

Abstract
Objective
On December 8–9, 2014, the Pennington Biomedical Research Center convened a scientific symposium to review the state-of-the-science and future directions for the study of developmental programming of obesity and chronic disease. The objectives of the symposium were to discuss: (i) past and current scientific advances in animal models, population-based cohort studies and human clinical trials, (ii) the state-of-the-science of epigenetic-based research, and (iii) considerations for future studies.
Results
The overarching goal was to provide a comprehensive assessment of the state of the scientific field, to identify research gaps and opportunities for future research in order to identify and understand the mechanisms contributing to the developmental programming of health and disease.
Conclusions
Identifying the mechanisms which cause or contribute to developmental programming of future generations will be invaluable to the scientific and medical community. The ability to intervene during critical periods of prenatal and early postnatal life to promote lifelong health is the ultimate goal. Considerations for future research including the use of animal models, the study design in human cohorts with considerations about the timing of the intrauterine exposure and the resulting tissue specific epigenetic signature were extensively discussed and are presented in this meeting summary.
Introduction
Human epidemiological studies and dietary interventions in animal models have suggested that maternal nutritional imbalance and metabolic disturbances during critical time periods of development may have long term health consequences on offspring (1, 2, 3). Such phenomena are widely referred to as ‘developmental programming’. There is growing acceptance of the notion that epigenetic changes associated with chromatin structure and regulation of gene expression provide a basis to contribute to developmental programming and establishment of predispositions to later in life chronic disease (4, 5, 6).
In addition to developmental programming, epigenetic changes can occur throughout an individual’s life as a result of acute or chronic exposure to an environment or intervention. Indeed, epigenetic modifications whether acquired during ontogenesis or later in life can influence the way an individual responds to an environmental exposure or intervention, which may in part explain the individual variation in heritability of complex traits not explained by DNA sequence (7, 8).
Both developmental programming and the potential for epigenetic changes later in life suggest new possibilities for the prevention and treatment of obesity and related chronic diseases (9). This symposium summary was constructed to: 1) provide an overview of epigenetics as it relates to nutritional and developmental programming; 2) discuss considerations for the current technologies, study design, and data interpretation and; 3) discuss the limitations of current knowledge and the need for future research.
Developmental Programming: Review and Current State-of-the-Science
The classical definition of ‘developmental programming’ refers to the ability of exposures during prenatal or early postnatal development to cause permanent changes to the physiology, metabolism and epigenome of an individual which subsequently will affect health and increase risk of disease (2, 10). Such exposures include, but are not limited to, undernutrition, overnutrition, malnutrition; teratogens including pollutants, drugs, alcohol, etc.; altered hormonal milieus resulting from maternal overweight, obesity, excess gestational weight gain, diabetes mellitus; maternal stress; oxidative stress from hypertension or placenta insufficiencies. Extensive effort over the past twenty five years has been expended to identify and understand the relationships between early exposure and disease, as well as the contributing mechanisms. Thus far, links have been identified through epidemiological, prospective and intervention studies in animal models and humans.
Early epidemiological work identified the relationship between intrauterine exposures and adult development of disease including obesity (11, 12) and coronary heart disease (13, 14). Since these observations, additional links between low birthweight and adult chronic diseases, including impaired glucose tolerance, hypertension, cardiovascular disease and obesity have been also identified (15, 16, 17, 18). Epidemiological studies of additional cohorts such as the Uppsala, Sweden, Helsinki, Finland and the Nurse’s Health Study, USA have also revealed correlations between low birthweight or low size at birth and the development of diabetes (19, 20, 21, 22, 23, 24), cardiovascular disease (25, 26, 27, 28, 29, 30, 31, 32, 33), unfavorable alterations in body composition (34, 35), and hypertension (36, 37) later in life.
Even though the observed association between low birth weight and adult chronic disease has been reproduced in many cohorts around the world, the supportive evidence for the developmental programming hypothesis of adult chronic disease relies heavily on the assumption that infant growth/size at birth is an indicator of adverse exposures in utero. However in the case of fetal exposure to inadequate nutrition, poor nutrition induced by either insufficient maternal intake or adverse fetoplacental development, is not the only cause for low birth weight. The developmental programming hypothesis has also been supported by epidemiological studies in populations with known exposures. For instance, studies of individuals exposed to the Dutch Famine in utero between December 1944 and April 1945, when the daily ration for adults was 400–800 calories, have observed relationships between famine exposure in utero and the development of diabetes (38), high blood pressure (39), and unfavorable body composition (12) in the offspring later in life.
The ability of researchers to obtain longitudinal data from individuals within large cohort studies with known in utero exposures has also proven to substantially advance the field. Data collected in addition to the important time of conception include prospective assessments of body composition, blood pressure, cardiovascular risk factors, food intake, medical history, glucose tolerance, and biospecimens allowing for the study of epigenetics (6, 40). The Quebec Ice Storm, which occurred in 1998 and led to a loss of electrical power for up to 6 weeks in 1,400,000 households, provided a unique opportunity for longitudinal data collection from offspring of a known in utero exposure, i.e. objective and/or subjective maternal stress (41). The offspring of mothers who were pregnant during this storm have participated in studies of their epigenomes, metabolism, cognitive function, motor skills, and body size in an effort to understand the consequences of intrauterine exposure to objective and subjective stress.
Observational and interventional clinical trials of populations experiencing adverse intrauterine exposures are also contributing to the understanding of developmental programming. Gambian women have been part of such research because The Gambia experiences two agricultural and thus nutritional seasons annually, the rainy (hungry) season and the dry (harvest) season, leading to fluctuation in nutritional status. Research groups have established relationships with communities in The Gambia, and clinical trials particularly in childbearing women have been ongoing since the 1970’s to study the effects of nutritional status in this population (42).
Mechanisms of Developmental Programming
Three principal, non-mutually exclusive mechanisms by which adverse intrauterine exposures developmentally program offspring were extensively discussed (Figure 1): 1) permanent organ or tissue structural changes, 2) accelerated cellular aging, and 3) epigenetic programming of gene expression.
Figure 1.
