

Abstract
Drug discovery has produced many successful therapeutic agents; however, most of these drugs were developed without a deep understanding of the systems-wide mechanisms of action responsible for their indications. Gene-disease associations produced by molecular and genetic studies of complex diseases provide great opportunities for a system-level understanding of drug activity. In this study, we focused on acute myocardial infarction (MI) and conducted an integrative network analysis to illuminate drug actions. We integrated MI drugs, MI drug interactors, drug targets, and MI disease genes into the human interactome and showed that MI drug targets are significantly proximate to MI disease proteins. We then constructed a bipartite network of MI-related drug targets and MI disease proteins and derived 12 drug-target-disease (DTD) modules. We assessed the biological relevance of these modules and demonstrated the benefits of incorporating disease genes. The results indicate that DTD modules provide insights into the mechanisms of action of MI drugs and the cardiovascular (side) effects of non-MI drugs.
Graphical Abstract
Introduction
Although drug discovery and development have produced many effective therapeutic agents during the past several decades, most of these drugs were developed without a deep knowledge of the systems-wide molecular mechanisms of action responsible for their indications. This knowledge gap of drug activity limits our molecular-level understanding of their therapeutic effects and adverse side effects. Drugs exert their effects by modulating molecular pathways rather than affecting a single specific target in isolation. It is, therefore, of great importance to investigate how drugs achieve their therapeutic functions via underlying signaling pathways and network modules. Indeed, drug discovery is moving from the conventional “one-effect/one-cause/one-target” magic bullet-type paradigm, to a systems biology paradigm, which considers the effect(s) of a drug as the result of perturbations of molecular network interactions 1.
Many computational approaches have been developed for predicting drug-target interactions and drug-disease associations from chemical structure and genome features 2–6, but few studies focus on drugs used for specific diseases. One of the rare exceptions is the study conducted by Azuaje and colleagues 7, who assembled the myocardial infarction (MI) drug-target interactome network. An important resource overlooked by this study, however, is the compilation of MI disease genes, which could provide rich information about the mechanisms underlying the therapeutic effects of MI drugs. Early relevant studies focusing on cardiovascular drugs include the work of Ivanov and colleagues 8 who developed a computational approach for identifying drug-induced MI-related proteins by predicting drug-target interactions, and the work of Li et al. 9 who constructed networks of different layered interactions underlying the universes of cardiovascular drugs, targets, genes, and disorders to reveal comprehensive insights into cardiovascular pharmacology.
With advances in genotyping and phenotyping, many gene-disease associations have been produced over recent decades 10. Increasing evidence indicates that most human diseases cannot be attributed to a single gene but are a result of complex interactions among multiple genetic variants and environmental factors. Some databases have been developed to store gene-disease associations 11–13, which provide great opportunities for a better understanding of the molecular mechanisms of drug action responsible for their specific diseases. Our goal in this study is to examine the molecular basis for the association between drug targets and disease genes and understand how drugs act in the context of complex biological pathways. We focus on a highly prevalent disease, acute myocardial infarction (MI). Although a few select drugs for MI have been widely used, little is known about the underlying mechanisms of action at a systems-level 7. We propose that integrating MI disease genes, MI drugs, and drug targets into the comprehensive human interactome, a network of all ascertainable protein-protein interactions in a cell, can provide a better understanding of systems-level molecular activities of MI drugs. This approach may also be helpful for repositioning some non-MI drugs or discovering their cardiovascular adverse effects.
The scheme of this study is shown in Figure 1(A–B). We construct a disease proteins using the interactions between them, and derive drug-target-disease (DTD) modules. We assessed the biological relevance of these modules from different perspectives and demonstrate the benefits of incorporating disease genes in the analysis of a drug-target network. The results also indicate that the DTD modules are biologically significant and represent potential signaling pathways of drug action. This study shows that network-based integrative analysis of MI drugs, targets and MI disease genes can help facilitate an understanding of the systems-wide action of MI drugs and identify the molecular basis for the cardiovascular side effects of some non-cardiovascular drugs.
Figure 1. The closeness relationships between MI(-related) drug targets and MI disease proteins in the human interactome.

(A) MI-related drugs, drug targets, and MI disease proteins are mapped onto the human interactome. (B) A bipartite network of MI-related drug targets and MI disease proteins is constructed, and the dense associations between them are identified as drug-target-disease (DTD) modules. (C) The overlap of MI-related drug targets and MI disease proteins. (D) MI-related drug targets (MI drug target, inset) and MI disease proteins have significantly more interactions in the interactome than expected by chance. (E) There are significantly more pairs of MI-related drug targets (MI drug target, inset) and MI disease proteins with common neighbors than expected by chance. (F) The average shortest path length between MI-related drug targets (MI drug target, inset) and MI disease proteins is significantly smaller than that between two random gene sets.
Materials and Methods
Drug and target datasets
We obtained 38 drugs used for MI and 330 non-MI drugs that have interactions with MI drugs from a previous study 7. Drug-drug interactions are mediated by common targets, transforming enzymes, transporters, or common underlying pharmacokinetics or pharmacodynamics according to DrugBank 14. We denoted non-MI drugs that interact with MI drugs as MI drug interactors. Only those drugs (300 MI drug interactors and 30 MI drugs) that have target information in the database will be considered for future analyses. In total, there are 425 drug targets for all of these drugs. For convenience, we denoted the drug targets in this study as MI-related drug targets, as the drugs are either MI drugs or MI drug interactors. Among all of the drug targets, 67 (denoted MI drug targets) are targeted specifically by MI drugs. We further collected MI disease genes or gene products from Phenopedia in HuGE navigator 11. To obtain a reliable list of MI genes, we only considered those with at least two publications that support the association. 431 MI disease genes satisfy this criterion. We then mapped MI-related drug targets and MI disease gene products (proteins) onto the human interactome.
Compiling a human protein interactome
Biological processes reflecting drug actions involve different types of molecular interactions. We, therefore, used a consolidated human interactome combining physical interactions from different databases and sources, including protein-protein interactions, protein complexes, protein-DNA interactions, kinase-substrate interactions, metabolic interactions, and signaling pathways. This interactome has been used in a previous study 15 and also enhanced by incorporating the latest additional data sources (Supplementary File 1); it has 14,174 proteins with 170,303 interactions, after removing duplicate interactions and self-loops. We mapped drug targets and disease genes onto the interactome, and found that 361 MI-related drug targets (including 65 MI drug targets) and 398 MI disease proteins overlap with the proteins in the interactome. A total of 256 MI drug interactors and 30 MI drugs whose targets can be found in the interactome were included in the overall analysis.
