

Abstract
Alzheimer’s disease (AD) is the most common underlying cause of dementia, and novel drugs for its treatment are needed. Of the different theories explaining the development and progression of AD, “amyloid hypothesis” is the most supported by experimental data. This hypothesis states that the cleavage of amyloid precursor protein (APP) leads to the formation of amyloid beta (Aβ) peptides that congregate with formation and deposition of Aβ plaques in the frontal cortex and hippocampus. Risk factors including neurotransmitter modulation, chronic inflammation, metal-induced oxidative stress and elevated cholesterol levels are key contributors to the disease progress. Current therapeutic strategies abating AD progression are primarily based on anti-acetylcholinesterase (AChE) inhibitors as cognitive enhancers. The AChE inhibitor, donepezil, is proven to strengthen cognitive functions and appears effective in treating moderate to severe AD patients. N-Methyl-D-aspartate receptor antagonist, memantine, is also useful, and its combination with donepezil demonstrated a strong stabilizing effect in clinical studies on AD. Nonsteroidal anti-inflammatory drugs delayed the onset and progression of AD and attenuated cognitive dysfunction. Based upon epidemiological evidence and animal studies, antioxidants emerged as potential AD preventive agents; however, clinical trials revealed inconsistencies. Pharmacokinetic and pharmacodynamic profiling demonstrated pleiotropic functions of the hypolipidemic class of drugs, statins, potentially contributing towards the prevention of AD. In addition, targeting the APP processing pathways, stimulating neuroprotective signaling mechanisms, using the amyloid anti-aggregants and Aβ immunotherapy surfaced as well-tested strategies in reducing the AD-like pathology. Overall, this review covers mechanism of inducing the Aβ formation, key risk factors and major therapeutics prevalent in the AD treatment nowadays. It also delineates the need for novel screening approaches towards identifying drugs that may prevent or at least limit the progression of this devastating disease.
Keywords: Donepezil, memantine, antioxidants, statins, screening
Introduction
Dementia is a neurodegenerative condition marked by diminished cognitive and thinking ability, altered personality and loss of reasoning [1]. Alzheimer's Disease (AD) is the best known and the most prevalent cause of dementia, accounting for about 60-80% of all dementia cases [2]. Owing to the increasing average life span, the prevalence of AD is sharply on the rise, with the WHO predicting above 20 million cases by 2020, and Delphi consensus study projecting about 70-80 million AD sufferers by 2050 [3-5]. Though AD is an aging-associated disorder mainly reported in patients aged 65-85 years, it can affect younger people too, with environmental and genetic factors playing a leading contributory role in such cases [6,7].
Presence of several pathophysiologic mechanisms in the progression of AD hinders development of a single potential treatment for the disease [8]. Although, a few therapeutic agents have gained prominence in the recent years and have reached the late stages of clinical trials [9], an overall lack of any suitable disease-modifying therapy prevents its effective management. This leads to severe damage and death of the functioning brain cells, which ultimately proves fatal [10]. In this review, we will briefly discuss the current mechanisms, rationales and targets for therapeutic interventions in AD. We will focus on the drugs that are known to suppress AD symptoms, specifically the neurotransmitter modulators, anti-inflammatory compounds, antioxidants and cholesterol-lowering statins. We also highlight the need for new drugs that may slow the disease progression, overcoming the deficiencies of existing therapies. Unlike earlier reviews that generally focus on either causes of AD or its treatment, we concisely provide a comprehensive idea about all the essential factors promoting AD pathogenesis, as well as potential approaches to the disease prevention and therapy.
AD hypotheses and Aβ Generation
Although the molecular mechanisms of AD pathogenesis are well-investigated, the key reason that governs the pathology is yet contentious. The most accepted “amyloid hypothesis” points to the deposition ofamyloid beta (Aβ) in the neurons and parenchyma as major reason for synaptic, axonal and neural dysfunctions and the consecutive cognitive impairment in patients [11-13] (Figure 1). Aβ formation is largely regulated by a shift in the balance between amyloidogenic and non-amyloidogenic amyloid precursor protein (APP) processing [14]. The APP, predominant in the brain as 695, 751 and 770 amino acid isoforms, undergoes non-amyloidogenic cleavage at the α-secretase site at position17 of the 40-42-Aβ amino acid domain. This results in the formation of two fragments, sAPPα and a C-terminal fragment (CTFα). sAPPα, proved to be neuroprotective, is secreted. CTFα undergoes further proteolysis by γ-secretase, yielding p3 peptide and a C-terminal fragment, CTFγ. A down-regulation of this non-amyloidogenic processing or a shift towards the amyloidogenic pathway causes β-secretase to cleave APP before the Aβ amino acid site, releasing CTFβ and sAPPβ. CTFβ is further cleaved by γ-secretase, generating Aβ40 and Aβ42. The Aβ42 isoform is deemed more toxic, participating more in the plaque formation [15,16]. Intracellular accumulation of hyperphosphorylated and aggregated tau in the form of neurofibrillary tangles is also closely related to neuronal death in AD [17,18]. It is believed that Aβ deposition precedes tau tangle formation, with the latter triggered by Aβ-activated calpain and an increased tau proteolysis [19,20]. An association of AD with several vascular and mitochondrial risk factors, e.g. diabetes mellitus, hypertension, atherosclerosis, hypercholesterolemia, metabolic syndrome and obesity, led to a view that vascular pathologies may trigger AD [21]. It is believed that apolipoprotein E (Apop E) genotype is linked with hypercholesterolaemia [22]. Amyloid and vascular mechanisms are closely related and both culminate in Aβ deposition via disruption and perturbation of Aβ transporters, especially in the blood-brain barrier (BBB) [23]. Familial AD is governed by the autosomal, dominantly inherited, rare mutations in APP and its processing molecules, such as the γ-secretase components, presenilin (PSEN)1 and PSEN2, and accounts for less than 1% of AD cases overall [24]. The present review will focus specifically on the amyloid-based Aβ concept of AD, highlighting the major risk factors and therapeutics.
Figure 1.