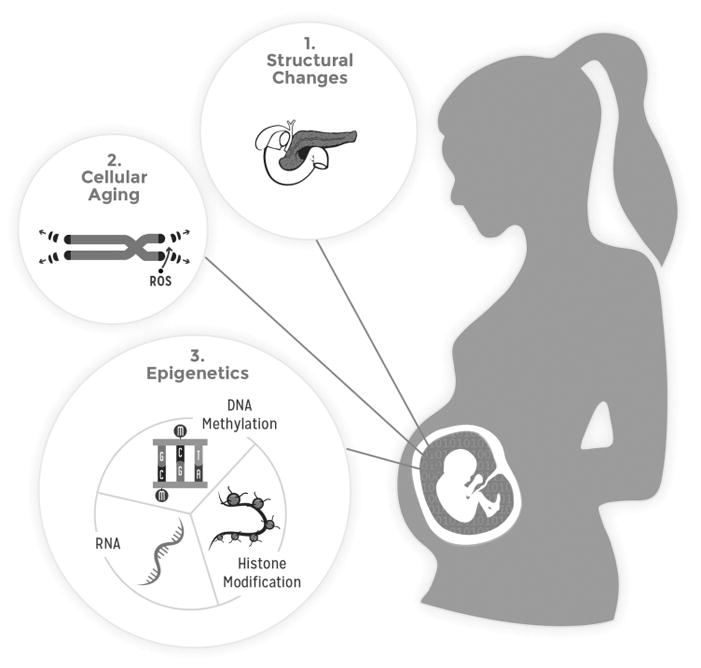
Mechanisms of developmental programming
a. Permanent organ or tissue structural changes
Adverse intrauterine exposures during organ and system development can cause permanent structural changes within the fetus and consequently contribute to the development of chronic disease later in life. During organogenesis, scarce available nutrients are shunted to vital organs, such as the brain, causing an exacerbated limitation of nutrients to visceral organs, such as the pancreas, liver, and kidneys. If a nutrient supply to an organ is restricted within a critical period of organ development, such nutritional challenge could result in permanent alterations of cellular function, organ structure, and consequently organ function. Reviewed here to demonstrate this susceptibility are the pancreas, liver, skeletal muscle, and adipose tissue. Similar observations include reduced nephron number in the kidney (43), remodeling of cardiac cells in the heart, remodeling of extracellular matrices of blood vessels (44), and altered neuronal projections (45).
i. Pancreas
Models of intrauterine growth restriction (IUGR) and maternal protein restriction have both demonstrated a reduction in pancreatic β cell mass in exposed offspring (46, 47). Furthermore, exposure to maternal low protein intake during gestation and lactation as well as during lactation alone results in fewer pancreatic islets with irregular shape and size in exposed rat offspring (48). Since the pancreatic β cell population is maintained in adulthood by the self-replication of existing β cells, a reduction in β cell mass resulting from adverse intrauterine exposure could alter metabolism and increase the risk for diabetes later in life.
ii. Liver
Models of maternal protein restriction have also elucidated the effects of adverse intrauterine exposure on liver and hepatic glucose homeostasis. The liver of rats exposed to protein restriction in utero have been shown to have a 50% reduction in glucokinase and a 100% increase in phosphoenolpyruvate. As these enzymes are well known to be located in different zones of liver, it is hypothesized that protein restriction exposure in utero alters the periportal and perivenous cells and therefore the liver structure. This shift of structure and hepatic enzymatic profile biases the liver to a 4-fold shift toward glucose production rather than glucose utilization in rats, indicating that in utero exposure could also alter liver metabolism and increase the risk for diabetes later in life (49, 50, 51, 52).
iii. Skeletal Muscle and Adipose Tissue
Skeletal muscle and adipose tissue are lower priority for nutrient partitioning during development, thus leaving these tissues vulnerable to nutrient deficiency (53, 54, 55). Nutrient restriction and/or IUGR create a hypoxic and hypoglycemic intrauterine environment resulting in reduced fetal muscle and satellite cell development. To adapt to the hypoxic, hypoglycemic, and hypoinsulinemic environment, skeletal muscle decreases glucose oxidation to spare glucose and oxygen for more critical organs such as the brain and the heart (54, 55). Growth restricted infants are born with less skeletal muscle mass and endure slower muscle mass growth rates than infants with appropriate weight for gestational age (15, 56). Reduced muscle mass is thought to contribute to positive energy balance due to decreased energy expenditure. In addition, glucose is redistributed from skeletal muscle to adipose tissue which results in adipocyte hypertrophy and adipose tissue expansion (57). The increased fat-to-muscle ratio seen in the postnatal period continues into adulthood resulting in obesity and metabolic syndrome (15, 56). In addition to increased adipocyte hypertrophy in the postnatal period, developmental programming contributes to adulthood obesity through structural and functional modifications made to the adipocytes during development. Adipocyte development and programming is sensitive to both maternal over- and under-nutrition (58). For example, maternal obesity and/or high fat diet during pregnancy leads to increased fetal adipocyte differentiation, increased genome-wide DNA methylation of CpG sites (59), and histone modifications related to leptin and adiponectin production (58).
b. Accelerated cellular aging
Studies have demonstrated adverse exposure in utero can program longevity and impact lifespan. Hales et al. showed in utero exposure to maternal protein restriction reduced the lifespan of male offspring in rats, while postnatal protein restriction prolonged lifespan (51). Accelerated cellular aging is another mechanism to consider by which developmental programming occurs.
