

Abstract
Background
Bevacizumab (BV) is broadly used to treat a number of cancers; however, BV resistance mechanisms and strategies to overcome this resistance are yet to be determined.
Methods
We established xenograft mice models harboring Kirsten rat sarcoma oncogene homolog (KRAS) mutations based on the A549 cell line, and tested the responses of xenograft tumors to a series of drugs in ex vivo and in vivo experiments. Changes in transcriptive level were analyzed by ribonucleic acid (RNA) sequencing and the expressions of connexins were determined by immunohistochemistry staining.
Results
A549 cell mutation type (KRAS G12S) was confirmed by sequencing. After treating the xenograft tumors with BV, the median interval time from BV administration to tumor volume more than 2.5‐fold of the original was 37 days, compared with 21 days in the control (P = 0.025). A549 cells showed resistantance to selumitinib (MEK inhibitor) but were sensitive to selumitinib plus BEZ235 (phosphoinositide 3‐kinase/mammalian target of rapamycin dual inhibitor). However, selumitinib could effectively reverse the resistance to BV in in vivo experiments. RNA sequencing showed that mouse genes, but not human genes, activated the mitogen‐activated protein kinase signaling pathway, accompanied by activation of the Wnt and Hedgehog pathways. Connexin43 (S261) was phosphorylated before and during BV treatment, and subsequently transitioned to negative phosphorylated‐connexin 43‐S261 after resistance to BV.
Conclusion
Combining an MEK inhibitor with BV was a potential strategy to reverse initial BV resistance. Phosphorylated‐connexin 43 might be associated with the response to BV.
Keywords: Bevacizumab, connexin, MEK inhibitor, non‐small‐cell lung cancer, resistance
Introduction
Angiogenesis contributes to tumorous pathologies and plays a key role in tumor growth, survival, invasion, and metastatic spread.1 Tumoral neovascularization is essential to provide oxygen and other nutrients when tumor size reaches beyond 1–2 mm3, meaning that antiangiogenesis may be a potential and promising therapy for cancer treatment.2 Blocking the dynamic balance between pro‐angiogenic and anti‐angiogenic factors is a critical pilot strategy in inhibiting neovascularization. Vascular endothelial growth factor (VEGF), especially VEGF‐A, is a predominant pro‐angiogenic factor. The activation of the VEGF‐VEGF receptor (VEGFR) pathway stimulates the development of vascular endothelial cells.3 The effectiveness of bevacizumab (BV), an antibody to VEGF‐A specific to angiogenesis, has been proven. Whether combined with chemotherapy or administered alone as maintenance therapy, BV has shown meaningful anti‐tumor activity, with prolonged progression‐free survival (PFS) and overall survival (OS).4, 5, 6 Consequently, it is broadly administered for numerous cancers, such as non‐small‐cell lung cancer (NSCLC), colorectal and breast cancers, and renal cell carcinoma. However, after a period of time benefiting from BV therapy, almost all patients experienced progressive disease (PD), and some even present primary resistance to BV. Despite comprehensive investigation, a poor understanding of the reasons for BV resistance remains.
The empirical criteria for defining a clinical subtype of cancer are gradually transitioning from histopathology to genetic variations in driver genes. Epidermal growth factor receptor (EGFR) and Kirsten rat sarcoma oncogene homolog (KRAS) mutations are the main driver gene variations of lung adenocarcinoma in Asian and Caucasians, respectively. Targeting EGFR mutations has brought significant survival benefits to lung cancer patients with these mutations.7, 8, 9, 10, 11 However, to date, no molecular drug targeting KRAS mutations is available in conventional clinical practice. Chemotherapy combined with and followed by BV remains one of the major strategies for treating lung adenocarcinoma patients harboring KRAS mutations. As a result of inevitable PD after BV treatment, emphasis needs to be placed on how to reverse resistance to BV beyond changing to second‐line therapy. VEGF‐A can be secreted by both tumor and stromal cells. With the exception of the VEGFR‐VEGFR pathway, activation of alternative signaling pathways, such as EGFR and mitogen‐activated protein kinase (MAPK), can promote the release of VEGF‐A; the addition of MAPK inhibitors could reverse the resistance to BV.12, 13, 14 However, as a combination of MAPK inhibitors after resistance to BV has not been attempted, the internal mechanism remains unclear.
