

Abstract
Agarose gel electrophoresis is the most effective way of separating DNA fragments of varying sizes ranging from 100 bp to 25 kb1. Agarose is isolated from the seaweed genera Gelidium and Gracilaria, and consists of repeated agarobiose (L- and D-galactose) subunits2. During gelation, agarose polymers associate non-covalently and form a network of bundles whose pore sizes determine a gel's molecular sieving properties. The use of agarose gel electrophoresis revolutionized the separation of DNA. Prior to the adoption of agarose gels, DNA was primarily separated using sucrose density gradient centrifugation, which only provided an approximation of size. To separate DNA using agarose gel electrophoresis, the DNA is loaded into pre-cast wells in the gel and a current applied. The phosphate backbone of the DNA (and RNA) molecule is negatively charged, therefore when placed in an electric field, DNA fragments will migrate to the positively charged anode. Because DNA has a uniform mass/charge ratio, DNA molecules are separated by size within an agarose gel in a pattern such that the distance traveled is inversely proportional to the log of its molecular weight3. The leading model for DNA movement through an agarose gel is "biased reptation", whereby the leading edge moves forward and pulls the rest of the molecule along4. The rate of migration of a DNA molecule through a gel is determined by the following: 1) size of DNA molecule; 2) agarose concentration; 3) DNA conformation5; 4) voltage applied, 5) presence of ethidium bromide, 6) type of agarose and 7) electrophoresis buffer. After separation, the DNA molecules can be visualized under uv light after staining with an appropriate dye. By following this protocol, students should be able to: 1. Understand the mechanism by which DNA fragments are separated within a gel matrix 2. Understand how conformation of the DNA molecule will determine its mobility through a gel matrix 3. Identify an agarose solution of appropriate concentration for their needs 4. Prepare an agarose gel for electrophoresis of DNA samples 5. Set up the gel electrophoresis apparatus and power supply 6. Select an appropriate voltage for the separation of DNA fragments 7. Understand the mechanism by which ethidium bromide allows for the visualization of DNA bands 8. Determine the sizes of separated DNA fragments
Keywords: Genetics, Issue 62, Gel electrophoresis, agarose, DNA separation, ethidium bromide
Protocol
1. Preparation of the Gel
Weigh out the appropriate mass of agarose into an Erlenmeyer flask. Agarose gels are prepared using a w/v percentage solution. The concentration of agarose in a gel will depend on the sizes of the DNA fragments to be separated, with most gels ranging between 0.5%-2%. The volume of the buffer should not be greater than 1/3 of the capacity of the flask.
Add running buffer to the agarose-containing flask. Swirl to mix. The most common gel running buffers are TAE (40 mM Tris-acetate, 1 mM EDTA) and TBE (45 mM Tris-borate, 1 mM EDTA).
Melt the agarose/buffer mixture. This is most commonly done by heating in a microwave, but can also be done over a Bunsen flame. At 30 s intervals, remove the flask and swirl the contents to mix well. Repeat until the agarose has completely dissolved.
Add ethidium bromide (EtBr) to a concentration of 0.5 μg/ml. Alternatively, the gel may also be stained after electrophoresis in running buffer containing 0.5 μg/ml EtBr for 15-30 min, followed by destaining in running buffer for an equal length of time.
Note: EtBr is a suspected carcinogen and must be properly disposed of per institution regulations. Gloves should always be worn when handling gels containing EtBr. Alternative dyes for the staining of DNA are available; however EtBr remains the most popular one due to its sensitivity and cost.
Allow the agarose to cool either on the benchtop or by incubation in a 65 °C water bath. Failure to do so will warp the gel tray.
Place the gel tray into the casting apparatus. Alternatively, one may also tape the open edges of a gel tray to create a mold. Place an appropriate comb into the gel mold to create the wells.
Pour the molten agarose into the gel mold. Allow the agarose to set at room temperature. Remove the comb and place the gel in the gel box. Alternatively, the gel can also be wrapped in plastic wrap and stored at 4 °C until use (Fig. 1).
2. Setting up of Gel Apparatus and Separation of DNA Fragments
Add loading dye to the DNA samples to be separated (Fig. 2). Gel loading dye is typically made at 6X concentration (0.25% bromphenol blue, 0.25% xylene cyanol, 30% glycerol). Loading dye helps to track how far your DNA sample has traveled, and also allows the sample to sink into the gel.
Program the power supply to desired voltage (1-5V/cm between electrodes).
Add enough running buffer to cover the surface of the gel. It is important to use the same running buffer as the one used to prepare the gel.