Several mechanisms have been postulated as causes of accelerated aging, including oxidative stress, inflammation, and an altered hormonal milieu; all of which are also known to be influenced by early nutritional exposures. For example, oxidative stress can be induced in utero by maternal obesity and over- or undernutrition. Such oxidative stress can lead to DNA damage, point mutations, and direct cleavage of DNA resulting in increased DNA methylation, altered cellular function, accelerated cellular aging, and increased cell turnover (60, 61, 62). Frequently, maternal protein restriction and/or preeclampsia can contribute to IUGR which leads to increased production of reactive oxygen species associated with higher lipid peroxidation (60). Due to their reduced antioxidant capacity and high metabolic activity, pancreatic islets are especially sensitive to oxidative damage. Increased oxidative damage leads to shorter telomeres, advanced cellular aging, and more fibrotic islets. Subsequently, islet fibrosis leads to β cell dysfunction, resulting in accelerated progression of insulin intolerance and hyperglycemia in the offspring of low protein fed mothers (63). Surgically induced IUGR due to uteroplacental insufficiency in rodents shows similar results as protein restricted IUGR. An increase in reactive oxygen species production results in increased damage to the mitochondrial DNA in islets of offspring as well as impaired insulin secretion and progressive β cell dysfunction when compared to controls (64). This work provides another possible mechanism linking poor maternal diet and inadequate in utero growth to the increased risk of diseases such as diabetes later in life.
c. “Epigenetics”
The literal meaning of the word “epigenetics” is “outside conventional genetics” or “above or beyond genetics” (65, 66). Russo and colleagues defined epigenetics as, “mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” (67). Bird (68) cites phenomena where “epigenetic” changes occur but do not fall under this strict definition of heritability. Practically speaking, locus-specific alterations in histone marks are widely considered ‘epigenetic’ but would not be considered directly mitotically heritable due to the lack of a known mechanism for mitotic heritability (69). On the other hand, DNA methylation, which occurs in mammals predominantly at CpG dinucleotides, can be maintained very stably due to the semiconservative replication of CpG methylation during mitosis via the action of the maintenance methylase DNMT1 (70). Transgenerational heritability of epigenetic modifications is possible through modification of the epigenome of germ cells of the developing fetus (58, 71). Therefore, a more encompassing definition of epigenetics proposed by Bird is, “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states” (68). The specific mechanisms of epigenetic programming of gene expression include DNA methylation, various histone modifications, autoregulatory DNA binding proteins, and noncoding RNAs.
i. DNA methylation
DNA methylation was discovered in 1948 and was originally understood as a static, permanent change in the genome. However, DNA methylation is now known to be reversible and thus methylation and demethylation of DNA is in a dynamic state that allows additional control of gene expression and phenotypes. DNA methylation occurs most commonly at the cytosine residue of dinucleotide sequence CpG and most often the methylated gene is silenced. Metastable epialleles are genomic regions at which DNA methylation is established stochastically in the embryo and stably maintained in the differentiated tissues. Throughout the mammalian genome, CpG sites are relatively depleted. CpG islands are concentrated regions (~1000 base pairs) that are CpG rich, often found in promotor regions, and typically resistant to methylation. In general, highly methylated promoter regions reduce the ability of transcription factors to bind and thus impede gene expression, with notable exceptions in the genome. Differential methylation of CpG islands is associated with various disease states including cancer and diabetes (72), but correlation versus causation remains under debate. However, evidence that DNA methylation can cause phenotypic variability in isogenic animals due to methylation of metastable epialleles is clearly demonstrated in the varying coat colors and tail characteristics of mice due to the epigenetic state of the agouti viable yellow and axin fused alleles (73, 74).
Data suggest that the nutritional status of a mother at the time of conception can permanently influence the epigenome of her offspring, consequently impacting lifelong health (40, 73). Metastable CpG methylation resulting in phenotypic variability for example, has been shown to be influenced by methyl donor dietary supplementation beginning prior to conception and continuing through weaning (75). Furthermore, hypomethylation and decreased Dnmt1 expression have been observed in offspring of protein restricted rats; an effect rescued by folate (methyl donor) supplementation (76, 77). Prospective work done in Gambian women (78) has demonstrated similar effects at human metastable epialleles. Due to increased consumption of certain foods and associated changes in concentrations of methyl-donor pathway metabolites (e.g. vitamin B2, homocysteine, and cysteine) in maternal plasma, infants conceived during the rainy (hungry) season had increased methylation at metastable epialleles in peripheral blood leukocytes and hair follicles when compared to those infants conceived in the dry (harvest) season. Finding the same nutritionally-associated epigenetic effect in tissues derived from different germ layers of the embryo indicates that the effect was established in the early embryo (prior to gastrulation) and maintained during subsequent differentiation (78).
Changes in methylation patterns (hypo- and hypermethylation) are observed during periods of inadequate nutrition. Individuals conceived during the Dutch Famine had decreased methylation in CpG islands in whole blood when compared to sibling pairs conceived before or after the Famine. Individuals conceived early during the Famine had higher birth weights which corresponded to unfavorable metabolic profiles and higher BMI later in life (40). In both human and rodent IUGR offspring, alterations in DNA methylation and gene expression related to glucose homeostasis, insulin secretion, and pancreatic islet cell turnover are observed. These alterations in DNA methylation may explain the increased long-term risk for development of type 2 diabetes and cardiovascular disease seen in offspring with IUGR (16, 79, 80).
In addition to nutritional status, maternal behavior and stress can influence DNA methylation patterns in offspring. In rodents and humans alike, maternal care can affect the DNA methylation of the glucocorticoid receptor in the hippocampus of offspring and is associated with anxiety and impaired responsivity to stress later in life (81, 82). Maternal stress endured during the Quebec Ice Storm was shown to be correlated to DNA methylation changes in the offspring that remain well into adolescence (4).
ii. Histone Modification
Modifications of histones, including acetylation, methylation, phosphorylation, and ubiquitination, are direct mechanisms of transcriptional regulation. Frequently, histone acetylation results in active chromatin and subsequently increased transcription of the target genes while histone methylation may result in an inactive or active chromatin structure (83). In addition to influencing DNA methylation, IUGR can result in the deacetylation of histones H3 and H4. These traditionally heavily acetylated regions in β cell islets are associated with Pdx1, a critical regulator of β cell growth. In relation to this dysregulation, islet cell function decreases and insulin resistance increases. Once diabetes occurs, methylation of the CpG island in the promoter region of Pdx1 occurs resulting in the suppression of Pdx1 transcription and further β cell dysfunction (72).
iii. Noncoding RNA
Other epigenetic regulators are noncoding RNAs such as long noncoding RNA (lncRNA) and microRNA (miRNA). With great allelic specificity, lncRNA can recruit proteins involved in gene transcription and can interact with chromatin remodeling complexes to up- and down-regulate gene expression through other epigenetic modifications, i.e. DNA methylation and histone modifications (84, 85, 86, 87). MicroRNA on the other hand influence gene expression post-transcriptionally by binding to mRNA and inhibiting translation or targeting the transcript for degradation (88). Altered maternal nutrition during pregnancy, such as high fat, low protein, and obesogenic diets, has been shown in animal models to impact the expression in offspring of specific microRNA and subsequently regulation of the translation of their target genes (89). For example, upregulation of miR-126 and down regulation of its target, IRS-1, was observed in adipose tissue of male offspring of dams fed an obesogenic diet compared to offspring of dams fed standard chow (90). Phenotypically, these animals exhibited elevated fasting insulin at 8 weeks of age (90). Finally, the roles of noncoding RNAs in epigenetic programming are only recently becoming realized and require further investigation.