Apart from blocking the development of new vessels, an emerging concept of BV delivery is the “normalization” of tumor vasculature.15, 16 Normalized tumor vessels may increase the efficacy of chemotherapy. The circulating levels of short VEGF‐A isoforms and the expressions of neuropilin‐1 and VEGFR‐1 in tumor or plasma were candidate markers predicting the response to BV; however, they failed to imply the renormalization of tumor vasculature. Connexins play an important role in the connection of vascular endothelial cells, and the de‐phosphorylation of connexins might influence the “normalization” of tumor vasculature.17 Therefore, detecting the phosphorylation status of connexins might be associated with the response to BV.
Herein, we utilized A549 cells, a stable cell line harboring a KRAS mutation, to establish xenografts and found that an MAPK/extracellular‐signal‐regulated kinase (ERK) kinase (MEK) inhibitor (selumetinib) could reverse the resistance to BV in in vivo experiments. The mechanism might be the development of stromal cells; most likely vascular endothelial progenitor cells were inhibited by selumetinib. Connexin (Cx)43 might be related with and predictive of resistance to BV.
Methods
Cell line and chemicals
Human NSCLC cell line A549 was provided by the National Institute of Biologic Sciences (Beijing, China). BV (anti‐VEGF antibody), AZD6244 (MEK inhibitor), and BEZ235 (phosphoinositide 3‐kinase [PI3K]/mammalian target of rapamycin [mTOR] dual inhibitor) were purchased from Selleck Chemicals, LLC (Houston, Texas, USA).
Detection of Kirsten rat sarcoma oncogene homolog mutation in A549 cells
Genomic DNA of A549 cells was extracted according to the manual (Axygen AxyPrep‐96 Blood Genomic DNA Kit, Union City, CA, USA). The exon 1 of KRAS was amplified by polymerase chain reaction (forward primer: 5′‐GGTACTGGTGGAGTATTTGATAG‐3′; reverse primer: 5′‐TGGTCCTGCACCAGTAATATG‐3′) and was then sequenced using the Sanger method to check the KRAS mutation status in A549 cells.
Cell viability analysis
Cell viability tests were conducted using a cell counting kit‐8 (CCK8) (Dojindo, Mashikimachi, kamimashiki gun Kumamoto, Japan). Briefly, cells were seeded in sextuplicate in 96‐well plates containing 100 μl media at a density of 3 × 103 cells/well for 24 hours, and then cultured with increased concentrations of the indicated drugs for an additional 72 hours. Then, 10 μl of water soluble tetrazolium salt (WST‐8) was added to each well and incubated for three hours. The absorbance was measured via scanning with a microplate reader at 450 nm. Relative viability was calculated as follows: (%/control) = (A450 [treated]‐ A450 [blank])/(A450 [control]‐ A450 [blank]).
Western‐blot analysis
The expression of protein in cells was evaluated by Western blotting. Whole cell lysates (WCL) were prepared by extraction in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA), followed by protein quantification using a bicinchoninic acid assay (Applygen, Beijing, China) and lysis in Laemmli sample buffer. A total of 30 μg of the protein sample was run on a 10% Tris‐glycine gel and transferred to nitrocellulose. Primary antibodies were added and incubated overnight at 4°C, and secondary antibodies were conjugated to horseradish peroxidase for two hours at room temperature. Blots were developed by enhanced chemiluminescence and photographed using Fujifilm Dark Box II and Image Reader LAS‐1000 Plus software (Minato‐ku, Tokyo, Japan). Primary antibodies included protein kinase B (AKT), ERK1/2, beta‐actin (antibodies at 1:2000 dilutions), and phosphorylated (p)AKT, pERK1/2 (antibodies at 1:1000 dilutions; Cell Signaling Technology). Peroxidase labeled anti‐rabbit or anti‐mouse secondary antibodies were used (Cell Signaling Technology).