Network analysis and implementation
In this study, most of the network analyses were performed using Python, with the assistance of a Python package, NetworkX 16. It bipartite network of MI-related drug targets and MI contains many built-in network analysis algorithms, such as shortest path algorithms, subgraph induction, random graph generators, and module detection, etc. We readily used these algorithms for examining the proximity between drug targets and MI disease proteins (Supplementary File 1). In addition, we used a null model to assess the significance of emerging properties. The null model keeps the human interactome unchanged and randomly selects 1,000 pairs of random protein sets of the same size as MI drug targets and MI disease proteins respectively (Supplementary File 1). The topological properties of random protein sets are then compared with those of real drug targets and disease proteins.
To find modules densely connecting MI-related drug targets and MI disease proteins, we constructed a bipartite network using the interactions between them, and used the Louvain method to maximize a modularity function Q 17, 18 and, thereby, identify drug-target-disease (DTD) modules (Supplementary File 1):
where m is the total number of edges; the Ai,j are the adjacency matrix elements; ki and kj are the degrees of node i and node j, respectively; ci and cj are the module indices of node i and node j, respectively; and δ, the delta function, is equal to 1 if ci = cj, and is otherwise equal to 0. Modularity function Q is defined as the fraction of the edges that fall within modules minus the expected fraction in a random network with the same node degree distribution. Since modules in a network are conceptually defined as groups of nodes that are more densely connected internally than with the rest of the network, modularity optimization is one of the most effective methods for module identification. Maximization of Q is known to be an NP-hard problem which means that there are no exact algorithms for finding global optimal solutions within acceptable time 19. The Louvain method is a fast greedy heuristic algorithm that attempts to optimize the modularity of a partition of the network approximately 18. The method first searches for small communities by optimizing modularity locally. It then merges nodes belonging to the same community and creates a new network whose nodes are the communities. These two steps are repeated iteratively until a maximum of modularity is attained 18.
Statistical tests and tools
All network visualization was performed with Cytoscape 20. GO-based functional similarity of pairs of MI-related drug targets and MI disease proteins was quantified by GS2 (GO-based similarity of gene sets) developed in a previous study 21. The daily snapshot of the GO tree and human gene annotations was downloaded from the GO web site (http://www.geneontology.org) 22. Unless otherwise specified, when we assess the significance of emergent properties of observations by comparing them with null models, all P-values are obtained by fitting the histograms to normal distributions (Supplementary File 1). All error bars in the figures are standard errors.
Results
MI drug targets are significantly proximate to MI disease proteins in the interactome
Most drugs exert their therapeutic effects through binding to one or more protein targets. Thus, we hypothesize that drug targets should not be very far from proteins associated with the indicated diseases in the interactome. For convenience, we refer to the targets of the drugs in this study as MI-related drug targets since the drugs are either MI drugs or MI drug interactors. In order to assess the proximity between MI-related drug targets and MI disease genes, we mapped them onto the consolidated human interactome and identified 361 MI-related drug targets (including 65 MI drug targets) and 398 MI disease proteins in the network. As shown in Figure 1 (C), 66 of MI-related drug targets and 23 of MI drug targets are also MI disease proteins. The intersecting sets of proteins are significantly larger than expected by chance (hypergeometric test, P<2.5×10−35 and P<8.4×10−20, respectively). This result indicates that MI-related drug targets overlap significantly with MI disease proteins.
In addition to this gene overlap analysis, we also assessed the proximity between MI drug targets and MI disease proteins at the network level. We identified 1,029 interactions between MI-related drug targets and MI disease proteins, which is significantly greater than the number of interactions between two random sets of the same size (P<1.0×10−16), as shown in Figure 1 (D). Previous studies have used the number of common interacting neighbors of two proteins to predict whether they have functional associations or not 23. Similarly, we next checked the number of pairs of MI-related drug targets and MI disease proteins that have common neighbors. As shown in Figure 1 (E), there are significantly more pairs of MI-related drug targets and MI disease proteins with common neighbors than protein pairs from two random sets (P<1.0×10−16). The conclusion holds as well if we focus on MI drug targets only (P=2.8×10−12). We also calculated the average shortest path length between MI-related drug targets and MI disease proteins in the interactome. As shown in Figure 1 (F), the average shortest path length between MI-related drug targets and MI disease proteins is 4.19, significantly smaller than that between two random sets of the same size (4.33±0.02, P =6.9×10−10). All of these results, therefore, support the conclusion that MI-related drug targets are significantly proximate to MI disease proteins in the human interactome than random expectation, which, in turn, provides evidence for the utility of incorporating MI disease proteins in a network-based analysis to provide insights into the mechanisms of action of MI drugs.
To avoid the potential concern that the targets of drugs for use in other diseases may also be close to MI disease proteins, we performed a control experiment in which the same number of drug targets was randomly selected from the pool of all drug targets 14 while excluding MI-related drug targets. We assessed the closeness relationship between control drug targets and MI disease proteins, in terms of overlap, the number of interactions, the number of protein pairs that have common neighbors, and the average shortest path length. The results, shown in Figure 2 (A–D), indicate that MI-related drug targets have significantly greater overlap with MI disease proteins and are closer to MI disease proteins than control drug targets, confirming that MI-related drug targets are truly proximate to MI disease proteins in the interactome. These results, after removing MI drug targets from MI-related drug targets, support the same conclusion (Supplementary Figure S1). Similarly, to avoid the potential concern that MI-related drug targets may also be close to proteins associated with other diseases, we performed another control experiment wherein we randomly selected the same number of disease proteins from the pool of all proteins involved in other diseases 15 while excluding MI disease proteins. The results, shown in Figure 3 (A–D), indicate that MI disease proteins have significantly greater overlap with MI-related drug targets and are closer to MI-related drug targets than control disease proteins in the human interactome. The comparative closeness analysis of control disease proteins and MI disease proteins with MI drug targets yields very similar results (Supplementary Figure S2).
Figure 2. The proximity between control drug targets and MI disease proteins.