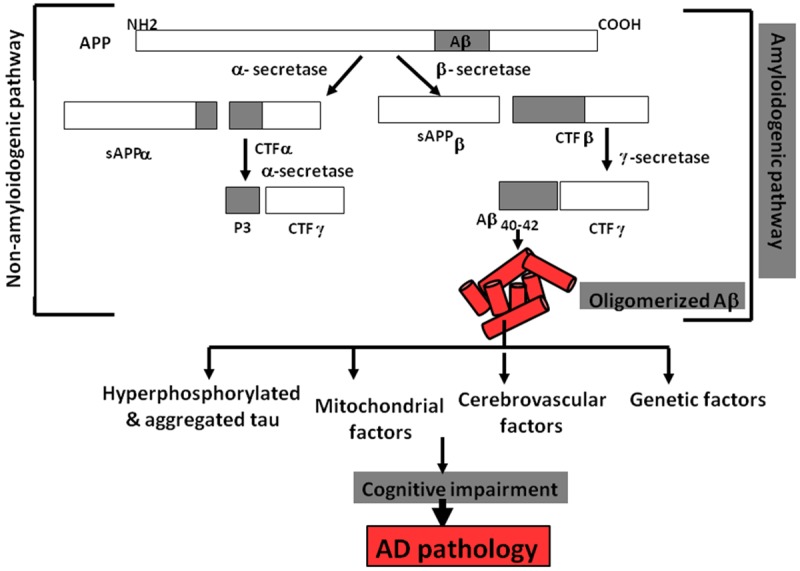
APP processing pathway stimulating Aβ plaque deposition and AD pathology.
AD therapeutics
Risk factors, such as neurotransmitter modulation, chronic inflammation, metal-induced oxidative stress and elevated cholesterol, primarily contribute to AD progression (Figure 2). Altered neurotransmission involving cholinergic dysfunction, increased glutamate release and N-methyl-D-aspartate (NMDA) receptor action, and aberrant gamma-aminobutyric acid (GABA), histamine and serotonin functioning participate in the pathogenesis [25-29]. Elevated levels of brain-specific inflammatory cytokines and a pronounced increase in the metals, iron, copper and zinc, in the amyloid plaques of the AD brain suggest inflammation and metal-induced oxidative stress as key mechanisms in AD pathology [30,31]. Hypercholesterolemia and cholesterol-linked pathways play an important role in AD [32]. Thus, here we elaborate on these risk factors and their mitigation approach in reducing the Aβ generation and, thereby, AD pathology.
Figure 2.
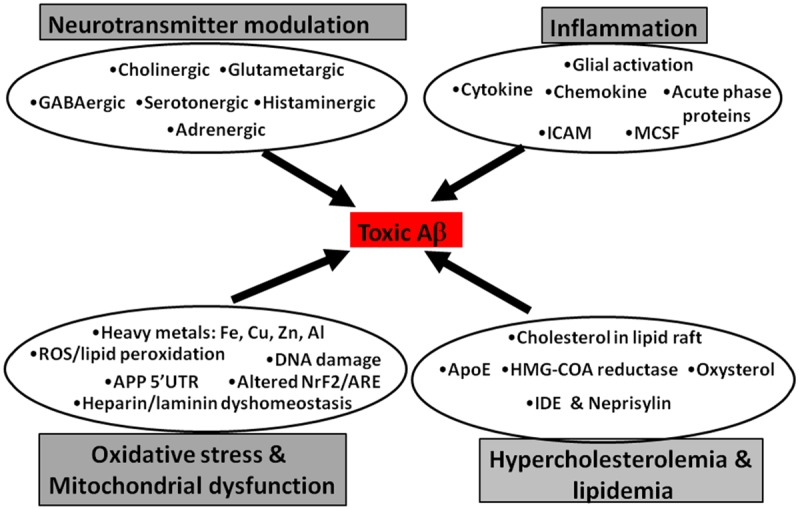
Major risk factors promoting Aβ plaque deposition in AD.
Targeting neurotransmitters (Table 1)
Table 1.
Important publications on Neurotramitter Modulators as AD therapeautics
| Neurotransmitters | Therapeutics | References |
|---|---|---|
| Cholinergic | Tacrine | Nagabukuro et al., 2005; Minarini et al., 2013; Alonso et al., 2005; Guzior et al., 2015 |
| Donepezil | Cacabelos, 2007 | |
| Galanthamine | Hopkins et al., 2012; Albuquerque et al., 2001 | |
| Rivastigmine | Rodrigues Simoes et al., 2014; Toda et al., 2003 | |
| Xanthostigmine | Belluti et al., 2005 | |
| Pyrrolo-isoxazole derivatives | Anand and Singh, 2012; Razavi et al., 2013; | |
| Glutametargic | Memantine | Golde, 2006; Farlow et al., 2008; Cummings, 2007; Danysz and Parsons, 2012; Dominguez et al., 2011; Prokselj et al., 2013 |
| GABAergic | Etazolate | Marcade et al., 2008 |
| Gabapentin | Cooney et al., 2013 | |
| Serotonergic | Lecozotan | Schechter et al., 2005 |
| Tandospirone and Buspirone | Miller et al., 1992; Sumiyoshi et al., 2007 | |
| PRX-3140, PF-04995274 and RQ-00000009 (RQ-9) | Ramirez et al., 2014 | |
| SB-742457 | Maher-Edwards et al., 2010 | |
| Lu-AE-58054 (SGS-518) | Ramirez, 2013 | |
| Dimebon | Bezprozvanny, 2010 | |
| SSRI | Nagy et al., 2004 | |
| Histaminergic | Carebastine and mepyramine | Mizuguchi et al., 2012 |
| R-(alpha)-methylhistamine | Motawaj et al., 2010 | |
| Pyrilamine | Kharmate et al., 2007 | |
| Thioperamide and ondansetron | Passani and Blandina, 1998 | |
| Chlorpheniramine | Baronio et al., 2014 | |
| Adrenergic | SCH58261 | Melani et al., 2003 |
Cholinesterase inhibitors
Cholinergic hypothesis of AD development focuses on the increased hydrolysis of acetylcholine by acetylcholinesterase (AChE). This leads toa reduction in synaptic acetylcholine levels detected in AD brain [33]. The resulting modulation in cholinergic neurotransmission and functions prompts learning-memory impairments and altered intellectual, behavioral and emotional responses. Clinical findings reveal significant cortical and hippocampal atrophy associated with the cholinergic modulations [33]. This cholinergic damage is promoted by butyrylcholinesterase (BuChE) that functions as a co-regulator and enhancer of AChE. However, since the effect of BuChE is more prominent at peripheral tissues, AChE gains predominance as the cholinergic neuromodulator in AD [34]. Of the known clinically-used AChE drugs, tacrine, donepezil, rivastigmine and galantamine (Figure 3) are the most commonly used and considered promising for AD treatment. Further, these drugs have been derivatized to improve their efficacy and potency and reduce toxic side effects [35]. Tacrine (Figure 3) is the foremost approved of the first-generation AChE and BuChE inhibitors that has cholinomimetic properties [36]. Owing to tacrin’s hepatotoxic effects, ring structure-modified derivatives of tacrine were synthesized [37]. Dual-binding site, homo- and hetero-dimeric derivatives capable of binding to both active and peripheral AChE sites were generated based on homodimers of two tacrine moieties linked by oligomethylene chains [38]. Of these, linkers with a heptamethylene tether were found to be significantly more potent than tacrine [39]. Incorporation of a protonable amino group in the middle of the tether enhanced AChE selectivity and activity, and an amide group in place of the central methylene group of a heptamethylene linker increased BuChE selectivity [40]. Some derivatives with chloro and iodo moieties were also designed [41]. Heterodimeric derivatives with indanone and phthalimide tagged to tacrine also proved suitable [42]. Tacrine heterodimers were designed by connecting the tacrine moiety with