Considerations for Study Design and Data Analyses and Interpretation
a) Human Studies
Retrospective studies with Unknown Exposure(s) have extensively demonstrated the association between small size at birth and chronic disease later in life. However, as previously stated, the primary weakness of such studies is the reliance on the assumption that small birth size is indicative of adverse intrauterine exposure (Table 1).
Retrospective studies with Known Exposure(s) allow for the study of human populations with available birth and death records, with valid documentation of known adverse intrauterine exposures during development, e.g. exposed to maternal famine or nuclear radiation in utero. Since adverse intrauterine exposure (e.g. maternal famine or nuclear radiation) is known, such model permits stronger causative arguments for the relationship between adverse intrauterine exposures and disease later in life. Both designs provide strong, supporting evidence of developmental programming, yet are limited in their conclusions by the data available in medical records.
Retrospective studies with the ability to add Prospective follow-up consider retrospective data (e.g. birth records and known adverse intrauterine exposures) collected in research studies designed to genetically and metabolically phenotype individuals, e.g. measurement of glucose tolerance during adulthood in individuals exposed to famine in utero. Such design provides insight into the in vivo metabolic dysregulation and developmental adversities which can result from small birth size or adverse intrauterine exposures, yet conclusions can be limited by the lack of data on or during the specific intrauterine environment exposures.
Prospective Longitudinal Clinical Trials allow for the evaluation of adverse intrauterine exposures in real time as well as the respective fetal outcomes and infant and adolescent development. Such observational studies are the most ideal, although feasibility and ethical considerations must be examined. First, the feasibility of these study designs are hindered by the logistics, cost and potential poor retention for conducting a research study which lasts the life time of its participants, e.g. 80 years. Moreover, the Code of Federal Regulations (Title 45, Part 46, Subpart B) strictly outlines regulations for conducting research in pregnant women, human fetuses and neonates, allowing only for research to be conducted in vulnerable populations under stringently specified risk-to-benefit ratios for mother and/or fetus. Therefore, the study of adverse intrauterine exposures must take place in an observational environment (e.g. studying mothers in developing countries experiencing food insecurity, mothers with diabetes, mothers with excess gestational weight gain, etc.) as the implementation of intentional adverse intrauterine exposures is unethical.
Randomized Clinical Trial: While randomization to a known adverse exposure during pregnancy and development is unethical in humans, randomization to a generally regarded-as-safe intervention (e.g. micronutrient supplementation (folic acid), exercise, etc.) is routinely done. Well controlled, randomized clinical trials are the gold standard for intervention research, but often include multiple variables that cannot be controlled such as individual response to intervention and genetic variability. Recently, the rapidly evolving field of genetics of complex diseases has revealed hundreds of common variants associated with human diseases or traits, allowing us to use this knowledge to apply the concept of Mendelian Randomization (MR) to strengthen causality inference (91). MR is based on the fact that genetic variants are randomly selected during meiosis and conception, and are not influenced by confounding factors typical of observational studies. In brief, we apply MR by creating instrumental variables using known genetic variants as exposure to test “un-bias” associations with outcomes of interest. MR has shown to reproduce some randomized controlled trial findings (92) – both positive and negative – and today’s pharmacologic industry is using genetic information and MR to make decisions in their molecules selection pipelines. In the field of Developmental Origins of Health and Disease, we can use MR to mimic randomized clinical trials for exposure that would be unethical to conduct – for example smoking versus non-smoking – or for exposures that are difficult to untangle – excess weight vs hyperglycemia (91). This should help us to focus our interventions on exposures that are more likely causal.
Table 1.
Considerations for experimental models and study designs commonly used in developmental programming research
| Experimental Model | Considerations | Advantages | Disadvantages |
|---|---|---|---|
| Animal |
|
|
|
| Human |
|
|
|
|
Unknown Exposure Retrospective |
|
|
|
|
Known Exposure Retrospective |
|
|
|
| Prospective and Retrospective |
|
|
|
| Longitudinal |
|
|
|
| Interventional |
|
|
|
Epigenetic patterns can also inform interventions. If our investigations show that epigenetic adaptations are truly mediating and part of the causal link between in utero exposure and long term outcomes, we can use identified epigenetic marks to identify children that would benefit from early life interventions to ‘re-normalize’ their epigenetic profile. Also, having predictive epigenetics markers measureable at birth in cord blood would help to evaluate impact of interventions during pregnancy without the need to follow-up the offspring for decades to assess the risks/benefits on long term programming.
Another way to control for genetic variability within an intervention study is to use a crossover study design. Crossover designs allow for an individual to be exposed to both intervention scenarios (e.g. high-fat vs. low fat diet) and act as their own control, and they often require fewer participants for equivalent statistical power than parallel randomized clinical trials. Considerations for crossover designs include epigenetic and genetic history of an individual, the order of exposure, the length of the exposure and the washout period. While many changes in DNA methylation in early embryonic development have been thought to be mitotically stable, environment and lifestyle (i.e. interventions) can induce changes in DNA methylation patterns throughout adulthood. For example, it has been reported that acute exercise in a dose dependent manner induces gene-specific (FTO, THADA, TCF7L2, and KCNQ1) hypomethylation in human skeletal muscle (93). We have much to learn about epigenetic responses to acute versus chronic exposures such as change in diet or physical activity to know in which scenario we can use a crossover design adequately and determine the necessary wash-out period for each specific study question.
b) Animal Studies
The use of animal models versus humans in epigenetic research has several benefits such as decreased cost and length of gestation (20 days in mice versus 270 days in humans), the ability to enforce tight environmental conditions (sleep cycles, energy balance, exercise, stress, etc.) as well as genetics (breeding pairs, knockdowns, knockouts, transgenics, etc.), and the ability to implement adverse intrauterine exposures, analyze tissues, and progressively study fetal development by sacrificing animals throughout gestation. The most commonly studied species of developmental programming include rats and mice, non-human primates, sheep and guinea pigs.