(A) Compared to control drug targets, MI(-related) drug targets have larger overlap with MI disease proteins. (B) Compared to control drug targets, MI(-related) drug targets have more interactions with MI disease proteins. (C) Compared to control drug targets, there are more pairs of MI(-related) drug targets and MI disease proteins that have common neighbors in the interactome. (D) Compared to control drug targets, MI(-related) drug targets have a smaller average shortest path length with MI disease proteins.
Figure 3. The proximity between MI(-related) drug targets and control disease proteins.

(A) Compared to control disease proteins, MI disease proteins have significantly greater overlap with MI-related drug targets. (B) Compared to control disease proteins, MI disease proteins have more interactions with MI-related drug targets. (C) Compared to control disease proteins, there are more pairs of MI disease proteins and MI-related drug targets that have common neighbors in the interactome. (D) Compared to control disease genes, MI disease proteins have a smaller average shortest path length with MI-related drug targets in the interactome.
Bipartite network of drug targets and disease proteins
Based on the analyses performed above, we note that MI-related drug targets are, indeed, significantly proximate to MI disease proteins in the human interactome. Previous studies have shown that densely connected subgraphs in protein interaction networks indicate protein complexes and novel functional modules 24, 25. Therefore, the modules of MI-related drug targets and MI disease proteins may represent functional associations between them and define the complex biological pathways underlying the mechanisms of drug action. To understand the network-based mechanisms of action of MI drugs and the cardiovascular adverse effects of non-MI drugs, we constructed a network of MI-related drug targets and MI disease proteins by using the molecular interactions between them. The network comprises 244 MI-related drug targets, 246 MI disease proteins, and 1,029 interactions. As some MI-related drug targets are also MI disease proteins, we can duplicate them to represent the two roles (one as drug target and the other as disease protein) so that the network is mathematically bipartite.
We used the Louvain method to maximize the modularity function defined for characterizing the modular structure of networks 17, 18 and to identify modules of MI-related drug targets and disease proteins. A total of 12 modules comprising more than 5 proteins are detected in the bipartite network (Supplemental Figure S3). Note that the bipartite network does not contain the interactions between drug targets nor the interactions between disease proteins, except for those that are both drug targets and disease genes. Therefore, the derived modules truly represent the specific, direct associations between MI-related drug targets and MI disease proteins. After we derived the modules, we restored the interactions among drug targets, the interactions among disease proteins, and the drug-target interactions. These modules, called DTD (drugs, drug targets, and disease proteins) modules, reflect the mechanisms of drug action and underlying biological pathways of the corresponding drugs that target the modules.
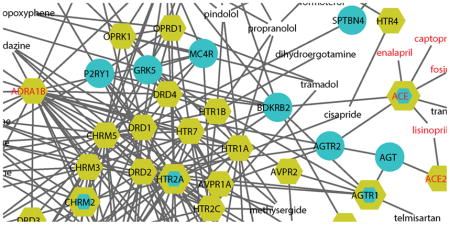
Figure 4 shows some of the DTD modules; a full list of modules is provided in Supplementary Figure S4. Although each DTD module only contains a few MI drugs and targets (owing to the limited number of MI drugs), many drug targets have dense connections with MI disease proteins. This finding may offer an explanation for why some non-MI drugs have adverse cardiovascular side effects. For example, disulfiram is a drug used for alcohol dependence. Previous studies have shown that when disulfiram is used at very high doses or in individuals with cardiovascular disease, severe reactions can occur, including myocardial infarction, arrhythmias, and congestive heart failure 26. In Module 1, the target of disulfiram is ALDH2, which is a drug target for myocardial protection from ischemia-reperfusion injury 27, providing a plausible mechanism by which to account for disulfiram’s cardiovascular side effects. Minocycline is a tetracycline antibiotic used to treat several different bacterial infections; it is located in Module 5 and has been associated with some cardiovascular side effects, including vasculitis and myocarditis, according to the Sider database 28. Imatinib is a drug used to treat certain types of leukemia and has severe cardiovascular side effects, such as congestive heart failure, tachycardia, pulmonary fibrosis, and thrombosis 28. In Module 11, the target of imatinib has interactions with many MI disease proteins, providing a plausible basis for its cardiovascular side effect profile.
Figure 4. The DTD modules.

The blue nodes represent MI disease proteins, and the yellow nodes denote MI-related drug targets. The nodes with both colors are both drug targets and MI disease proteins. The nodes with only labels (without node shapes) are drugs. MI drugs and MI drug targets are denoted in red.
We next assessed specific features of the bipartite network of MI-related drug targets and MI disease proteins. Drug targets and disease proteins in the bipartite network have similar degree distributions (Figure 5 (A)): most MI-related drug targets are connected to few MI disease proteins, whereas a few MI-related drug targets are connected to many MI disease proteins. The robustness of the DTD modules is vital since the modules are used as the basis for interpreting mechanisms underlying drug action and drug side effects in this study. We, therefore, assessed their robustness from different perspectives. To demonstrate that the constructed bipartite network is truly modular, we tested it against a set of shuffled bipartite networks (1,000) of the same size by randomly rewiring existing interactions between MI-related drug targets and MI disease proteins while maintaining the same number of interacting partners. The average modularity of the shuffled bipartite networks is 0.436±0.005 (Figure 5 (B)). None of the randomized networks achieved the same modularity observed from the bipartite network we constructed, confirming the significance of these modules (P<1.0×10−16). Although the human interactome we used in this study is very comprehensive, it may still be prone to false positives. To examine the robustness of our DTD modules in the presences of false positives, we randomly removed a certain percentage of nodes and edges (from 5% to 20%) from the human interactome to determine if the best partitions are similar to those obtained from the original network. The results of this analysis, shown in Figure 5 (C), demonstrate that the partitions obtained from the perturbed networks are very close to the original best partitions (the Normalized Mutual Information (NMI) measure 29 is over 0.7 even when we remove 20% of the nodes and edges). In addition, the modularity of the perturbed networks after removing a high percentage of nodes and edges remains as high as the original network, as shown in Figure 5 (D). All of these results indicate that the DTD modules derived from the best partition of the original network are robust and can serve as a reasonable basis upon which to interpret the potential mechanisms and signaling pathways of drug action.
Figure 5. The robustness of the DTD modules.

(A) The degree distributions of MI-related drug targets and MI disease proteins. (B) The constructed bipartite network is significantly modular compared to randomized networks with the same degree distributions (P<1.0×10−16). (C) The NMI value between the best partitions obtained from the original bipartite network and that obtained from the perturbed networks after removing a certain percentage of nodes and edges. (D) The modularity of the perturbed networks after removing a certain percentage of nodes and edges.