Attach the leads of the gel box to the power supply. Turn on the power supply and verify that both gel box and power supply are working.
Remove the lid. Slowly and carefully load the DNA sample(s) into the gel (Fig. 3). An appropriate DNA size marker should always be loaded along with experimental samples.
Replace the lid to the gel box. The cathode (black leads) should be closer the wells than the anode (red leads). Double check that the electrodes are plugged into the correct slots in the power supply.
Turn on the power. Run the gel until the dye has migrated to an appropriate distance.
3. Observing Separated DNA fragments
When electrophoresis has completed, turn off the power supply and remove the lid of the gel box.
Remove gel from the gel box. Drain off excess buffer from the surface of the gel. Place the gel tray on paper towels to absorb any extra running buffer.
Remove the gel from the gel tray and expose the gel to uv light. This is most commonly done using a gel documentation system (Fig. 4). DNA bands should show up as orange fluorescent bands. Take a picture of the gel (Fig. 5).
Properly dispose of the gel and running buffer per institution regulations.
4. Representative Results
Figure 5 represents a typical result after agarose gel electrophoresis of PCR products. After separation, the resulting DNA fragments are visible as clearly defined bands. The DNA standard or ladder should be separated to a degree that allows for the useful determination of the sizes of sample bands. In the example shown, DNA fragments of 765 bp, 880 bp and 1022 bp are separated on a 1.5% agarose gel along with a 2-log DNA ladder.
 Figure 1. A solidified agarose gel after removal of the comb.
Figure 1. A solidified agarose gel after removal of the comb.
 Figure 2. A student adding loading dye to her DNA samples.
Figure 2. A student adding loading dye to her DNA samples.
 Figure 3. A student loading the DNA sample into a well in the gel.
Figure 3. A student loading the DNA sample into a well in the gel.
 Figure 4. An example of a gel documentation system.
Figure 4. An example of a gel documentation system.
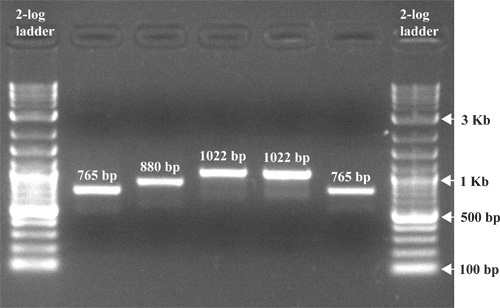 Figure 5. An image of a gel post electrophoresis. EtBr was added to the gel before electrophoresis to a final concentration of 0.5 μg/ml, followed by separation at 100 V for 1 hour. The gel was exposed to uv light and the picture taken with a gel documentation system.
Figure 5. An image of a gel post electrophoresis. EtBr was added to the gel before electrophoresis to a final concentration of 0.5 μg/ml, followed by separation at 100 V for 1 hour. The gel was exposed to uv light and the picture taken with a gel documentation system.
Discussion
Agarose gel electrophoresis has proven to be an efficient and effective way of separating nucleic acids. Agarose's high gel strength allows for the handling of low percentage gels for the separation of large DNA fragments. Molecular sieving is determined by the size of pores generated by the bundles of agarose7 in the gel matrix. In general, the higher the concentration of agarose, the smaller the pore size. Traditional agarose gels are most effective at the separation of DNA fragments between 100 bp and 25 kb. To separate DNA fragments larger than 25 kb, one will need to use pulse field gel electrophoresis6, which involves the application of alternating current from two different directions. In this way larger sized DNA fragments are separated by the speed at which they reorient themselves with the changes in current direction. DNA fragments smaller than 100 bp are more effectively separated using polyacrylamide gel electrophoresis. Unlike agarose gels, the polyacrylamide gel matrix is formed through a free radical driven chemical reaction. These thinner gels are of higher concentration, are run vertically and have better resolution. In modern DNA sequencing capillary electrophoresis is used, whereby capillary tubes are filled with a gel matrix. The use of capillary tubes allows for the application of high voltages, thereby enabling the separation of DNA fragments (and the determination of DNA sequence) quickly.
Agarose can be modified to create low melting agarose through hydroxyethylation. Low melting agarose is generally used when the isolation of separated DNA fragments is desired. Hydroxyethylation reduces the packing density of the agarose bundles, effectively reducing their pore size8. This means that a DNA fragment of the same size will take longer to move through a low melting agarose gel as opposed to a standard agarose gel. Because the bundles associate with one another through non-covalent interactions9, it is possible to re-melt an agarose gel after it has set.