imidazole, piperidine and ferulic acid. A potent tacrine heterodimer, carbacrine, was generated by combining tacrine with the carbazole moiety of carvedilol that had IC50 values of 2.15 nM and 296 nM for AChE and BuChE respectively [43]. Combination of tacrine with the hepatoprotective nitric oxide (NO) donors appeared safe [44]. Interestingly, to reduce Ca2+ toxicity, Ca2+ channel blockers, such as 1,4-dihydropyridine (DHP) were inserted forming tacripyrines, of which the one with a cycloheximide ring in DHP moiety appeared the most potent as AcHE inhibitor (IC50 = 0.37 µM) [45]. Tacrine-phenyl-benzoheterocyclic derivative demonstrated AChE inhibitory property. Moreover, this benzo derivative functioning was also dependent on the methylene linker chains [46]. Donepezil (IC50 = 5.7 nm) (Figure 3) is considered less toxic and physiologically well-accepted and was the second FDA-approved drug for treating AD. The drug enhances cholinergic transmission and attenuates neuronal damage [47]. Interaction of its benzyl piperidine and indanone groups with the indole rings at the peripheral anionic site (PAS) proved useful. N-benzylpiperidine derivatives with aroylthiourea, fluoro and a chloro incoroporation at indanone system were also designed and demonstrated to have 30-50% of donepezil’s IC50 value of [48]. A combination of 3-amino-6-phenylpiridazine with N-benzylpiperidine units yielded a compound that was several times more potent than donepezil. Combining piperidine, indanone and methylene groups resulted in a compound with the highest potency, with an IC50 of 0.0018 μM [49]. Galanthamine (IC50 = 800 nM) (Figure 3), a tertiary alkaloid drug, manifests AChE activity reduction and modulates nicotinic acetylcholine receptors (nAChR) towards enhancing acetylcholine generation [50]. The drug is a proven allosteric modulator of nAChR [51]. Though less toxic, its reduced potency for acetylcholine release compared to tacrine led to the designing of few derivatives using alkyl linkers, especially eight to ten methylene groups, and a terminal ammonium or phthalimido group with several fold increased efficacy [52]. N-substituted galanthamine derivatives with incorporated benzylpiperidines and alkyl linkers, specifically with six methylene units, appeared to have highest AChE efficacy amongst all derivatives [43]. Rivastigmine (IC50 = 4.15 µM) (Figure 3), with a carbamate moiety, emerged as a new generation of AChE inhibitor which is long-acting and reversible. Benzopyrano[4,3-b]pyrrole carbamate derivatives with further methyl derivatization at carbamoyl nitrogen showed a potent inhibitory property [53]. A combination of donepezil and rivastigmine linked through 5,6-dimethoxy-indan-1-one and dialkyl-benzylamine moieties demonstrated significantly higher AChE rather than BuChE inhibition, indicating selectivity towards the former [54]. For these compounds, variations in the meta- and para-substituted derivatives were evident [54]. A heterodimer of rivastigmine and the serotonin transport inhibitor, fluoxetine, appeared as a potent second-generation dual AChE-SERT inhibitor, emphasizing the importance of incorporating dual functions in drugs [55]. Xanthostigmine derivatives possess the amyloid pro-aggregatory property due to their binding at the AChE peripheral site. Its arylidenebenzocycloalkanone derivative targeted both the active and peripheral sites, and a further incorporation of three or seven methylene units alkoxy spacer chain and arylidene moiety into the arylidene aryl ring moiety enhanced contact with PAS [56]. Of the tested meta- and para-isoforms, the para-aminobenzoic acid derivative possessed a Ki value of 53 nM (AChE). Further molecular dynamics and docking studies confirmed their efficacy as AcHE inhibitors. Cis-isomers of pyrrolo-isoxazole derivatives with methoxy substitution, especially at the para-position were deemed highly potent, claiming an anti-amnestic and AChE inhibitory abilities higher than that of donepezil [57]. The polyphenolic compounds, coumarinand its derivatives, such as ensaculin (KA-672 HCl) containing a benzopyran ring and a piperazine substitution [58] and AP2238 having benzylamino group linked to coumarin via phenyl ring, are rising as AChE/BuChe inhibitors with peripheral and catalytic site-binding capacities [59]. Flavonoid derivatives linking flavonoid and benzylpiperidine through oxygen atom or alkoxyl group (-OCH2) spacers proved effective [60]. A replacement of benzyl piperidine moiety with amino alkyl or the conformationally restrained hydrophobic groups, pyrrolidine or piperidine at the meta- or para-positions was more potent, whith the latter two demonstrateinga greater effect [60]. Carbamate-substituted 5,7-dimethoxyflavanone, having an IC50 of around 10 nM or several folds greater respectively [61].