Biological significance of the DTD modules
In the DTD modules, MI-related drug targets and MI disease proteins are densely associated through the molecular interactions between them. We assessed the biological relevance of the DTD modules in several different ways. MI-related drug targets and MI disease proteins may participate in similar physiological and pathological processes as they cluster together in the interactome. We, therefore, evaluated the functional similarity of interacting pairs of MI-related drug targets and MI disease proteins that are located in the same modules. As a control for significance, we randomly selected an interaction set with the same number of interactions as the module and calculated its functional similarity. Figure 6 (A) shows that most of the identified DTD modules have higher functional similarity than randomized modules, suggesting that these modules are mechanistically rational and represent potential signaling pathways of drug action. The results based on all interacting pairs in modules are given in Supplementary Figure S5, which is similar to Figure 6 (A).
Figure 6. Biological relevance of the DTD modules.

(A). Functional similarity of MI-related drug targets and MI disease proteins in the DTD modules; *P<0.05. (B) Enrichment of drug pairs with similar side effects in each module. The red line represents P=0.05. (C) Enrichment of drug pairs with similar ATC codes in each module. The red line represents P=0.05.
Understanding adverse side effects of drugs is equally important as identifying the molecular mechanisms of drug action. Adverse side effects have been used to infer whether two drugs share a target protein 2. By contrast, the side-effect similarity of drugs could also be caused by the closeness of their target proteins in the interactome. Another hypothesis about the biological relevance of the DTD modules is that drugs targeting the same modules may have similar side effects. To test this hypothesis, we collected a set of drug pairs that have similar side effects from previous studies 30 and examined whether DTD modules are significantly enriched with drug pairs that have similar side effects. The results, shown in Figure 6 (B), indicate that compared with a random drug set, drugs that target the same modules are significantly enriched with drug pairs with similar side effects.
The Anatomical Therapeutic Chemical (ATC) Classification System is used to classify drugs into different groups according to the organ or system on which they act and/or their therapeutic and chemical characteristics. The DTD modules represent the potential underlying signaling pathways of drug action. We, thus, hypothesized that drugs targeting the same modules may have similar therapeutic effects. To this end, we retrieved ATC codes used to annotate the drugs from the DrugBank database and constructed two sets of drug pairs that have similar therapeutic effects based on the first-level and the second-level of ATC codes, respectively. The results indicate that compared with a random drug set, drugs in the same DTD modules are significantly enriched with drug pairs that have similar therapeutic effects (Figure 6 (C)). The similarity of therapeutic effects of drugs may provide insights into the mechanism of action of one drug from that of another which acts within the same molecular pathway in the interactome. Collectively, the observations in Figure 6 demonstrate the strong biological relevance of the DTD modules.
Cardiovascular adverse effects of unrelated drugs
Many drugs not used to treat cardiovascular diseases have adverse cardiovascular (side) effects 31. Since our DTD modules contain non-MI drugs that have interactions with MI drugs, we may obtain some insights into the cardiovascular (side) effects of these non-MI drugs. Among 300 non-MI drugs with target information, 224 were assigned to modules; these are denoted module drugs. Other non-MI drugs not located in the DTD modules are denoted non-module drugs. SIDER 2 (Side Effect Resource) contains information on marketed medicines and their recorded adverse reactions extracted from public documents and package inserts 28. Based on this database and the literature, 133 module drugs have cardiovascular side effects (at least two pieces of supportive evidence), which are provided in Supplemental File 2. Table 1 presents some typical examples of non-cardiovascular drugs that have cardiovascular side effects. For example, thioridazine is a typical antipsychotic drug that can cause an arrhythmia leading to sudden death. Thus, it has an FDA “black-box warning.” 31 Rosiglitazone is an antidiabetic drugs used for the treatment of type II diabetes that has been reported to increase the risk of cardiovascular complications, including myocardial infarction 7, 32. The derived DTD modules provide mechanistic insights into the cardiovascular side effects of these drugs.
Table 1.
Cardiovascular adverse effects of some non-cardiovascular drugs based on the Sider database and the literature.
| Drug name | Module ID | Cardiovascular side effects with references |
|---|---|---|
| Gemcitabine | 1 | Arrhythmia; atrial fibrillation; cardiac failure; myocardial infarction 28, 31 |
| Clozapine | 2, 3 | Tachycardia; Metabolic syndrome; myocardial infarction; ventricular fibrillation; venous thrombosis 28, 31 |
| Tizanidine | 2, 3 | Bradycardia; arrhythmia; myocardial infarction; coronary artery disease 31 |
| Escitalopram | 2, 3, 9 | Tachycardia; arrhythmia; cardiac failure; myocardial infarction; atrial fibrillation 31 |
| Vardenafil | 2, 6 | Coronary artery disease; myocardial infarction; tachycardia 28, 31 |
| Trazodone | 2, 3, 9 | Arrhythmia; cardiac arrest; ventricular tachycardia; myocardial infarction 31 |
| Celecoxib | 2, 7 | Risk of cardiovascular complications; cardiac failure; myocardial infarction; thrombosis 7, 31, 59 |
| Thioridazine | 3 | Torsades de Pointes; ventricular tachycardia 28, 31 |
| Aminophylline | 3, 6 | Atrial fibrillation; tachycardia; arrhythmia; cardiac flutter 28, 31 |
| Rosiglitazone | 5 | Adverse cardiac effects; cardiac failure 31, 32, 60 |
| Methylprednisolone | 5 | Atrial fibrillation; arrhythmia; cardiac arrest; cardiac failure; myocardial infarction 28, 31, 61 |
| Aprotinin | 9 | Arrhythmia; cardiac arrest; myocardial infarction; thrombosis 31 |
We also hypothesized that module drugs tend to have cardiovascular side effects compared to non-module drugs, as module drugs are functionally associated with MI disease proteins through their targets. We, therefore, also checked the side effects of non-module drugs as well, and found that only 26 of them have cardiovascular side effects. Table 2 provides the contingency table for the enrichment of non-MI drugs with cardiovascular effects. Chi-squared testing indicates that compared to non-module drugs, module drugs are significantly enriched with drugs that have cardiovascular side effects (P = 2.5×10−4). If we restrict the non-module drugs to those that have targets in the interactome, the result is still significant (Supplementary Table S1), indicating that incorporating MI disease proteins is helpful for understanding the cardiovascular side effects of module drugs.