Subcutaneous in vivo experiments
To generate tumor xenografts, A549 cells (3 × 106) in 100 ul phosphate buffered saline were injected into the subcutaneous flanks of six‐week‐old female BALB/c nude mice. Body weight and tumor volume were recorded twice weekly. Tumor volumes were calculated as π/6 × a2 × b, where a was the smaller measurement of the tumor and b was the larger one, expressed in cubic millimeters. When the tumor volumes reached an average of approximately 250–300 mm3, tumor bearing mice were randomly assigned to one of the following treatment groups: (i) control – oral (p.o.) administration of vehicle daily; (ii) BV – 10 mg/kg intraperitoneal injection (i.p.) twice weekly. When tumor volumes reached three‐fold the initial volumes, the mice were randomly assigned to one of the following treatment groups: (i) control – p.o. administration of vehicle daily; (ii) BV + AZD6244 – 50 mg/kg, p.o. daily; (iii) BV + AZD6244 + BEZ235 – 45 mg/kg, p.o. daily (n = 5 per group). Animals were sacrificed as a result of the tumor burden. Log‐rank tests were performed to compare survival curves between different treatment groups using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, USA). All animal studies reported were approved by the National Institute of Biologic Sciences' animal care committee (Beijing, China), and abided by the “Principles of Laboratory Animal Care” (http://grants.nih.gov/grants/guide/notice‐files/not96‐208.html).
Ribonucleic acid (RNA) sequencing analysis
Total ribonucleic acid (RNA) was extracted from A549 xenograft tumor tissues before and after BV resistance using TRIzol (Life Technologies, Waltham, MA USA), according to the manufacturer's protocol. Messenger RNA was purified for RNA‐Seq library construction and whole transcriptome analysis. The homo genome sequences and annotated gene models were downloaded from the University of California Santa Cruz Genome Bioinformatics website (http://genome.ucsc.edu/). TopHat v1.4.1 was used to align the raw reads to genome sequences. Cufflinks (v2.0.2 http://cufflinks.cbcb.umd.edu/) was used to assemble transcripts and to calculate transcript abundances. Differences in RNA transcript levels between cell types were identified using the Cuffdiff criterion. The criterion for gene detection was the presence of more than three fragments per kilobase of transcript per million mapped reads (FPKM) in the total exons of a gene.
Immunohistochemistry staining of connexins
The expressions of Cx 23, 30, and 43, Cx43‐S261, and S368 were analyzed in A549 xenograft tumor tissue specimens using immunohistochemistry (IHC). Briefly, dried 5‐micron slides with formalin‐fixed, paraffin embedded tissue were prepared. Combined sodium citrate (pH 6.0) and incubation in a pressure cooker (3 minutes, 125°C) were used for antigen retrieval. Slides were then incubated overnight at 4°C with primary monoclonal anti‐connexin antibodies, including anti‐Cx26 (ab65969), anti‐Cx43 (ab11370), anti‐Cx43 (phosphorylated [phos]‐S261) (ab62252; Abcam Co., Cambridge, UK), anti‐Cx30 (sc‐84801), and anti‐Cx43 (phos‐368) (sc‐101660; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at a dilution of 1:100. A two‐step polymer‐horseradish peroxidase method was used for detection (Dako, Carpinteria, CA, USA). No staining was observed for negative controls, which included incubation of lung tissue with a non‐immune primary antibody.
Positive IHC immunoreactivity was defined if more than 10% of cancer cells were stained. IHC staining was evaluated independently by different investigators and a pathologist.
Statistical analysis
Median PFS was calculated using the Kaplan–Meier method, and comparisons between groups were made using log‐rank tests. For comparison of continuous variables between two groups, two‐tailed Student's t‐tests and Mann–Whitney Wilcoxon tests were used. All statistical tests were two‐tailed, with significance defined as P less than 0.05. All analyses were performed using SPSS, version 17.0 (SSPS, Chicago, IL, USA).
Results
A549 xenograft tumors exhibited moderate resistance to bevacizumab (BV)
To investigate the response of KRAS mutant NSCLC cells to BV, we initially used A549, an established lung adenocarcinoma cell line harboring KRAS G12S mutation (Fig 1a), to perform the in vitro and in vivo experiments. First, we tested the response of A549 cells in vitro to BV and illustrated that no significant improvement of response was observed using BV compared with the control (Fig 1b). We then injected female BALB/c nude mice with A549 cells and established the A549 xenografts. Approximately three weeks after A549 cell injection, A549 xenografts with a mean volume of about 300 mm3 were randomized to receive either the control or BV. Animals were treated until they were euthanized as a result of the tumor burden. Tumors were considered to be resistant when they increased 2.5‐fold in volume (i.e. tumor progression) compared with the pretreatment tumor size, and PFS was measured as the time from initiation of treatment until tumor progression. The median PFS after BV was 37 days (95% confidence interval [CI]: not applicable [NA]), compared with the control (21 days, 95% CI: 18–29 days; P = 0.025) (Fig 1c). No tumor shrinkage was observed and the tumors presented a grossly slow progressive pattern after BV treatment (Fig 1d).