Figure 3.
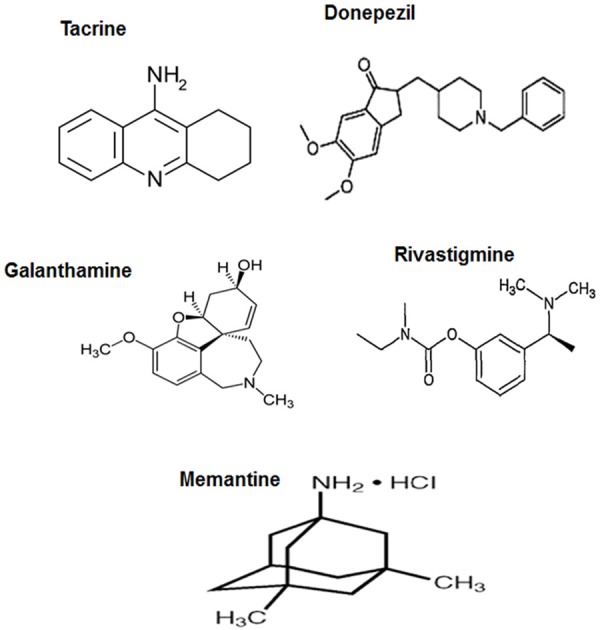
Prevalently used AD-drugs targeting neurotransmitters.
However, cholinesterase inhibitors are not recommended for patients with advanced AD and are prescribed rather for moderate or mild AD cases. Other side effects of these drugs include unwanted cholinergic stimulation in the intestine, heart, muscle, kidney, and other organs. Thus, AChE inhibitors specifically targeting the cholinergic system of the brain are desired.
Glutametargic alteration
Modulation in the functioning of glutamatergic neurons is generally viewed as a property of mature AD pathology, mediated strongly by the altered levels of the synaptic glutamate neurotransmitter [62]. NMDA receptor activation inducing excitotoxicity by enhanced synaptic glutamate accumulation damages the glutamatergic neurons and adversely impacts the neuronal proliferation and differentiation, causing learning, memory and cognitive impairments [62]. Further, a direct interaction between the NMDA receptors and APP is also reported, indicating the importance of glutamatergic synaptic transmission in AD [63]. Memantine (1-amino-3,5-dimethyl-adamantane) (Figure 3), an adamantane derivative, is the most used and cost-efective drug targeting the NMDA receptor for AD treatment, especially in the USA and Europe [64,65]. It is proved through clinical studies that the drug has symptomatic effectiveness and is known to treat moderate to severe AD [66]. As detected through cultured neuronal whole-cell patch clamp recordings, memantine has a modest affinity of about 1 mM at 70 mV [67]. The proposed mechanism appears uncompetitive, fast, voltage-dependent and with reduced tendency for entrapment in the receptor channel [67]. Memantine is capable of blocking the NMDA receptor channel and in preventing glutamate-mediated excitotoxicity by interacting with Mg2+ or binding to NMDA channel close to the magnesium-binding site, and thereby reducing Ca2+ entry in the post-synaptic neurons [68]. Other than NMDA receptor-mediated functioning, memantine also reduced Aβ-induced neuronal apoptosis, marked by an attenuated DNA fragmentation and altered Bcl-2 immunostaining [69]. A non-specific neurotransmitter targeting is also detected with memantine, via its ability to antagonize human α7nAChR, indicating a concern for studies involving the co-existence of NMDA and nACh receptors [70]. However, for situations that involve aberrant functioning of both nAcHER and NMDAR in AD, memantine appears very effective. A modulation of dopaminergic and serotonergic/histaminergic neurotransmission is also reported with memantine [71,72]. Interestingly, it was claimed that a combination of memantine and cholinesterase inhibitor with different and interconnected activities is preferred: the former takes care of agitation/aggression and delusions, and the latter reduces depression, anxiety and apathy thus indicating complementary activities [73,74].
GABAergic alteration
It is proven that transgenic mice with early-stage amyloid pathology manifest modulations in the cholinergic neurons, followed by glutamatergic and lastly the inhibitory GABAergic neurons [75]. By inducing GABAA receptor subunit endocytosis, Aβ impedes synaptic inhibition [76]. GABAA undergoes a compensatory increase in a few hippocampal regions and sub-regions where the NMDA and non-NMDA type α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA)-receptors are reduced at the late AD stages, probably in order to maintain hippocampal functions [77]. The pyrazolopyridine compound etazolate with anxiolytic-like properties at nanomolar tolow micromolar pharmacological doses selectively affected the GABAA receptor and enhanced the propagation of α-secretase pathway towards sAPPα [78]. Another anticonvulsant drug, gabapentin, structurally related to GABA demonstrated prominent positive responses to treatment at low doses in mixed vascular/Alzheimer dementia patients [79]. However, the drug caused sedation at a high dose, and appears to affect other neurotransmitter systems, including serotonin and glutamate [80].
