.1323106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calon A, Tauriello DVF, Batlle E. TGF-β in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 2014; 25:15-22; PMID:24412104; http://dx.doi.org/ 10.1016/j.semcancer.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, Pavlides S, Lamb R, Sneddon S, Howell A, et al.. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle 2012; 11:4402-13; PMID:23172369; http://dx.doi.org/ 10.4161/cc.22776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 52.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3–new capabilities and interfaces. Nucleic Acids Res 2012; 40:e115; PMID:22730293; http://dx.doi.org/ 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bräutigam C, Raggioli A, Winter J. The Wnt/β-catenin pathway regulates the expression of the miR-302 cluster in mouse ESCs and P19 cells. PLoS One 2013; 8:e75315; http://dx.doi.org/ 10.1371/journal.pone.0075315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.