Figure 1.
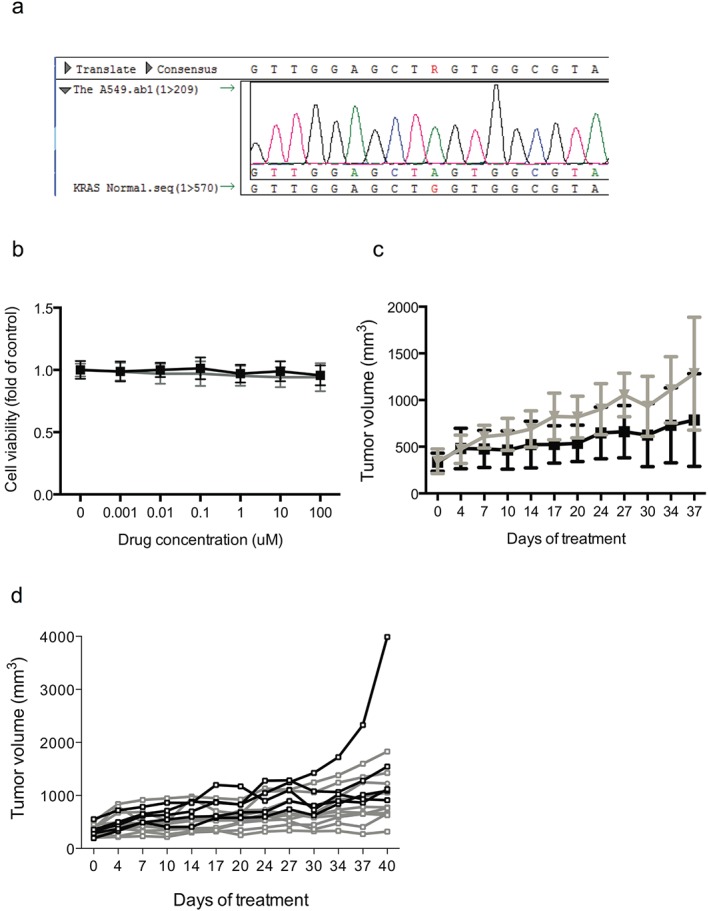
A549 xenograft tumors exhibited moderate resistance to bevacizumab (BV). (a) DNA sequence showed that the A549 cell carried Kirsten rat sarcoma oncogene homolog (KRAS) G12S mutation. (b) A549 cells showed resistance to BV in vitro compared with the control. (c) BV was effective on A549 xenografts compared with the control, but with slow progression. (d) No tumor shrinkage of individual tested xenografts was observed and all tumors showed a slow progressive pattern after BV treatment.
Inhibitors to MEK can reverse the resistance of A549 xenograft to BV
Considering that MEK is the downstream kinase of KRAS, we selected an MEK inhibitor, selumitinib, as a potential agent to reverse BV resistance. We first tested the response of A549 cells to selumitinib in an in vitro cell variability experiment. A549 cells showed resistantance to selumitinib with an inhibitory concentration (IC)50 value of 198 μM (Fig 2a). Western‐blot tests showed that AKT was phosphorylated after selumitinib treatment in A549 cells (Fig 2b). Therefore, we combined the PI3K/mTOR dual inhibitor, BEZ235, with selumitinib to treat A549 cells and observed that A549 cells were significantly inhibited compared with the control and selumitinib (Fig 2c). Western blot tests showed that both pAKT and pERK were blocked after the treatment of selumitinib plus BEZ235 in A549 cells (Fig 2b).
Figure 2.