Table 2.
Contingency table for the enrichment of non-MI module drugs with cardiovascular side effects, P = 2.5×10−4 (Chi-squared test).
| With cardiovascular side effects | No cardiovascular side effects | Total | |
|---|---|---|---|
| Module drugs | 133 | 91 | 224 |
| Non-module drugs | 26 | 50 | 76 |
| Total | 159 | 142 | 300 |
Similarly, we classified the drug targets into two groups as well. The drug targets assigned to DTD modules are module targets, and others are non-module targets. We hypothesized that module drug targets tend to be associated with cardiovascular function or cardiovascular diseases compared to non-module drug targets, as module drug targets are functionally associated with MI disease proteins. We, therefore, examined the overlap of module targets and non-module targets with the cardiovascular-associated proteins downloaded from the Cardiovascular Gene Annotation Initiative (http://www.ebi.ac.uk/GOA/CVI). Table 3 provides the contingency table for the enrichment of cardiovascular-associated proteins among module drug targets. Chi-squared testing indicates that compared to non-module drug targets, module drug targets are significantly enriched with cardiovascular-associated proteins (P = 1.7×10−4). Supplementary Table S2 shows similar results after we remove MI drug targets. Once again these results confirm the benefits of incorporating MI disease proteins for understanding drug action and the functionality of drug targets.
Table 3.
Contingency table test for the enrichment of cardiovascular-associated proteins in drug targets, P = 1.7×10−4 (Chi-squared test).
| Cardiovascular proteins | Not cardiovascular proteins | Total | |
|---|---|---|---|
| Module targets | 180 | 52 | 232 |
| Non-module targets | 75 | 54 | 129 |
| Total | 255 | 106 | 361 |
Pharmacological insights derived from network analysis
The DTD modules consist of the connections from MI drugs and MI drug interactors to MI disease proteins via drug targets in the human protein-protein interaction network. These modules represent the potential signaling pathways of drug action and provide insights into the mechanisms of action of MI drugs as well as the cardiovascular side effects and new therapeutic indications of non-MI drugs. A detailed review of DTD modules offers insights for potential repurposing of some drugs in the treatment of MI (Table 4). For example, valproic acid is a fatty acid with anticonvulsant properties used in the treatment of epilepsy. It is also a histone deacetylase inhibitor and is under investigation for treatment of HIV and various cancers14. The mechanisms of its therapeutic actions are not well understood. We predict that valproic acid may be repurposed for treatment of MI. In Module1, ABAT, the target of valproic acid, is densely connected to aldehyde dehydrogenase (ALDH) family proteins, which have a role in cardioprotection 33. Recent epidemiological studies indicate that valproic acid is associated with a reduced risk of myocardial infarction 34, 35, supporting the potential repositioning of valproic acid for MI. Minocycline is a tetracycline analog effective against several bacterial infections. One of its drug targets is MMP9 whose increased level in diabetes causes vascular remodelling, which contributes the cardiovascular complications of diabetes. Recent studies suggest that the combination of minocycline and aspirin prevents worsening of acute MI in diabetic rats by inhibiting MMP2 and MMP936. In addition, encouraging evidence exists for minocycline’s protective role in cardiovascular pathology and its activity against myocardial ischemia-reperfusion injury37. Ginseng has been used as one of the traditional Chinese medicines for treating a variety of disorders. North American ginseng (Panax quinquefolius) has been increasingly attracting interests in western societies for its therapeutic potential in cardiovascular diseases 38, 39. Although its pharmacological action is not very clear, ginseng has been found to target PTGS2, IL6, and AHR 14, 40. PTGS2 (COX-2) contains a polymorphism as an inherited protective factor against myocardial infarction and stroke 41. It is also the drug target of several MI drugs including naproxen, ibuprofen, diclofe, and aspirin. Thus, ginseng has potential for treating myocardial infarction as well via this common mechanistic target. In addition, in Module 7, the drug targets of acetaminophen are PTGS1 and PTGS2 which are also MI disease proteins. Several studies have shown that acetaminophen is functionally cardioprotective and can attenuate the damaging effects of peroxynitrite and hydrogen peroxide during ischemia-reperfusion through an underlying antioxidant (free-radical scavenging) mechanism42, 43. Collectively, the DTD modules offer pharmacological insights into mechanistic bases for the repurposing of select drugs. As we illustrate here, one can identify drugs not previously known to treat the disease, ascertain their targets and targeted pathways within the module, identify the molecular mechanisms related to the disease that involve the targets and targeted pathways, and explore the literature for any prior epidemiological or clinical case-based evidence supporting an association between the drug and the disease (MI). This network-based analysis, therefore, offers a mechanistic rationale for pursuing these drugs as potential treatments for MI.
Table 4.
Pharmacological insights for some drugs based on analysis of the DTD modules.