EtBr is the most common reagent used to stain DNA in agarose gels10. When exposed to uv light, electrons in the aromatic ring of the ethidium molecule are activated, which leads to the release of energy (light) as the electrons return to ground state. EtBr works by intercalating itself in the DNA molecule in a concentration dependent manner. This allows for an estimation of the amount of DNA in any particular DNA band based on its intensity. Because of its positive charge, the use of EtBr reduces the DNA migration rate by 15%. EtBr is a suspect mutagen and carcinogen, therefore one must exercise care when handling agarose gels containing it. In addition, EtBr is considered a hazardous waste and must be disposed of appropriately. Alternative stains for DNA in agarose gels include SYBR Gold, SYBR green, Crystal Violet and Methyl Blue. Of these, Methyl Blue and Crystal Violet do not require exposure of the gel to uv light for visualization of DNA bands, thereby reducing the probability of mutation if recovery of the DNA fragment from the gel is desired. However, their sensitivities are lower than that of EtBr. SYBR gold and SYBR green are both highly sensitive, uv dependent dyes with lower toxicity than EtBr, but they are considerably more expensive. Moreover, all of the alternative dyes either cannot be or do not work well when added directly to the gel, therefore the gel will have to be post stained after electrophoresis. Because of cost, ease of use, and sensitivity, EtBr still remains the dye of choice for many researchers. However, in certain situations, such as when hazardous waste disposal is difficult or when young students are performing an experiment, a less toxic dye may be preferred.
Loading dyes used in gel electrophoresis serve three major purposes. First they add density to the sample, allowing it to sink into the gel. Second, the dyes provide color and simplify the loading process. Finally, the dyes move at standard rates through the gel, allowing for the estimation of the distance that DNA fragments have migrated.
The exact sizes of separated DNA fragments can be determined by plotting the log of the molecular weight for the different bands of a DNA standard against the distance traveled by each band. The DNA standard contains a mixture of DNA fragments of pre-determined sizes that can be compared against the unknown DNA samples. It is important to note that different forms of DNA move through the gel at different rates. Supercoiled plasmid DNA, because of its compact conformation, moves through the gel fastest, followed by a linear DNA fragment of the same size, with the open circular form traveling the slowest.
In conclusion, since the adoption of agarose gels in the 1970s for the separation of DNA, it has proven to be one of the most useful and versatile techniques in biological sciences research.
Disclosures
We have nothing to disclose.
References
- Sambrook J, Russell DW. Molecular Cloning. 3rd 2001.
- Kirkpatrick FH. Overview of agarose gel properties. Electrophoresis of large DNA molecules: theory and applications. 1991. pp. 9–22.
- Helling RB, Goodman HM, Boyer HW. Analysis of endonuclease R•EcoRI fragments of DNA from lambdoid bacteriophages and other viruses by agarose-gel electrophoresis. J. Virol. 1974;14:1235–1244. doi: 10.1128/jvi.14.5.1235-1244.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Aldridge PK, Callis JB. Observation of individual DNA molecules undergoing gel electrophoresis. Science. 1989;243:203–206. doi: 10.1126/science.2911733. [DOI] [PubMed] [Google Scholar]
- Aaji C, Borst P. The gel electrophoresis of DNA. Biochim. Biophys. Acta. 1972;269:192–200. doi: 10.1016/0005-2787(72)90426-1. [DOI] [PubMed] [Google Scholar]
- Lai E, Birren BW, Clark SM, Simon MI, Hood L. Pulsed field gel electrophoresis. Biotechniques. 1989;7:34–42. [PubMed] [Google Scholar]
- Devor EJ. IDT tutorial: gel electrophoresis. 2010. Available from: http://cdn.idtdna.com/Support/Technical/TechnicalBulletinPDF/Gel_Electrophoresis.pdf.
- Serwer P. Agarose gels: properties and use for electrophoresis. Electrophoresis. 1983;4:375–382. [Google Scholar]
- Dea ICM, McKinnon AA, Rees DA. Tertiary and quaternary structure and aqueous polysaccharide systems which model cell wall adhesion: reversible changes in conformation and association if agarose, carrageenan and galactomannans. J. Mol. Biol. 1972;68:153–172. doi: 10.1016/0022-2836(72)90270-7. [DOI] [PubMed] [Google Scholar]
- Sharp PA, Sugden B, Sambrook J. Detection of two restriction endonuclease activities in H. parainfluenzae using analytical agarose-ethidium bromide electrophoresis. Biochemistry. 1973;12:3055–3063. doi: 10.1021/bi00740a018. [DOI] [PubMed] [Google Scholar]