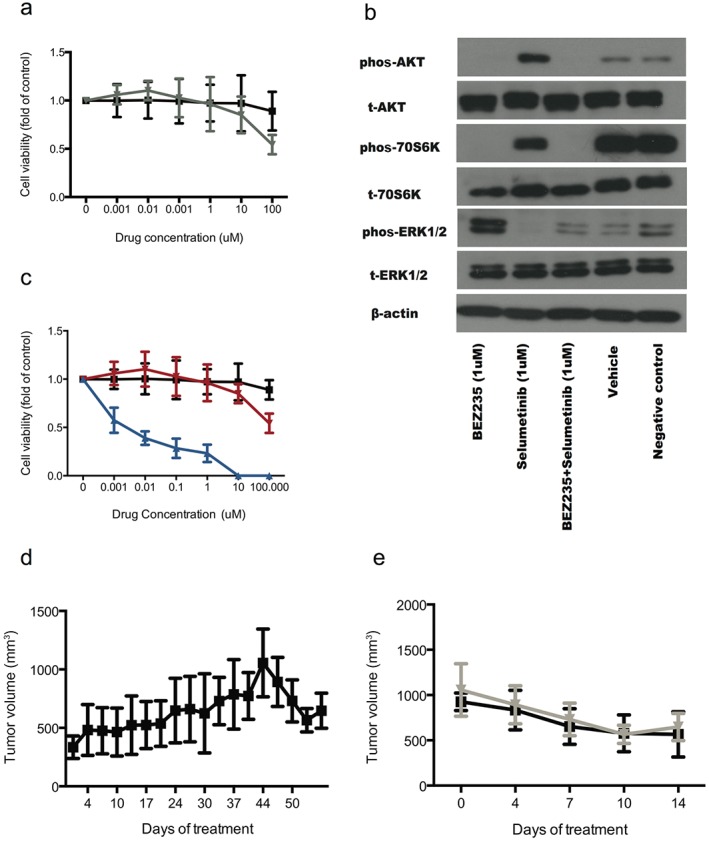
An inhibitor to MEK can reverse the resistance of A549 xenografts to bevacizumab (BV). (a) A549 cells showed resistance to selumitinib in the in vitro cell variability experiment. (b) Western blot tests showed that protein kinase B (AKT) was phosphorylated after the treatment of selumitinib in A549 cells. (c) BEZ235 combined with selumitinib showed significant activity to A549 cells compared with the control and selumitinib alone. (d) After A549 xenograft tumors showed resistance to BV, additional selumitinib treatment reversed the resistance. (e) Selumitinib alone showed similar activity to reverse resistance to BV compared with selumitinib plus BEZ235. ERK, extracellular‐signal‐regulated kinase; phos, phosphorylated.
We then tested the efficacy of selumitinib and selumitinib plus BEZ235 in A549 xenografts to reverse the resistance to BV. After A549 xenograft tumors showed resistance to BV, additional treatment (selumitinib or selumitinib plus BEZ235) was applied. After approximately 14 days, tumors shrank to the initial volumes before BV treatment in both groups (Fig 2d,e); however, mice presented more frequent and severe side effects (e.g. anorexia, fatigue, etc.) and mortality in the selumitinib plus BEZ235 than in the selumitinib group.
RNA sequencing revealed the mechanism of BV resistance
To identify changes in tumor and stromal gene expression associated with resistance to BV, we performed RNA sequencing analyses comparing the A549 control and BV resistant xenograft tumors using mouse‐specific and human‐specific expression arrays. We screened and selected the genes with at least two‐fold elevated RNA transcriptive levels in BV resistant xenograft tumors compared with controls. We found that a much larger number of mouse genes were significantly elevated in BV resistant xenograft tumors compared with human genes (230 vs. 72).
The human genes elevated after BV resistance were mainly involved in hemostasis of the extracellular region, such as chemokine (CCL24, CCL28, CXCL14, and CX3CL1), proangigenesis factor (ANGPTL2), and proteinaceous extracellular matrix (EMID1, COL12A1, FBLN2, MMP2, MFAP4, MUC2, NID1, POSTN, TNC, and TF). Functional analyses of gene features, using Kyoto Encyclopedia of Genes and Genomes Pathway analysis, showed that the chemokine signaling pathway was activated (CCL24, CCL28, CXCL14, and CX3CL1). The mouse genes with increasing expression after BV resistance were mainly cellular components, including extracellular matrix, proteinaceous extracellular matrix, collagen, and fibril (Table 1). We then analyzed the pathway activations in elevated mouse genes and found that several signaling pathways were activated, composed of focal adhesion, extracellular matrix‐receptor interaction, MAPK, Hedgehog, and Wnt signaling pathways, and pathways in cancer (Fig 3).
Table 1.