Serotonergic alteration
The hippocampal serotonin 5-hydroxytryptamine (5-HT) receptor, especially 5-HT1A receptor, and the cortical 5-HT6 serotonergic receptors and their metabolites, particularly 5-HIAA, play key role in the memory and cognition [81]. Post-mortem AD brain demonstrated a decrease in 5-HT, and its reduction in the cortex correlated with the neuronal loss at the raphe nuclei [82]. This serotonergic dysfunction is a property of the early-onset AD, reportedly with the mis-regulation of α-secretase processing [83]. Given the location of many of these receptors on terminals of other neurons, it is possible that these changes reflect the loss of cholinergic synapses as well [84,85]. Thus, preclinical studies on 5-HT1A receptor antagonists reported pro-cognitive effects with the significant abetment of glutamatergic and cholinergic transmission [86]. The 5-HT1A receptor antagonist lecozotan (SRR-333) initially appeared promising, safe and well tolerated in clinical pharmacokinetic studies at a single drug dose [87]. However, adverse side effects prevented its progress into phase II clinical trials. Few partial agonists, such as tandospirone and buspirone, are being considered useful in preventing dementia [88,89]. Agonists of the 5-HT4 G-protein-coupled receptor, such as PRX-3140, PF-04995274 and RQ-00000009 (RQ-9), that act by enhancing the acetylcholine release, are in the phase I or II clinical studies [90]. Along with the memory enhancement, RQ-9 was capable of reducing Aβ deposition in the transgenic Tg2576 mice [90]. 5-HT6 receptor antagonist, SB-742457, completed phase II clinical studies [91]. SB-742457 went through four phase II trials for mild-to-moderate AD, and was claimed comparable to donepezil [92]. Studies also revealed an additive effect of the 5-HT6 receptor antagonist, Lu-AE-58054 (SGS-518) with donepezil [85]. An antihistamine drug, dimebon, that binds to the 5-HT6 receptor, appeared very promising in phase II, but failed in phase III [93]. However, a dimebon derivative, P7C3, seemed to have neuroprotective properties and is being taken up for clinical observations [94]. In addition to antagonists, the 5-HT6 receptor agonist E-6801 also proved effective in improving cognition synergistically with donepezil [95]. Serotonin reuptake inhibitors (SSRIs), eg. Fluoxetine, sertraline and citalopram, proved useful with AChE inhibitors such as rivastigmine, donepezil, etc. in dementia [96]. It is well accepted that these SSRIs may attenuate Aβ levels and promote neuronal survival and functioning [97]. Overall, serotonergic therapy, via targeting 5-HT1A, 5-HT4 and especially 5-HT6 receptors, is being considered increasingly important, although mostly for mild-to-moderate AD patients on the early stages of AD.
Histaminergic modulation
It is reported that short-term and long-term memory is regulated by histamine, and a degeneration of histaminergic neurons impairs the cognition [98]. Studies on null-mutations of the excitatory receptors, H1R and H2R, in frontal cortex, amygdala and hippocampus revealed histidine participation in maintaining the normal synaptic plasticity and learning-memory [99]. For the histaminergic synaptic functioning, an interaction with aminergic and peptidergic systems was also reported [100]. An enhanced activity of the inhibitory H3Rs attenuated the cholinergic system as well [101]. Histamine is known to contribute to dendritic cell (DC) functioning and regulates immune responses via increased interleukin IL-6 and IL-10, decreased IL-12, and enhanced secretion of chemokines and matrix metalloproteases-9 and -12, the latter participating in DC migration via the H2R binding [102]. H1R and H4R triggered the T helper 2 (Th2)-type immune responses and modulated Ca2+ influx, involving store-operated calcium entry (SOCE) [103]. Aβ-dependent alteration in T helper cell memory with altered expression of cytokines and chemokines clearly indicated a link with histidine. An aberrant postreceptor signaling in AD involving phosphoinositide hydrolysis and adenylate cyclase pathways that are constitutively suppressed by the cortical H3R substantiated association between histidine and AD [102]. Inverse agonists of histidine receptor, carebastine and mepyramine, suppressed H1R and also histamine generation, and prevented an alteration in Ca2+-signaling via alteration of protein kinase A (PKA) and cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) [104]. The H3R agonist, R-(alpha)-methylhistamine, was found to distinctly attenuate muscarinic acetylcholine receptor-dependent phospholipase C (PLC) activity and calcium-calmodulin- and cAMP-induced protein kinases, thereby predicting an attenuation of the presynaptic excitatory functions of acetylcholine in AD [105]. The H3R antagonists that promote the generation of histamine, acetylcholine, dopamine and norepinephrine, and suppress the adenyl cyclase-PKA pathways are being promoted as drug targets for AD. The H1R antagonist, pyrilamine, inactivated His-dependent STAT6 hyperphosphorylation, while H2R antagonist, ranitidine, and H3R/H4R antagonist, thioperamide, failed to do so [106]. Rather, H3R antagonists that stimulate the cognitive domains were proposed as new therapeutic agents for AD treatment. Chlorpheniramine, an H1R antagonist, that functioned as a serotonin-norepinephrine reuptake inhibitor altered the cortical and hippocampal cholinergic tone and affected learning and memory in AD [107]. Furthermore, the H3R antagonist, thioperamide, and 5-HT3 antagonist, ondansetron, healed cholinergic deficits, suggesting their probable role in inhibiting the AD pathogenesis [108].
Adenosine receptor
Activation of adenosine receptor of A2A subtype induced the anti-inflammatory IL-10 and prevented Aβ deposition [109,110]. An interaction of Aβ with β2-adrenergic receptors induced internalization and degradation of the latter, causing adrenergic and glutamatergic aberrations and reduction in β2-adrenergic-stimulated cAMP [111,112]. It was found that both caffeine and adenosine receptor antagonists averted Aβ build-up via increased striatal PKA activity and p-CREB levels, and decreased p-JNK and p-ERK [113,114]. The reduction in A2a adenosine receptors, therefore, stimulated pro-survival anti-apoptotic cascades and cognitive impa irments induced by Aβ [115]. A selective adenosine A(2A) receptor antagonist SCH58261 also demonstrated a similar effect [116].
Targeting inflammation (Table 2)
Table 2.