| Potential drug repurposing | ||||
|---|---|---|---|---|
| Drug names | Original use | Targets in the DTD modules | Mechanism of action | Supportive evidence |
| Valproic acid | Epilepsy | ABAT; ACADSB; HDAC9 | Fatty acid Histone deacetylase inhibitor | Sodium valproate exposure was associated with the risk of MI35. There is a consistent association between valproate treatment and a reduced risk of MI in patients with epilepsy34. |
| Minocycline | Antibacterial drug | ALOX5; CASP1, CASP3; CYCS; IL1B; MMP9 | A tetracycline analog | The combination of minocycline and aspirin prevent worsening of AMI in diabetic rats36. Minocycline has protective roles in cardiovascular pathology and the activity against myocardial ischemia-reperfusion injury 37. |
| Ginseng | Adaptogen | PTGS2; IL6; AHR | Unclear | Ginseng has a marked ability to reverse cardiac hypertrophy, myocardial remodeling, and heart failure38. Ginseng protects rodent hearts from acute myocardial ischemia-reperfusion injury 62. |
| Acetaminophen | Analgesic and antipyretic drug | PTGS2;PTGS1 | Inhibiting primarily COX-2 (PTGS2) vs. COX-1 (PTGS1) and possibly COX-3 (in CNS); Central effects via NMDA receptor inhibition | Acetaminophen shows positive cardioprotective effects In myocardial infarction and arrhythmia 42, 63. Acetaminophen is both functionally cardioprotective and antiarrhythmic against H2O2-induced oxidative injury 43; |
| Tetracycline | Antibacterial drug | PRNP | Interfering with protein synthesi | Tetracycline may be of use in suppressing the development of infarction caused by myocardial ischemia 64. |
| Cardiovascular side effects | ||||
| Disulfiram | Alcohol dependence | ALDH2 | Inhibiting aldehyde dehydrogenase | When disulfiram is used at very high doses, severe reactions can occur, including myocardial infarction, arrhythmias, and congestive heart failure 26 |
| Danazol | Endometriosis and breast disorders | AR; CCL2; ESR1; PGR | Inhibiting the pituitary output of gonadotropins | Danazol inhibits the activity of CCL2 and also enhances hemostasis, increasing the risk of MI with chronic use 46, 47. |
| Miconazole | Antifungal | KCNMA1; KCNMB1; NOS3 | Interacting with 14-α demethylase | Miconazole may induce rat cardiotoxicity via a ROS-mediated pathway, which is initiated by the inhibition of APE/Ref-1 expression 65. Miconazole has been implicated as the causes for QT interval prolongation 66. |
| Triazolam | Insomnia | GABRB1; GABRB2; GABRG2; GABRR1 | Binding bezodiazepine receptors | Triazolam overdose can cause life-threatening cardiotoxicity 52. |
| Rituximab | Lymphoma | C1QA ; C1QB; C1QC ; C1R; C1S; FCGR1A; FCGR2A; FCGR2B ; FCGR2C; FCGR3A ; MS4A1 | A murine/human chimeric monoclonal antibody directed against the CD20 antigen | Rituximab can induce polymorphic ventricular tachycardia 67. Two patients presented several cardiovascular risk factors the administration of rituximab 68. |
DTD modules can offer insights into the cardiovascular side effects of some drugs as well (Table 4). As mentioned earlier, disulfiram is a drug used in the treatment of chronic alcoholism. It acts by inhibiting aldehyde dehydrogenase and targets ALDH2 which has a role in protecting the heart and brain from ischemia-reperfusion injury 27. Therefore, we predict that disulfiram may have adverse cardiac side effects. Although the Sider database does not include the cardiovascular side effects of disulfiram, severe reactions including myocardial infarction, arrhythmias, and congestive heart failure, can occur when disulfiram is used at very high doses 26. Danazol is a drug used for the treatment of endometriosis and breast disorders. Danazol inhibits the expression of monocyte chemotactic protein-1 (MCP-1/CCL2) 44. As MCP-1/CCL2 is important in the development of collateral vessels following acute myocardial infarction 45, Danazol may increase the risk of MI and its consequences with chronic use 46, 47. Triazolam targets various gamma-aminobutyric acid (GABA) A receptors, such as GABRB1, GABRB2, GABRG2, and GABRR1, which are the main inhibitory neurotransmitters in the brain. These receptors also mediate cardiovascular regulation48, 49. In addition, GABAergic input affects blood vessels and participate in the control of vascular tone, blood pressure, and heart rate50, 51. A previous study has shown that the overdose of triazolam can cause life-threatening cardiotoxicity 52. Table 4 provides more examples of predicted cardiovascular adverse effects of non-cardiovascular drugs. The DTD modules provide the potential mechanisms of cardiotoxicity of these drugs based on their network links.
In addition, network integration analysis also provides other insights regarding the connectivity of MI drug targets. For example, in Module 1, known MI drug targets, PPP2CA, PPP2CB, and ANXA2, are all peripheral nodes in the network, while in Module 2, PRKCA is a highly connected node (hub). Of note, PPP2CA, PPP2CB, and PRKCA are all targeted by vitamin E, suggesting that drugs with multiple targets are connected to the network with varying degrees of centrality. Interestingly, highly useful MI drugs, such as the angiotensin converting enzyme (ACE) inhibitors, enalapril, captopril, lisinopril, fosinpril, and trandolapril, or the purinergic receptor P2Y, G-Protein coupled 12 (P2RY12) inhibitors, clopidogrel and prasugrel, target peripheral nodes, i.e., ACE and P2RY12 are weakly connected in Module 3. Yet, these weakly linked nodes have important effects on module function, and likely do so via their associations with other nodes of greater centrality, such as ACE’s link to the angiotensin receptor 1 (AGTR1) pathway and its downstream signaling pathways governing vascular tone and vascular cell proliferation, or the interaction of the P2RY12 receptor and the beta1-adrenergic receptor (ADRB1) governing cross-talk in signaling pathways modulating platelet function. A clear benefit of this integrated network analysis is derived from the inclusion of non-MI drugs that have interactions with MI drugs via common targets, transforming enzymes, transporters, or underlying pharmacokinetics or pharmacodynamics as indicated in DrugBank 14. This expanded MI drug set coupled with the physical protein-protein association links generated from the comprehensive human interactome provides a unique and rich network data set for exploratory analysis and hypothesis testing regarding drug action and benefit. The summary provided here only begins that exploratory process, which can evolve much more comprehensively in future studies.
Conclusions
In this study, we assessed the closeness relationships between MI-related drug targets and MI disease proteins and sought to decipher the molecular basis of drug action and drug side effects through drug-target-disease (DTD) modules. We assessed the biological relevance of the DTD modules in different ways. The results demonstrate the benefits of incorporating diseases genes for gaining insight into the mechanisms of drug action, the functionality of drug targets, and the pathobiology of diseases. There are other published studies examining the distance between drug targets and disease genes in the interactome 53, 54 or predicting adverse side effects of drugs 55. These earlier studies considered all diseases or major disease categories, and did not provide insights into the mechanisms of action of individual drugs. We took an additional step by examining whether the interactome-based localization of disease genes facilitates an understanding of drug actions and drug side effects through DTD modules. Through assessing the biological relevance of the DTD modules, we confirmed that the DTD modules identify potential signaling pathways of drug actions.
In this study, we used snapshot static networks to analyze the functional relationships of MI-related drugs, drug targets, and MI disease proteins. It would be interesting to explore how to incorporate drug-induced time-series gene expression data into the analyses once available. In addition, drugs may have potential off-target effects and different efficacies, and the molecular interactions in the network may have different strengths (weights). Considering such detailed information may complicate the topological analyses and require the construction of a dynamic model for optimal insights.