Main mouse genes with increasing expression after bevacizumab resistance
| Gene ontology | Genes |
|---|---|
| Extracellular matrix | Adamtsl5, Emid1, Adam12, Cilp, Col1a1, Col3a1, Col5a1, Col5a3, Col6a1, Col6a2, Col6a3, Col8a1, Col11a1, Col12a1, Col16a1, Crispld2, Eln, Fbln2, Gpc3, Lox, Mmp10, Mmp3, Mmp9, Mfap2, Mfap4, Muc2, Postn, Ptn, Fbn2, Spon1, Tnc, Tnn, Thsd4, Wnt11, Wnt5a, Wnt9a |
| Proteinaceous extracellular matrix | Adamtsl5, Emid1, Adam12, Cilp, Col1a1, Col3a1, Col5a1, Col5a3, Col6a1, Col6a2, Col8a1, Col11a1, Col12a1, Col16a1, Eln, Fbln2, Gpc3, Lox, Mmp10, Mmp3, Mmp9, Mfap2, Mfap4, Muc2, Postn, Ptn, Fbn2, Spon1, Tnc, Tnn, Thsd4, Wnt11, Wnt5a, Wnt9a |
| Collagen | Col1a1, Col3a1, Col5a1, Col5a3, Col11a1, Lox |
| Fibril | Mfap2, Fbn2 |
Figure 3.
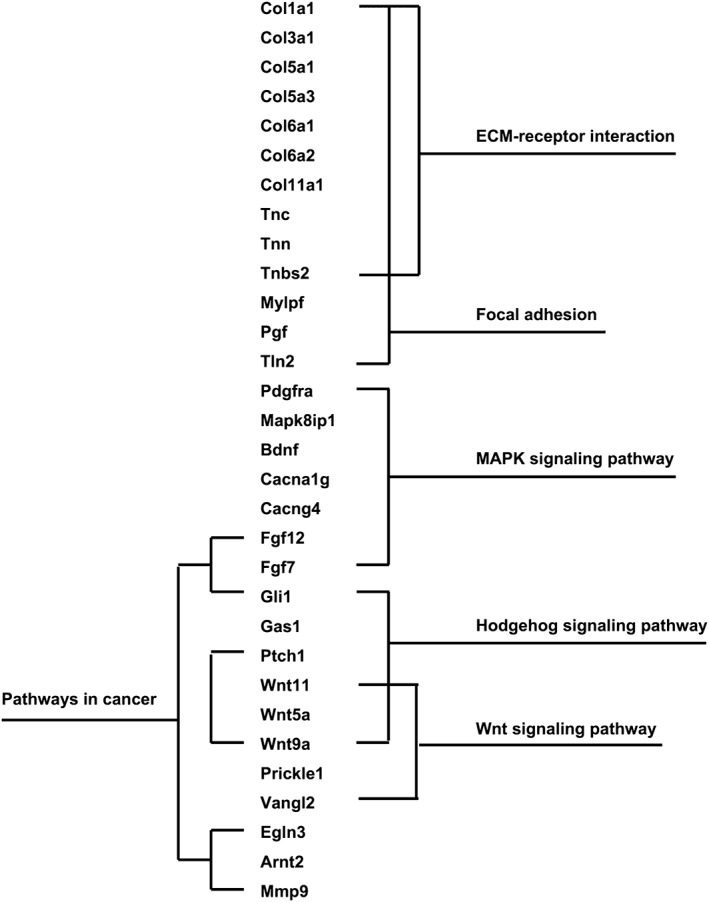
The related pathways involved in bevacizumab resistance. Pathway activations were analyzed in elevated mouse genes and several signaling pathways were found activated, including focal adhesion, extracellular matrix (ECM)‐receptor interaction, mitogen‐activated protein kinase (MAPK), Hedgehog, and Wnt signaling pathways and pathways in cancer.
Expressions of connexin43 and phosphorylated‐connexin43 can predict the efficacy of BV
Connexins might be related to normalization and and could be potential predictive markers of response to BV. Therefore, we detected the immunochemistry staining of different connexins, including Cx 23, 30, 43 and phos Cx43 (S261 and S368) before BV administration and after resistance. Cx43‐S261 was positively expressed and maintained positive expression during non‐progressive status in the tumor tissue before BV treatment, but converted to negative when resistant to BV. Cx43 converted from positive to negative whether responding or resistant to BV, suggesting that the Cx43 change was a reflection of BV treatment. However, phos‐Cx 43 (Cx43‐S261) was predictive of a response to BV. We assessed Cx23 and Cx30 expressions, but failed to find a relationship between the changes in these two connexins and response of A549 xenograft tumors to BV (Fig 4).