When a study strives to most closely model human beings, non-human primates are the model of choice because the genetic background is similar, pregnancies are usually singleton or twin, the length of gestation is similar (and therefore the length of intrauterine exposure), and offspring are fully developed at birth. Sheep are also an excellent experimental model for epigenetic studies for many of the same reasons yet sheep studies are less expensive and have the capability for surgical operations to advance modeling of adverse intrauterine exposure. Mice, rats and guinea pigs are smaller models commonly used because costs are lower and gestation, time to maturation, and life span are shorter. The obvious limitation of studying small animals, when compared to humans, are genetic differences, altricial offspring (guinea pigs excluded), and polytocous pregnancies.
c) Timing
The timing of the intrauterine exposure can produce different programming effects. For example, due to the strict food transport embargo, precise timing of famine exposure was determined in the Dutch Famine Cohort based on birth dates. Epigenetic modifications induced by food restriction during World War II followed by frozen canals and waterways during the winter of 1944–1945 have been observed as a result of this exposure occurring during early gestation (6, 40) and permanent structural changes have been observed as a result of exposure occurring during mid to late gestation. Timing of exposure should also play a role in intervention studies. A better understanding of the critical periods of development during which exposures are most detrimental as well as the effects of said exposures (e.g. epigenetic, pancreatic function, etc.) will allow for intervention and hopefully prevention of adverse developmental consequences later in life.
d) Tissue specific epigenetic signature
While epigenetic modifications that occur in early embryonic development can be passed on to various tissues, not all modification patterns are identical across all tissue types. Animal models allow for unlimited tissue access, however due to limited access in humans, the most common tissue analyzed is blood (peripheral or cord). While specific methylation patterns in differing tissues may correlate with one another (e.g. placenta and cord blood) (94, 95), researchers should use caution when extrapolating changes seen in one tissue to other tissues as each cell and cell type has its own epigenetic signature (9).
e) Epigenetic analysis and interpretation
Like many molecular techniques, methods to assess epigenetic signatures are continually being improved. Genome-wide analysis of epigenetic patterns is generating a large amount of data that require sophisticated bioinformatics to draw conclusions. Global methylation analysis is costly, and may not provide an accurate assessment of the effect of methylation patterns on specific metabolic pathways. Other methods include measurement of methylation of specific candidate genes and genomic regions of interests or taking averages across genomic regions of interest. When choosing an analysis technique it is important to consider the reproducibility of the analysis and how processing the sample may alter the original epigenetic pattern. In addition, seasonal variation, length of exposure, and other population characteristics can impact the reproducibility of a study and should be taken into consideration when interpreting or comparing study results.
Conclusion
Future Directions
Needs for future research include: a) identification of predictors, such as epigenetic biomarkers, to predict risk of development of disease or to monitor success of intervention (validated predictors could lead to prevention with earlier detection), b) advances in epigenetics including the study of cellular mechanisms by which epigenetic changes are regulated and implemented, c) exploration of the causative relationship between epigenetic changes and phenotype (is epigenetics a cause or consequence?), d) studies in evolutionary plasticity by comparing epigenetic modifications within tissues within species, tissues between species, as well as transgenerational inheritance including paternal inheritance, e) studies focusing on those who are protected from disease (e.g., the small percentage of mice who do not become obese during high fat feeding), f) studies that investigate the mechanisms underlying sex-specific epigenetic modifications, g) increase in studies and identification of cohorts for epigenetic epidemiology, h) study of long term follow up of infants enrolled in established trials, particularly those with well phenotyped pregnancies, and finally h) furthering the understanding of the effects of timing of exposure during gestation, including the timing to intervene and timing of proper postnatal intervention. Identifying the mechanisms which cause or contribute to developmental programing of future generations will be invaluable to the scientific and medical community. The ability to intervene during critical periods of prenatal and early postnatal life to promote lifelong health is the ultimate goal.
What is known
Intrauterine exposures are associated with fetal epigenetic modifications and have been linked to increased risk for diseases later in life.
The epigenome can be altered throughout the lifecycle due to nutritional and environmental exposures.
What the study adds
Assesses the current state of science in developmental and nutritional programming and epigenetics throughout the lifecycle.
Discusses the current considerations and limitations of study designs and applications within the field of developmental programming.
Discusses areas for future research to expand epigenetic research within the field of developmental programming.
Acknowledgments
Funding: The Symposium was funded by NORC Center Grant P30DK072476 from the NIDDK. LAG1 is supported by T32DK064584 from the NIDDK. Work in MS laboratory8 was supported by a grant from the Canadian Institute of Health Research MOP-42411. Work in JAM laboratory6 was supported by the Center for Nutrition Research at the University of Navarra in Pamplona Spain. RAW9 is supported by a grant from the U.S. Department of Agriculture (USDA) [CRIS 3092-5-001-059]. Work in BTH laboratory3 was supported by the National Institutes of Health (R01AG042190) and the European Union’s Seventh Framework Program IDEAL (FP7/2007-2011; grant agreement No. 259679). Work funded in CL laboratory5 was funded by The Swedish Research Council and The Novonordisk Foundation. SEO2 is a member of the University of Cambridge MRC Metabolic Diseases Unit. Dr. Hivert4 is the recipient of an American Diabetes Association (ADA) Pathways To Stop Diabetes Award. Work in ER1 and LMR1 laboratory was partially funded by a NORC grant entitled “Nutritional Programming: Environmental and Molecular Interactions” to ER (P30DK072476).