Choosing protein interactomes and module-finding techniques may have an impact on this study. There are several other protein interaction databases with higher coverage, e.g., STRING v1056. However, these databases contain a large number of predicted functional associations, rather experimentally ascertained physical interactions. Neverthess, we used the interactions from STRING v10 to assess the closeness relationships between MI-related drug targets and MI disease proteins to attempt to validate our findings with other interactomes. We also used another modularity optimization method57, 58 to detect modules and assess the robustness of DTD modules. As shown in Supplementary File 1, the results confirm the reliability of our conclusions and DTD modules.
Supplementary Material
Acknowledgments
This work was supported by NIH grants HL61795, HL108630 (MAPGen Consortium), and HL048743 (to J.L.). The authors thank A.-L. Barabasi’s laboratory at Northeastern University for providing access to the consolidated human interactome data they compiled, and Ms. Stephanie Tribuna for excellent editorial assistance.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Csermely P, Korcsmaros T, Kiss HJ, London G, Nussinov R. Pharmacol Ther. 2013;138:333–408. doi: 10.1016/j.pharmthera.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- 3.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang YF, Yeh HY, Soo VW. BMC Med Genomics. 2013;6(Suppl 3):S4. doi: 10.1186/1755-8794-6-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng F, Liu C, Jiang J, Lu W, Li W, Liu G, Zhou W, Huang J, Tang Y. PLoS Comput Biol. 2012;8:e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamanishi Y, Kotera M, Kanehisa M, Goto S. Bioinformatics. 2010;26:i246–254. doi: 10.1093/bioinformatics/btq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azuaje FJ, Zhang L, Devaux Y, Wagner DR. Sci Rep. 2011;1:52. doi: 10.1038/srep00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov SM, Lagunin AA, Pogodin PV, Filimonov DA, Poroikov VV. Chem Res Toxicol. 2014;27:1263–1281. doi: 10.1021/tx500147d. [DOI] [PubMed] [Google Scholar]
- 9.Li P, Fu Y, Ru J, Huang C, Du J, Zheng C, Chen X, Lu A, Yang L, Wang Y. BMC Syst Biol. 2014;8:141. doi: 10.1186/s12918-014-0141-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.T. W. T. C. C. Consortium. Nature. 2007;447:661–678. [Google Scholar]
- 11.Yu W, Clyne M, Khoury MJ, Gwinn M. Bioinformatics. 2010;26:145–146. doi: 10.1093/bioinformatics/btp618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos EM, Hoffman D, Junkins HA, Maglott D, Phan L, Sherry ST, Feolo M, Hindorff LA. Eur J Hum Genet. 2014;22:144–147. doi: 10.1038/ejhg.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Nucleic Acids Res. 2005;33:D514–517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. Nucleic Acids Res. 2014;42:D1091–1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabasi AL. Science. 2015;347:1257601. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagberg AA, Schult DA, Swart PJ. presented in part at the Proceedings of the 7th Python in Science Conference (SciPy2008); Pasadena, CA USA. August 2008; 2008. [Google Scholar]
- 17.Newman ME. Proc Natl Acad Sci U S A. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Journal of Statistical Mechanics: Theory and Experiment. 2008;10:P10008. [Google Scholar]
- 19.Brandes U, Delling D, Gaertler M, Gorke R, Hoefer M, Nikoloski Z. IEEE Transactions on Knowledge and Data Engineering. 2008;20:172–188. [Google Scholar]
- 20.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruths T, Ruths D, Nakhleh L. Bioinformatics. 2009;25:1178–1184. doi: 10.1093/bioinformatics/btp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samanta MP, Liang S. Proc Natl Acad Sci U S A. 2003;100:12579–12583. doi: 10.1073/pnas.2132527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spirin V, Mirny LA. Proc Natl Acad Sci U S A. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitra K, Carvunis AR, Ramesh SK, Ideker T. Nat Rev Genet. 2013;14:719–732. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huffman JC, Stern TA. Prim Care Companion J Clin Psychiatry. 2003;5:41–44. doi: 10.4088/pcc.v05n0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo XJ, Liu B, Ma QL, Peng J. Curr Drug Targets. 2014;15:948–955. [PubMed] [Google Scholar]
- 28.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. Mol Syst Biol. 2010;6:343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harenberg S, Bello G, Gjeltema L, Ranshous S, Harlalka J, Seay R, Padmanabhan K, Samatova N. Wiley Interdisciplinary Reviews: Computational Statistics. 2014;6:426–439. [Google Scholar]
- 30.Brouwers L, Iskar M, Zeller G, van Noort V, Bork P. PLoS One. 2011;6:e22187. doi: 10.1371/journal.pone.0022187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raj SR, Stein CM, Saavedra PJ, Roden DM. Circulation. 2009;120:1123–1132. doi: 10.1161/CIRCULATIONAHA.107.728576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C, Kelman JA. JAMA. 2010;304:411–418. doi: 10.1001/jama.2010.920. [DOI] [PubMed] [Google Scholar]
- 33.Chen CH, Sun L, Mochly-Rosen D. Cardiovasc Res. 2010;88:51–57. doi: 10.1093/cvr/cvq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olesen JB, Hansen PR, Abildstrom SZ, Andersson C, Weeke P, Schmiegelow M, Erdal J, Torp-Pedersen C, Gislason GH. Pharmacoepidemiol Drug Saf. 2011;20:146–153. doi: 10.1002/pds.2073. [DOI] [PubMed] [Google Scholar]