Figure 4.
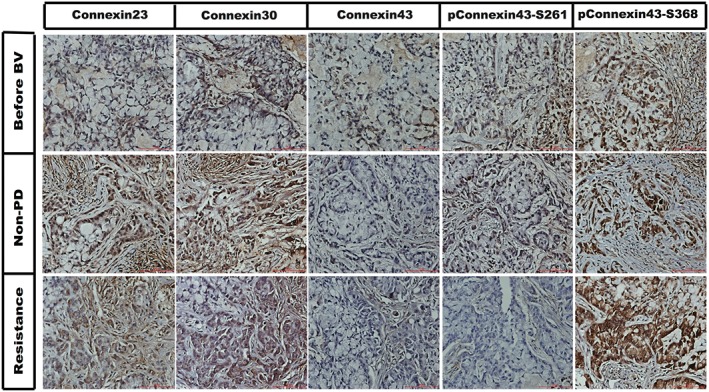
The immunochemistry staining of different connexins including connexin 23, 30, 43 and phosphorylated (p) connexin 43 (S261 and S368) before bevacizumab (BV) delivery, during BV response, and after resistance to BV. PD, progressive disease.
Discussion
Although BV has been broadly utilized for numerous cancers, the resistance mechanisms of BV and the strategies overcoming this resistance remain poorly explored. Herein, we established xenograft mice models harboring KRAS mutations, and, for the first time, proved that the addition of selumetinib, an MEK inhibitor to BV, was a potential strategy to reverse initial BV resistance. This effect might be a result of selumetinib inhibiting the MAPK signaling pathway to stromal cells, most likely vascular endothelial progenitor cells.
In the present study, we observed that A549 xenograft tumors did show a moderate response to BV, although they were relatively insensitive to BV. We also tested the response of PC9 (a stable lung adenocarcinoma cell line with EGFR 19del) xenograft tumors to BV and found that PC9 xenograft tumors seemed to be more sensitive to BV than A549 xenograft tumors (data not shown), similar to the results of a previous study by Cascone et al., which suggested that the cancer genotype might influence efficacy after BV administration.18 However, after BV treatment, Kim et al. failed to determine whether KRAS mutations are predictive of PFS in metastatic colorectal cancers (mCRC).19 Bencsikova et al. also concluded that KRAS mutations could not interfere with the clinical benefits of first‐line treatment with BV plus chemotherapy in mCRC patients.20 However, these results were all based on mCRCs; therefore, any effects of genotyping on BV efficacy need to be explored in lung cancer patients.
Kirsten rat sarcoma oncogene homolog mutation can activate the MAPK signaling pathway. MEK is the downstream kinase of KRAS. Theoretically, blocking MEK may inhibit activation of the MAPK pathway and the growth of KRAS mutation driving lung cancer cells. However, we failed to observe the inhibition of selumetinib to A549 cells in the ex vivo cell viability test. A subsequent Western blot test showed that the PI3K‐AKT pathway was significantly activated after selumetinib treatment. Combined with the results of the superior inhibiting activity on A549 cells of selumetinib plus BEZ235, we concluded that selumetinib alone could not effectively inhibit the growth of A549 cancer cells. Of interest, selumetinib reversed BV resistance with similar effect as selumetinib plus BEZ235, which suggested that the key target in the in vivo test was not the cancer cells. Our RNA sequencing results showed that the MAPK signaling pathway was activated in mouse but not human genes, which implied that the target of selumetinib was the MEK of stromal cells. We also found activation of stem cell related pathways, such as Wnt and Hedgehog pathways. Therefore, we speculate that the reversed resistance was a result of the inhibition of selumetinib to vascular endothelial progenitor cells that were activated gradually after BV treatment.
The “normalization” of tumor vasculature is now considered the main mechanism of BV, meaning that resistance to BV leads to de‐normalization of tumor vessels.15, 16 Phosphorylation of connexin protein leads to normal close and firm connection of vascular endothelial cells, while de‐phosphorylation of connexin leads to abnormal connection, which suggests that connexins could be a potential marker for predicting BV response. In the present study, we found that phos‐Cx43‐S261 was associated with the sensitivity and resistance of BV, and a dynamic change in the phosphorylated status of Cx43‐S261 could be a likely biomarker predicting resistance to BV. To the best of our knowledge, our study represents the first study illustrating the potential value of Cx43 in response to BV; however, further studies need to be performed to confirm our results.