Thank you to Dr. Phillip Brantley and Anne Hawes for their dedication to a very successful Symposium. This work was partially supported by a NORC Center Grant # P30DK072476 entitled “Nutritional Programming: Environmental and Molecular Interactions” sponsored by NIDDK. LAG1 is supported by T32DK064584 from the NIDDK. Work in MS laboratory8 was supported by a grant from the Canadian Institute of Health Research MOP-42411. Work in JAM laboratory6 was supported by the Center for Nutrition Research at the University of Navarra in Pamplona Spain. RAW9 is supported by a grant from the U.S. Department of Agriculture (USDA) [CRIS 3092-5-001-059]. Work in BTH laboratory3 was supported by the National Institutes of Health (R01AG042190) and the European Union’s Seventh Framework Program IDEAL (FP7/2007-2011; grant agreement No. 259679). Work funded in CL laboratory5 was funded by The Swedish Research Council and The Novonordisk Foundation. SEO2 is a member of the University of Cambridge MRC Metabolic Diseases Unit. Dr. Hivert4 is the recipient of an American Diabetes Association (ADA) Pathways To Stop Diabetes Award. Work in ER1 and LMR1 laboratory was partially funded by a NORC grant entitled “Nutritional Programming: Environmental and Molecular Interactions” to ER (P30DK072476).
Footnotes
Disclosure Statement: The authors have nothing to disclose.
References
- 1.Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, et al. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006;118:e1644–1649. doi: 10.1542/peds.2006-1379. [DOI] [PubMed] [Google Scholar]
- 2.Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr. 1999;69:179–197. doi: 10.1093/ajcn/69.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Waterland RA. Does nutrition during infancy and early childhood contribute to later obesity via metabolic imprinting of epigenetic gene regulatory mechanisms? Nestle Nutrition workshop series. Paediatric programme. 2005;56:157–171. doi: 10.1159/000086298. discussion 171–154. [DOI] [PubMed] [Google Scholar]
- 4.Cao-Lei L, Massart R, Suderman MJ, Machnes Z, Elgbeili G, Laplante DP, et al. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS One. 2014;9:e107653. doi: 10.1371/journal.pone.0107653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milagro FI, Mansego ML, De Miguel C, Martinez JA. Dietary factors, epigenetic modifications and obesity outcomes: progresses and perspectives. Molecular aspects of medicine. 2013;34:782–812. doi: 10.1016/j.mam.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsen SC, Gillberg L, Bork-Jensen J, Ribel-Madsen R, Lara E, Calvanese V, et al. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia. 2014;57:1154–1158. doi: 10.1007/s00125-014-3198-8. [DOI] [PubMed] [Google Scholar]
- 9.Waterland RA. Epigenetic mechanisms affecting regulation of energy balance: many questions, few answers. Annu Rev Nutr. 2014;34:337–355. doi: 10.1146/annurev-nutr-071813-105315. [DOI] [PubMed] [Google Scholar]
- 10.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 11.Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 12.Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70:811–816. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- 13.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 14.Forsdahl A. Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease? British journal of preventive & social medicine. 1977;31:91–95. doi: 10.1136/jech.31.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayer AA, Syddall HE, Dennison EM, Gilbody HJ, Duggleby SL, Cooper C, et al. Birth weight, weight at 1 y of age, and body composition in older men: findings from the Hertfordshire Cohort Study. Am J Clin Nutr. 2004;80:199–203. doi: 10.1093/ajcn/80.1.199. [DOI] [PubMed] [Google Scholar]
- 16.Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kensara OA, Wootton SA, Phillips DI, Patel M, Jackson AA, Elia M. Fetal programming of body composition: relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen. Am J Clin Nutr. 2005;82:980–987. doi: 10.1093/ajcn/82.5.980. [DOI] [PubMed] [Google Scholar]
- 18.Osmond C, Barker DJ, Winter PD, Fall CH, Simmonds SJ. Early growth and death from cardiovascular disease in women. BMJ. 1993;307:1519–1524. doi: 10.1136/bmj.307.6918.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson JG. Early growth and coronary heart disease and type 2 diabetes: findings from the Helsinki Birth Cohort Study (HBCS) Am J Clin Nutr. 2011;94:1799S–1802S. doi: 10.3945/ajcn.110.000638. [DOI] [PubMed] [Google Scholar]
- 20.Lithell HO, McKeigue PM, Berglund L, Mohsen R, Lithell UB, Leon DA. Relation of size at birth to non-insulin dependent diabetes and insulin concentrations in men aged 50–60 years. BMJ. 1996;312:406–410. doi: 10.1136/bmj.312.7028.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forsen T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D. The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med. 2000;133:176–182. doi: 10.7326/0003-4819-133-3-200008010-00008. [DOI] [PubMed] [Google Scholar]
- 22.Pulizzi N, Lyssenko V, Jonsson A, Osmond C, Laakso M, Kajantie E, et al. Interaction between prenatal growth and high-risk genotypes in the development of type 2 diabetes. Diabetologia. 2009;52:825–829. doi: 10.1007/s00125-009-1291-1. [DOI] [PubMed] [Google Scholar]
- 23.Whincup PH, Kaye SJ, Owen CG, Huxley R, Cook DG, Anazawa S, et al. Birth weight and risk of type 2 diabetes: a systematic review. JAMA. 2008;300:2886–2897. doi: 10.1001/jama.2008.886. [DOI] [PubMed] [Google Scholar]
- 24.Rich-Edwards JW, Colditz GA, Stampfer MJ, Willett WC, Gillman MW, Hennekens CH, et al. Birthweight and the risk for type 2 diabetes mellitus in adult women. Ann Intern Med. 1999;130:278–284. doi: 10.7326/0003-4819-130-4_part_1-199902160-00005. [DOI] [PubMed] [Google Scholar]