- 35.Dregan A, Charlton J, Wolfe CD, Gulliford MC, Markus HS. Pharmacoepidemiol Drug Saf. 2014;23:759–767. doi: 10.1002/pds.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhatt LK, Veeranjaneyulu A. Arch Med Res. 2014;45:203–209. doi: 10.1016/j.arcmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 37.Thind GS, Agrawal PR, Hirsh B, Saravolatz L, Chen-Scarabelli C, Narula J, Scarabelli TM. Future Cardiol. 2015;11:61–76. doi: 10.2217/fca.14.76. [DOI] [PubMed] [Google Scholar]
- 38.Moey M, Gan XT, Huang CX, Rajapurohitam V, Martinez-Abundis E, Lui EM, Karmazyn M. Circ Heart Fail. 2012;5:504–514. doi: 10.1161/CIRCHEARTFAILURE.112.967489. [DOI] [PubMed] [Google Scholar]
- 39.Karmazyn M, Moey M, Gan XT. Drugs. 2011;71:1989–2008. doi: 10.2165/11594300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Kim SJ, Jeong HJ, Yi BJ, Kang TH, An NH, Lee EH, Yang DC, Kim HM, Hong SH, Um JY. Am J Chin Med. 2007;35:329–339. doi: 10.1142/S0192415X07004850. [DOI] [PubMed] [Google Scholar]
- 41.Cipollone F, Toniato E, Martinotti S, Fazia M, Iezzi A, Cuccurullo C, Pini B, Ursi S, Vitullo G, Averna M, Arca M, Montali A, Campagna F, Ucchino S, Spigonardo F, Taddei S, Virdis A, Ciabattoni G, Notarbartolo A, Cuccurullo F, Mezzetti A. JAMA. 2004;291:2221–2228. doi: 10.1001/jama.291.18.2221. [DOI] [PubMed] [Google Scholar]
- 42.Merrill GF. Am J Physiol Heart Circ Physiol. 2002;282:H1341–1349. doi: 10.1152/ajpheart.00716.2001. [DOI] [PubMed] [Google Scholar]
- 43.Jaques-Robinson KM, Golfetti R, Baliga SS, Hadzimichalis NM, Merrill GF. Exp Biol Med (Maywood) 2008;233:1315–1322. doi: 10.3181/0802-RM-68. [DOI] [PubMed] [Google Scholar]
- 44.Jolicoeur C, Lemay A, Akoum A. Am J Reprod Immunol. 2001;45:86–93. doi: 10.1111/j.8755-8920.2001.450204.x. [DOI] [PubMed] [Google Scholar]
- 45.Xia Y, Frangogiannis NG. Inflamm Allergy Drug Targets. 2007;6:101–107. doi: 10.2174/187152807780832265. [DOI] [PubMed] [Google Scholar]
- 46.Boos CJ, Dawes M, Jones R, Farrell T. J Obstet Gynaecol. 2003;23:327–328. doi: 10.1080/01443610310000106064. [DOI] [PubMed] [Google Scholar]
- 47.Cicardi M, Castelli R, Zingale LC, Agostoni A. J Allergy Clin Immunol. 1997;99:194–196. doi: 10.1016/s0091-6749(97)70095-2. [DOI] [PubMed] [Google Scholar]
- 48.Matsuyama S, Saito N, Taniyama K, Tanaka C. Am J Physiol. 1991;261:H1437–1442. doi: 10.1152/ajpheart.1991.261.5.H1437. [DOI] [PubMed] [Google Scholar]
- 49.Matsumoto RR. Brain Res Brain Res Rev. 1989;14:203–225. doi: 10.1016/0165-0173(89)90001-5. [DOI] [PubMed] [Google Scholar]
- 50.DiMicco JA, Gale K, Hamilton B, Gillis RA. Science. 1979;204:1106–1109. doi: 10.1126/science.451556. [DOI] [PubMed] [Google Scholar]
- 51.Zhang W, Mifflin S. Hypertension. 2010;55:201–206. doi: 10.1161/HYPERTENSIONAHA.109.146407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamagiwa T, Amino M, Morita S, Yamamoto R, Saito T, Inokuchi S. Clin Toxicol (Phila) 2010;48:149–152. doi: 10.3109/15563650903524126. [DOI] [PubMed] [Google Scholar]
- 53.Sun J, Zhu K, Zheng W, Xu H. BMC Bioinformatics. 2015;16(Suppl 5):S1. doi: 10.1186/1471-2105-16-S5-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 55.Huang LC, Wu X, Chen JY. BMC Genomics. 2011;12(Suppl 5):S11. doi: 10.1186/1471-2164-12-S5-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guimera R, Nunes Amaral LA. Nature. 2005;433:895–900. doi: 10.1038/nature03288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Zhang S, Wang RS, Zhang XS, Chen L. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;77:036109. doi: 10.1103/PhysRevE.77.036109. [DOI] [PubMed] [Google Scholar]
- 59.Gislason GH, Rasmussen JN, Abildstrom SZ, Schramm TK, Hansen ML, Fosbol EL, Sorensen R, Folke F, Buch P, Gadsboll N, Rasmussen S, Poulsen HE, Kober L, Madsen M, Torp-Pedersen C. Arch Intern Med. 2009;169:141–149. doi: 10.1001/archinternmed.2008.525. [DOI] [PubMed] [Google Scholar]
- 60.Azuaje FJ, Devaux Y, Wagner DR. Clin Transl Sci. 2012;5:111. doi: 10.1111/j.1752-8062.2011.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Hooft CS, Heeringa J, Brusselle GG, Hofman A, Witteman JC, Kingma JH, Sturkenboom MC, Stricker BH. Arch Intern Med. 2006;166:1016–1020. doi: 10.1001/archinte.166.9.1016. [DOI] [PubMed] [Google Scholar]
- 62.Zhou H, Hou SZ, Luo P, Zeng B, Wang JR, Wong YF, Jiang ZH, Liu L. J Ethnopharmacol. 2011;135:287–298. doi: 10.1016/j.jep.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 63.Merrill GF, Rork TH, Spiler NM, Golfetti R. Am J Physiol Heart Circ Physiol. 2004;287:H1913–1920. doi: 10.1152/ajpheart.00565.2004. [DOI] [PubMed] [Google Scholar]
- 64.Kagawa N, Senbonmatsu TA, Satoh K, Ichihara K, Yamagata N, Hatano O, Saito T, Nguyen VQ, Waterman MR, Price E, Jr, Atkinson JB, Inagami T. Front Biosci. 2005;10:608–619. doi: 10.2741/1557. [DOI] [PubMed] [Google Scholar]
- 65.Won KJ, Lin HY, Jung S, Cho SM, Shin HC, Bae YM, Lee SH, Kim HJ, Jeon BH, Kim B. Toxicol Sci. 2012;126:298–305. doi: 10.1093/toxsci/kfr347. [DOI] [PubMed] [Google Scholar]
- 66.Simko J, Csilek A, Karaszi J, Lorincz I. Infection. 2008;36:194–206. doi: 10.1007/s15010-007-7211-8. [DOI] [PubMed] [Google Scholar]
- 67.Poterucha JT, Westberg M, Nerheim P, Lovell JP. Tex Heart Inst J. 2010;37:218–220. [PMC free article] [PubMed] [Google Scholar]
- 68.Passalia C, Minetto P, Arboscello E, Balleari E, Bellodi A, Del Corso L, Molinari E, Ponassi I, Oneto C, Sicbaldi V, Ghio R. Tumori. 2013;99:288e–292e. doi: 10.1700/1390.15471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.