Important publications on anti-inflammatory agents as AD therapeautics
| Inflammation inhibitor | Therapeutics | References |
|---|---|---|
| NSAID | Indomethacin | Hoozemans et al., 2001 |
| BF389 | Blom et al., 1997 | |
| SC-560 | Choi et al., 2013 | |
| Nitro-flurbiprofen | Jantzen et al., 2002; Cole et al., 2004 | |
| SD-282 | Koistinaho et al., 2002 | |
| Targeting Rho-GTPases | Kubo et al., 2008 | |
| Targeting PPAR | Nenov et al., 2014 |
Neuroinflammation is established as intricately associated with AD, involving the participation of complement, cytokines, chemokines and acute phase proteins [117]. Chronic complement activation leading to membrane blebbing and endocytosis, and neurite opsonization was observed in the vicinity of Aβ [117]. Activated microglia and astrocyte-generated cytokines, such as the pro-inflammatory IL-1 and tumor necrosis factor alpha (TNFα), regulate cyclooxygenase (COX) activity and extensively participate in APP metabolism [118,119]. Supportively, cytokine and Transforming Growth factor beta polymorphism is observed in the AD brain [120]. In addition, these cytokines, alongside chemokine IL-8, intracellular adhesion molecule-1, macrophage colony stimulating factor and acute phase proteins such as C-reactive protein, serum amyloid A, and transthyretin coordinate the acute phase mechanisms [121-125]. The inflammatory mediators evidently activate the AChE enzyme activity and thereby aggravate the cholinergic dysfunction in AD [126]. Thus, targeting inflammation to reduce AD pathology appears to be advantageous.
Microglial generation of superoxides and oxidative intermediates is an early feature of AD [127]. Thus, therapeutics that target microglia and prevent microglial activation are being designed. Small molecules directed towards the 13-16 site of Aβ (HHQK domain) that binds within the microglia [128], and SD-282 that targets microglial P38-MAPK mechanism of inflammation [129] are also being assessed. Drugs aiming at the plaque-linked complement C3 and neurotoxic C5b-9 [130] and the brain opsonins integrated to the inflammatory cascade are hypothesized to suppress the membrane attack complex that mediate the complement cascade-mediated neuronal killing [131]. However, the most well-studied compounds are the non-steroidal anti-inflammatory drug (NSAID) that had undergone epidemiological studies. The studies revealed that consistent use of NSAID has positive effect in attenuating the AD risk [132]. Indomethacin inhibited astroglial IL-1-dependent IL-6 secretion involving a reduction in the prostaglandin-2 [133]. A specific cycloxygenase-2 (COX-2) inhibitor, BF389, followed almost a similar IL mechanism [134]; however, the COX-2 inhibitors failed to prevent cognitive decline. Cognitive restoration and reduction in the AD pathological features in triple transgenic mice by the anti-inflammatoy COX-1 inhibitor, SC-560, appeared hopeful [135]. Ibuprofen, rather than adopting the COX inhibition pathway, suppressed γ-secretase activity and reduced Aβ levels, as found in APP-transgenic mice [136]. A nitro-derivative of ibuprofen, nitro-flurbiprofen, released NO that promoted microglial Aβ clearance and also prevented the reduction of plasticity-related genes [137,138]. Very interestingly, the anti-inflammatory mode of action of nitro-ibuprofen was different from SD-282, that is used for suppressing microglial activation in AD [129]. Thus, a suitable NSAID tha balancies the two opposing properties in terms of microglial activation is essential. Via targeting the BACE activity, the peroxisome proliferator-activated receptor gamma agonists, pioglitazone, prevented cytokine-dependent Aβ production, while the antagonists, GW0072, prevented NSAID-mediated amyloid formation [139-141]. NSAID regulated Rho-GTPases and restrained the reduction in axonal functioning and astroglial migration and activation in AD [142]. Another very interesting aspect is that the targeting of the IL-1 responsive element of 5’-untranslated region (5’UTR) of APP proved responsible for driving APP translation [143]. These drugs are also conjectured to target the interaction of AU-rich protein with the 3’-UTR of IL-1 and TNFα, or the thalidomides that block the cytokine translation [143]. Thus, targeting the NSAID-dependent cytokines as well as APP translation appears quite promising. However, long duration treatments with COX inhibitors were found to cause damage to heart, kidney, intestine and other organs [144-146]. Moreover, the belief that NSAIDs function in the ApoEε4-carrying AD population restricts the use of NSAID in other AD patients [147]. Thus, these limitations associated with the anti-inflammatory targets enlighten the need for newer compounds and drugs for AD.
Targeting metals and oxidative stress (Table 3)
Table 3.
Important publications on Antioxidants and Mitochondrial targets as AD therapeautics and preventive agents
| Anti-Oxidative stress agents | Therapeutics | References |
|---|---|---|
| Antioxidants | Vitamin C | Sano et al., 1997, Zandi et al., 2004 |
| Carotenoids | Javed et al., 2012; Vijayapadma et al., 2014 | |
| Resveratrol | Joseph et al., 2003; Ho et al., 2009 | |
| Curcumin | Yang et al., 2005, Ringman et al., 2012 | |
| Neu-P11 | He et al., 2013 | |
| Green tea and food additives | Kim et al., 2009; Zhao et al., 1989; Goodman et al., 1994; Iuvone et al., 2006 | |
| Mitochondrial targeting | Coenzyme Q10 (CoQ10) | Lee et al., 2009 |
| a-lipoic acid | Siedlak et al., 2009 | |
| MitoQ and plastoquinone | Kapay et al., 2011; McManus et al., 2011 | |
| SS31 | Calkins et al., 2011 | |
| Nrf2/ARE pathway targets | Tertbutylhydroquinone | Ramsey et al., 2007; Kanninen et al., 2008; Dumont et al., 2012 |
| Adenovirus-dependent gene delivery | Kanninen et al., 2008; Dumont et al., 2012 | |
| Metal Chelation | Desferrioxamine, EDTA and Clioquinol | Mandel et al., 2007; Amit et al., 2008; Hegde et al., 2009 |
Trapping of Fe, Cu and Zn ions within the amyloid plaques and their interactions with APP and Aβ are prominent mechanisms accelerating the amyloid pathology [143]. Several experimental techniques, such as proton-induced X-ray emission, epifluorescence microscopy, immersion autometallography [148], synchrotron X-ray fluorescence (SXRF), magnetic resonance imaging (MRI), susceptibility weighted MR (SWI), and laser capture microdissection coupled with X-ray fluorescence microscopy confirmed the metal localization in concentrations of ~15 μM for Cu2+ and about 1 mM for iron and zink [149-152]. Aluminium is another metal that has emerged as an important participant in AD [152]. Catalytic reactions involving the generation of neurotoxic hydrogen peroxide (H2O2) and superoxide ion generation are the major metal-mediated mechanism of Aβ generation. Thus, superoxide dismutase-1 and co-enzyme Q that possess endogenous anti-oxidant properties are assessed for their usefulness in animal models exposed to oxidative stress, and are also being tested for efficacy in AD [153]. In the current review section, we will discuss the specific participation of these heavy metals in the AD pathogenesis, followed by the therapeutics targeting the metals and their induced mechanism.