- 25.Leon DA, Lithell HO, Vagero D, Koupilova I, Mohsen R, Berglund L, et al. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915–29. BMJ. 1998;317:241–245. doi: 10.1136/bmj.317.7153.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsen T, Eriksson JG, Tuomilehto J, Osmond C, Barker DJ. Growth in utero and during childhood among women who develop coronary heart disease: longitudinal study. BMJ. 1999;319:1403–1407. doi: 10.1136/bmj.319.7222.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ. 1999;318:427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eriksson JG, Forsen T, Tuomilehto J, Osmond C, Barker DJ. Early growth and coronary heart disease in later life: longitudinal study. BMJ. 2001;322:949–953. doi: 10.1136/bmj.322.7292.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barker DJ, Forsen T, Uutela A, Osmond C, Eriksson JG. Size at birth and resilience to effects of poor living conditions in adult life: longitudinal study. BMJ. 2001;323:1273–1276. doi: 10.1136/bmj.323.7324.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forsen T, Osmond C, Eriksson JG, Barker DJ. Growth of girls who later develop coronary heart disease. Heart. 2004;90:20–24. doi: 10.1136/heart.90.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frankel S, Elwood P, Sweetnam P, Yarnell J, Smith GD. Birthweight, body-mass index in middle age, and incident coronary heart disease. Lancet. 1996;348:1478–1480. doi: 10.1016/S0140-6736(96)03482-4. [DOI] [PubMed] [Google Scholar]
- 32.Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet. 1996;348:1269–1273. doi: 10.1016/s0140-6736(96)04547-3. [DOI] [PubMed] [Google Scholar]
- 33.Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, et al. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ. 1997;315:396–400. doi: 10.1136/bmj.315.7105.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yliharsila H, Kajantie E, Osmond C, Forsen T, Barker DJ, Eriksson JG. Body mass index during childhood and adult body composition in men and women aged 56–70 y. Am J Clin Nutr. 2008;87:1769–1775. doi: 10.1093/ajcn/87.6.1769. [DOI] [PubMed] [Google Scholar]
- 35.Yliharsila H, Kajantie E, Osmond C, Forsen T, Barker DJ, Eriksson JG. Birth size, adult body composition and muscle strength in later life. Int J Obes (Lond) 2007;31:1392–1399. doi: 10.1038/sj.ijo.0803612. [DOI] [PubMed] [Google Scholar]
- 36.Huxley RR, Shiell AW, Law CM. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens. 2000;18:815–831. doi: 10.1097/00004872-200018070-00002. [DOI] [PubMed] [Google Scholar]
- 37.Roseboom TJ, van der Meulen JH, Ravelli AC, van Montfrans GA, Osmond C, Barker DJ, et al. Blood pressure in adults after prenatal exposure to famine. J Hypertens. 1999;17:325–330. doi: 10.1097/00004872-199917030-00004. [DOI] [PubMed] [Google Scholar]
- 38.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–177. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 39.Roseboom TJ, van der Meulen JH, van Montfrans GA, Ravelli AC, Osmond C, Barker DJ, et al. Maternal nutrition during gestation and blood pressure in later life. J Hypertens. 2001;19:29–34. doi: 10.1097/00004872-200101000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. doi: 10.1038/ncomms6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King S, Laplante DP. The effects of prenatal maternal stress on children’s cognitive development: Project Ice Storm. Stress. 2005;8:35–45. doi: 10.1080/10253890500108391. [DOI] [PubMed] [Google Scholar]
- 42.Prentice AM, Whitehead RG, Roberts SB, Paul AA. Long-term energy balance in child-bearing Gambian women. The American journal of clinical nutrition. 1981;34:2790–2799. doi: 10.1093/ajcn/34.12.2790. [DOI] [PubMed] [Google Scholar]
- 43.Bagby SP. Maternal nutrition, low nephron number, and hypertension in later life: pathways of nutritional programming. J Nutr. 2007;137:1066–1072. doi: 10.1093/jn/137.4.1066. [DOI] [PubMed] [Google Scholar]
- 44.Khorram O, Momeni M, Desai M, Ross MG. Nutrient restriction in utero induces remodeling of the vascular extracellular matrix in rat offspring. Reproductive sciences. 2007;14:73–80. doi: 10.1177/1933719106298215. [DOI] [PubMed] [Google Scholar]
- 45.Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 46.Snoeck A, Remacle C, Reusens B, Hoet JJ. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate. 1990;57:107–118. doi: 10.1159/000243170. [DOI] [PubMed] [Google Scholar]
- 47.Styrud J, Eriksson UJ, Grill V, Swenne I. Experimental intrauterine growth retardation in the rat causes a reduction of pancreatic B-cell mass, which persists into adulthood. Biol Neonate. 2005;88:122–128. doi: 10.1159/000086136. [DOI] [PubMed] [Google Scholar]
- 48.Berney DM, Desai M, Palmer DJ, Greenwald S, Brown A, Hales CN, et al. The effects of maternal protein deprivation on the fetal rat pancreas: major structural changes and their recuperation. The Journal of pathology. 1997;183:109–115. doi: 10.1002/(SICI)1096-9896(199709)183:1<109::AID-PATH1091>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 49.Desai M, Crowther NJ, Ozanne SE, Lucas A, Hales CN. Adult glucose and lipid metabolism may be programmed during fetal life. Biochemical Society transactions. 1995;23:331–335. doi: 10.1042/bst0230331. [DOI] [PubMed] [Google Scholar]
- 50.Burns SP, Desai M, Cohen RD, Hales CN, Iles RA, Germain JP, et al. Gluconeogenesis, glucose handling, and structural changes in livers of the adult offspring of rats partially deprived of protein during pregnancy and lactation. J Clin Invest. 1997;100:1768–1774. doi: 10.1172/JCI119703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hales CN, Desai M, Ozanne SE, Crowther NJ. Fishing in the stream of diabetes: from measuring insulin to the control of fetal organogenesis. Biochemical Society transactions. 1996;24:341–350. doi: 10.1042/bst0240341. [DOI] [PubMed] [Google Scholar]
- 52.Desai M, Byrne CD, Zhang J, Petry CJ, Lucas A, Hales CN. Programming of hepatic insulin-sensitive enzymes in offspring of rat dams fed a protein-restricted diet. The American journal of physiology. 1997;272:G1083–1090. doi: 10.1152/ajpgi.1997.272.5.G1083. [DOI] [PubMed] [Google Scholar]
- 53.Zhu MJ, Ford SP, Means WJ, Hess BW, Nathanielsz PW, Du M. Maternal nutrient restriction affects properties of skeletal muscle in offspring. The Journal of physiology. 2006;575:241–250. doi: 10.1113/jphysiol.2006.112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yates DT, Macko AR, Nearing M, Chen X, Rhoads RP, Limesand SW. Developmental programming in response to intrauterine growth restriction impairs myoblast function and skeletal muscle metabolism. Journal of pregnancy. 2012;2012:631038. doi: 10.1155/2012/631038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan X, Zhu MJ, Dodson MV, Du M. Developmental programming of fetal skeletal muscle and adipose tissue development. Journal of genomics. 2013;1:29–38. doi: 10.7150/jgen.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gale CR, Martyn CN, Kellingray S, Eastell R, Cooper C. Intrauterine programming of adult body composition. The Journal of clinical endocrinology and metabolism. 2001;86:267–272. doi: 10.1210/jcem.86.1.7155. [DOI] [PubMed] [Google Scholar]