In summary, we successfully established xenograft mice models harboring KRAS mutations, and determined that a combined MEK inhibitor with BV was a potential strategy to reverse initial BV resistance. Using RNA sequencing, we found that the effect of selumetinib might result from the inhibition of MAPK signaling pathway activation in stromal cells, most likely vascular endothelial progenitor cells. Phos‐Cx43 might be associated with the response to BV. Our results provide a basis for further studies on BV resistance.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This work was supported by the National Natural Sciences Foundation, China (81101778 and 81472206).
References
- 1. Bartolotti M, Franceschi E, Poggi R, Tosoni A, Di Battista M, Brandes AA. Resistance to antiangiogenic therapies. Future Oncol 2014; 10: 1417–1425. [DOI] [PubMed] [Google Scholar]
- 2. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 82: 4–6. [DOI] [PubMed] [Google Scholar]
- 3. Kowanetz M1, Ferrara N. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin Cancer Res 2006; 12: 5018–5022. [DOI] [PubMed] [Google Scholar]
- 4. Johnson DH, Fehrenbacher L, Novotny WF et al Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non‐small‐cell lung cancer. J Clin Oncol 2004; 22: 2184–2191. [DOI] [PubMed] [Google Scholar]
- 5. Reck M, von Pawel J, Zatloukal P et al Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab a first‐line therapy for nonsquamous non‐small‐cell lung cancer: AVAil. (Published erratum appears in J Clin Oncol 2009; 27: 2415) J Clin Oncol 2009; 27: 1227–1234. [DOI] [PubMed] [Google Scholar]
- 6. Crinò L, Mezger J, Griesinger F et al MO19390 (SAiL): Safety and efficacy of first‐line bevacizumab (Bv)‐based therapy in advanced non‐smal‐cell lung cancer (NSCLC). 2009 ASCO Annual Meeting Proceedings. J Clin Oncol 2009; 27 (Suppl. 15S): Abstract 8043. [Google Scholar]
- 7. Mok T, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957. [DOI] [PubMed] [Google Scholar]
- 8. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–2388. [DOI] [PubMed] [Google Scholar]
- 9. Zhou CC, Wu YL, Chen GY et al Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): A multicentre, open‐label, randomised, phase 3 study. Lancet Oncol 2011; 12: 735–742. [DOI] [PubMed] [Google Scholar]
- 10. Rosell R, Carcereny E, Gervais R et al Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): A multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–246. [DOI] [PubMed] [Google Scholar]
- 11. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–128. [DOI] [PubMed] [Google Scholar]
- 12. Rak J, Yu JL. Oncogenes and tumor angiogenesis: The question of vascular “supply” and vascular “demand”. Semin Cancer Biol 2004; 14: 93–104. [DOI] [PubMed] [Google Scholar]
- 13. Giuliano S, Pagès G. Mechanisms of resistance to anti‐angiogenesis therapies. Biochimie 2013; 95: 1110–1119. [DOI] [PubMed] [Google Scholar]
- 14. Abdullah SE, Perez‐Soler R. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer 2012; 118: 3455–3467. [DOI] [PubMed] [Google Scholar]
- 15. De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat Rev Clin Oncol 2011; 8: 393–404. [DOI] [PubMed] [Google Scholar]
- 16. Mazzone M, Dettori D, Leite de Oliveira R et al Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009; 136: 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pries AR, Höpfner M, le Noble F, Dewhirst MW, Secomb TW. The shunt problem: Control of functional shunting in normal and tumour vasculature. Nature Rev Cancer 2010; 10: 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cascone T, Herynk MH, Xu L et al Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor‐resistant human lung adenocarcinoma. J Clin Invest 2011; 121: 1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim ST, Park KH, Shin SW, Kim YH. Dose KRAS mutation status affect on the effect of VEGF therapy in metastatic colon cancer patients? Cancer Res Treat 2014; 46: 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bencsikova B, Bortlicek Z, Halamkova J et al Efficacy of bevacizumab and chemotherapy in the first‐line treatment of metastatic colorectal cancer: Broadening KRAS‐focused clinical view. BMC Gastroenterol 2015; 15: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]