Fe
Cortical and hippocampal over-expression of hemeoxygenase (HO-1) promotes heme conversion to Fe2+ inducing the mitochondrial insufficiency, enhancing cytochrome C oxidase activity and H2O2 generation [154]. Furthermore, an involvement of Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH- + OH•) in neurons and astroglia triggers free radical generation and oxidative stress, which then activates neurotoxic mechanisms associated with nuclear factor-κB, p53, c-Jun transcription factors, DNA damage and apoptosis [155]. These oxidative stress mechanisms culminate in BBB disruption [156], myelin breakdown and eventually cognitive decline in AD [157]. An IRE-IRP binding that regulates APP translation is also altered due to excess iron accumulation via formation of an IRP-1 and [4Fe-4S] cluster that deregulate iron uptake and APP translation [143]. A resemblance of APP 5’-UTR sequence with the ferritin IRE stemloop strongly supportes the intense participation of iron in the pathology. Moreover, an iron binding site was also found in the APP 5’-UTR, corroborating the concept of Fe-regulated APP and thereby Aβ generation [158,159]. Furthermore, the iron-mediated down-regulation of α-secretase activator furin was also shown to participate in iron homeostasis [160].
Cu
The fact that copper ions bind to histidine, aspartate and tyrosine- residues in Aβ with a dissociation constant at the attomolar levels, and the Cu-promoted stimulation of Fenton reaction strongly support the participation of Cu in oxidative stress-mediated AD pathogenesis [161]. The released H2O2 interacts with copper tyrosinate that cross-links with Aβ, further enhancing Aβ generation [162]. The process of Cu-mediated toxicity during AD pathogenesis was proven to be stimulated by an interaction of copper ions with membrane lipid rafts, altering endocytosis of APP that bears the features of a Cu transporter [163].
Zn
A Micro-PIXE study revealed the Zn(II) levels of around 70-90 μg/g, amounting to about 1020-1060 μM concentration in the plaque rim, core and the total plaques [143]. The zink level as high as 1 mM was also reported in cerebral amyloid plaques, with a pre-synaptic vesicle to postsynaptic neuronal discharge [164]. The APP was found to have a Zn-binding domain located between the amino acids 181 and 200, leading to its binding to the amino acid 6-8 in Aβ [165]. The alteration in Zn release involvesan enhanced expression of zinc transporter proteins (ZNTs, ZnT2-8) that further promotes abnormal Zn deposition in the AD brain [166]. This change in Zn homeostasis alters its normal functions, influencing APP interaction with heparin-like molecules or laminin that participates in the biological process of neurite outgrowth [167]. Zinc deposition in Aβ promotes γ-secretase activity via enhanced presenilin formation, and also activates transcription factors nuclear factor kappa-B and Specificity protein-1 that bind to APP promoter, thereby promoting amyloidogenic APP synthesis [168].
Al
Studies in APP transgenic Tg2576 mice revealed an increase in insoluble Aβ deposition upon dietary Al supplementation and consumption of Al-treated drinking water [169]. This was supported by an increased Al levels within Aβ core plaques peptides of aluminium dust-exposed workers [170]. Apart from inducing oxidative stress, aluminium disrupts neuronal intracellular Ca2+ release and stimulates DNA damage during AD pathology [171]. Because of the explicit role of transition metals in AD, metal chelation in modifying the AD progression is being widely accepted. Desferrioxamine, EDTA and clioquinol were found to be effective in attenuating AD pathogenesis in transgenic AD mice, which also proved successful in clinical trials [172-174]. The metals function via an oxidative stress mechanism, and hence antioxidant treatments are deemed as a promising preventive approach in mitigating AD.