- 57.Cettour-Rose P, Samec S, Russell AP, Summermatter S, Mainieri D, Carrillo-Theander C, et al. Redistribution of glucose from skeletal muscle to adipose tissue during catch-up fat: a link between catch-up growth and later metabolic syndrome. Diabetes. 2005;54:751–756. doi: 10.2337/diabetes.54.3.751. [DOI] [PubMed] [Google Scholar]
- 58.Lecoutre S, Breton C. Maternal nutritional manipulations program adipose tissue dysfunction in offspring. Frontiers in physiology. 2015;6:158. doi: 10.3389/fphys.2015.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borengasser SJ, Zhong Y, Kang P, Lindsey F, Ronis MJ, Badger TM, et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology. 2013;154:4113–4125. doi: 10.1210/en.2012-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson LP, Al-Hasan Y. Impact of oxidative stress in fetal programming. Journal of pregnancy. 2012;2012:582748. doi: 10.1155/2012/582748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richter T, von Zglinicki T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Experimental gerontology. 2007;42:1039–1042. doi: 10.1016/j.exger.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 62.Kawanishi S, Oikawa S. Mechanism of telomere shortening by oxidative stress. Annals of the New York Academy of Sciences. 2004;1019:278–284. doi: 10.1196/annals.1297.047. [DOI] [PubMed] [Google Scholar]
- 63.Tarry-Adkins JL, Chen JH, Jones RH, Smith NH, Ozanne SE. Poor maternal nutrition leads to alterations in oxidative stress, antioxidant defense capacity, and markers of fibrosis in rat islets: potential underlying mechanisms for development of the diabetic phenotype in later life. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:2762–2771. doi: 10.1096/fj.10-156075. [DOI] [PubMed] [Google Scholar]
- 64.Simmons RA, Suponitsky-Kroyter I, Selak MA. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to beta-cell failure. J Biol Chem. 2005;280:28785–28791. doi: 10.1074/jbc.M505695200. [DOI] [PubMed] [Google Scholar]
- 65.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 66.Lecomte V, Youngson NA, Maloney CA, Morris MJ. Parental programming: how can we improve study design to discern the molecular mechanisms? Bioessays. 2013;35:787–793. doi: 10.1002/bies.201300051. [DOI] [PubMed] [Google Scholar]
- 67.Russo VEA, Riggs AD, Martienssen RA. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- 68.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 69.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends in genetics : TIG. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 70.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature reviews Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 71.Youngson NA, Morris MJ. What obesity research tells us about epigenetic mechanisms. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110337. doi: 10.1098/rstb.2011.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118:2316–2324. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351. doi: 10.1016/s0168-9525(02)02709-9. [DOI] [PubMed] [Google Scholar]
- 75.Cooper WN, Khulan B, Owens S, Elks CE, Seidel V, Prentice AM, et al. DNA methylation profiling at imprinted loci after periconceptional micronutrient supplementation in humans: results of a pilot randomized controlled trial. FASEB J. 2012;26:1782–1790. doi: 10.1096/fj.11-192708. [DOI] [PubMed] [Google Scholar]
- 76.Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–1073. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 78.Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746. doi: 10.1038/ncomms4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem. 2010;285:15111–15118. doi: 10.1074/jbc.M109.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quilter CR, Cooper WN, Cliffe KM, Skinner BM, Prentice PM, Nelson L, et al. Impact on offspring methylation patterns of maternal gestational diabetes mellitus and intrauterine growth restraint suggest common genes and pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014;28:4868–4879. doi: 10.1096/fj.14-255240. [DOI] [PubMed] [Google Scholar]
- 81.McGowan PO, Suderman M, Sasaki A, Huang TC, Hallett M, Meaney MJ, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One. 2011;6:e14739. doi: 10.1371/journal.pone.0014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suderman M, McGowan PO, Sasaki A, Huang TC, Hallett MT, Meaney MJ, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17266–17272. doi: 10.1073/pnas.1121260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 84.Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 85.Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 86.Saxena A, Carninci P. Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays. 2011;33:830–839. doi: 10.1002/bies.201100084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakagawa S, Kageyama Y. Nuclear lncRNAs as epigenetic regulators-beyond skepticism. Biochim Biophys Acta. 2014;1839:215–222. doi: 10.1016/j.bbagrm.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 88.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Casas-Agustench P, Iglesias-Gutierrez E, Davalos A. Mother’s nutritional miRNA legacy: Nutrition during pregnancy and its possible implications to develop cardiometabolic disease in later life. Pharmacological research. 2015;100:322–334. doi: 10.1016/j.phrs.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 90.Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes DE, Piekarz A, Ferland-McCollough D, et al. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab. 2014;3:325–333. doi: 10.1016/j.molmet.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41:161–176. doi: 10.1093/ije/dyr233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–411. doi: 10.1016/j.cmet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 94.Houde AA, Hivert MF, Bouchard L. Fetal epigenetic programming of adipokines. Adipocyte. 2013;2:41–46. doi: 10.4161/adip.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Houde AA, Legare C, Hould FS, Lebel S, Marceau P, Tchernof A, et al. Cross-tissue comparisons of leptin and adiponectin: DNA methylation profiles. Adipocyte. 2014;3:132–140. doi: 10.4161/adip.28308. [DOI] [PMC free article] [PubMed] [Google Scholar]