Oxidative stress attenuation: antioxidant treatment
Animal studies on APP and PS1 transgenic models, and Aβ over-expressing animals revealed an anti-oxidant-mediated cognitive restoration associated with a reduction in hippocampal and cortical Aβ deposition. The antioxidants mainly studied include the vitamins (carotenoids, vitamins C and E, lipoic acid, coenzyme Q10, N-acetylcysteine, polyphenols and Ginko biloba extract [175]. An attenuation in reactive oxygen species (ROS) generation and Aβ-induced neurotoxicity in cultured primary neuronal cultures and cell lines, such as PC12 cells, as well as in Tg 2576 AD transgenic mice, was observed using vitamin E [176,177]. The effect of vitamin E was more pronounced when combined with anti-inflammatory agents, as reported for indomethacin [178]. Patient studies revealed tocopherol-mediated attenuation in AD pathology, and its combination with vitamin C augmented the preventive effect [179,180]. Combining vitamin E with memantine and donepezil, however, failed to add to the protective effect of vitamin E alone [181,182]. Mitochondrial targeting aimed at mitigating the ROS generation appeared as a pertinent anti-oxidant mechanism, where coenzyme Q10 (CoQ10, ubiquinone) played a potent role [183]. However, the effect of ubiquinone or its water-soluble cognate, idebenone, though appealing in the pre-clinical studies, failed to prove effective in clinical trials [184,185]. Treatment with the mitochondrial antioxidant, α-lipoic acid, significantly improved cognition in Tg2576 transgenic AD mice [186]. This was supported by patient data, at a level comparable to AChE drug therapy [187]. A combination of lipoic acid with the anti-oxidant, omega-3-fatty acid, appeared potent in slowing down the functional cognitive decline [188]. In vitro effect of lipoic acid in inhibiting the Aβ oligomerization was also reported [189]. Other mitochondrial antioxidants, such as MitoQ and plastoquinone, prevented oxidative stress, the astroglia-induced neuroinflammation and, ultimately, AD pathology [190,191]. Plastoquinone also prevented hippocampal modulations of Long Time Potentiation in rats [191], and SS31 prevented alterations in the mitochondrial dynamics of transgenic AD mice neurons [192]. MitoVitE was found to be an advanced and more effective form of MitoQ [193]. Mitochondrial permeability transition pore (mPTP), that regulated mitochondrial functions, was also targeted. Dimebon, which appeared to be promising in this context, failed in clinical trials, probably because of its complex mechanism of action, which involves AChE reduction and 5-HT4 stimulation on one hand, and 5-HT6 and 5-HT3 activations that impaires cognition [194]. N-acetylcysteine (NAC) is another antioxidant that reduced malondialdehyde, increased glutathione and restored the LTP [195,196]. Interestingly, a study revealed that a combination of NAC and lipoic acid, along with curcumin, epigallocatechin gallate and the anti-oxidant vitamins is a prominent inducer of normal cognitive functioning in Tg2576 mice [197]. Flavonoids and carotenoids themselves were also widely studied as preventive agents in AD [194]. Natural flavonoid and carotenoid, namely rutin and lutein, prevented dementia [198,199]. Curcumin proved important in reducing Aβ and AChE in animal studies; however, the human studies were not so promising [200,201]. Resveratrol containing blueberry [202] and red grape [203] caused attenuation in Aβ plaque deposition, with epidemiological surveys proving the red wine-induced reduced memory loss in AD [204]. However, all these natural antioxidants could be claimed effective after completion of clinical trials [194]. Supplementation with the antioxidant melatonin that quenches free radicals reduced fibrillar amyloid burdens and prevented neurodegeneration via reduction of Aβ-induced neuronal apoptosis, with observed protection in human studies [205]. A melatonin agonist Neu-P11 was found to be effective in rats [206]. Alkaloid and a flavonoid derivative, Silibinin, Ginkgo biloba and a long-terncaffeine intake proved useful against Aβ-induced damage in transgenic animals [207-210]. Green tea and food additives, such as theanine, rosamarinic acid and nordihydroguiaretic acid had both antioxidant properties and anti-amyloid features as well [211-214]. An endogenous antioxidant mechanism targeting the nuclear receptor factor 2 (Nrf2)/antioxidant response element (ARE) pathway also seems to be a potential alternative approach to attenuate the AD pathology [215]. Tertbutylhydroquinone or adenovirus-dependent gene delivery along with the triterpenoid CDDO-methylamide that stimulated Nrf2 expression and translocation protected against AD pathology [216,217]. Overall, the general prevailing view is that although oxidative stress is a major risk factor for AD, the therapeutic effects of antioxidants alone in clinical trials appear less promising. Probably, the antioxidants bioavailability, water and lipid solubility, mechanism of action, time and duration of treatment, and combined use are to be taken into consideration for further preclinical and clinical studies.
Targeting hypercholesterolemia (Table 4)
Table 4.
Important publications on cholesterol-lowering drugs as AD therapeautics
| Anti-cholesterol agents | Therapeutics | References |
|---|---|---|
| Statin | Simvastatin | Shinohara et al., 2014 |
| Lovastatin | Friedhoff et al., 2001; Buxbaum et al., 2002 | |
| Atorvastatin, Cerivastatin, Fluvastatin, Pravastatin, Rosuvastatin | Barone et al., 2014 |
APP is a transmembrane protein, and cholesterol is an integral component of the lipid membrane. Hence, the changes in cholesterol level affect the lipid raft proteins, thereby impacting the APP as well [218]. The binding of cholesterol to CTFβ through the GXXXG motif is an important factor that brings APP, γ- and β-secretases close together and enhances the amyloidogenic cleavage [219]. Since the major non-amyloidogenic component, α-secretase, is not a part of the lipid raft, cholesterol contributes greater to the amyloidogenic cleavage pathway of APP [219]. Noticeably, APP is also closely associated with the cholesterol biosynthesis regulatory enzymes - sterol receptor element binding protein (SREBP) and HMG-CoA reductase [220]. Participation of cholesterol in Aβ aggregation was also reported through its interaction with the ganglioside GM1 found in lipid rafts of CNS [221]. Furthermore, localization of Aβ degrading enzymes, insulin-degrading enzyme (IDE) and neprisylin (NPE) and plasmin in the lipid rafts points towards the role of cholesterol in influencing the Aβ degradation [222]. Aβ clearance is also influenced by ApoE polymorphic allelles, ε4, ε3 and ε2, that participate in the cholesterol transport and impact the brain cholesterol homeostasis [223]. ApoE participates in neuroinflammation by modulating the toll like recetor and the nuclear factor kappaB pathways [224]. In fact, it is believed that the effect of APOE on BBB is inflammation-mediated, since its integral cell component, astrocytes, induce inflammation when activated [225]. Increased levels of oxysterols interact with APP and Aβ, with several hydroxylated cholesterol forms identified in the AD brain [226]. It is believed that the oxysterols in the form of 24- and 25-hydroxycholesterol (24-OH) that, unlike cholesterol, can cross the BBB, actually mediate the effects of cholesterol in the AD pathogenesis [226]. A significant overlap of cholesterol and metal-mediated AD pathology was also observed. Firstly, the capacity of both metals and cholesterol to bind monomeric Aβ and induce its oligomerization emerged as a good explanation [219]. Cholesterol itself undergoes oxidation that may enhance the Aβ generation [227]. Secondly, the IDE and NEP require metals for their functioning [219]. Generation of oxysterol was also conjectured to be metal-oxidative and stress-dependent [228]. Thus, anincreased dietary cholesterol and metal exposure, is hypothesized to promote Aβ generation and reduce Aβ degradation in a synergistc way, thereby enhancing plaque formation.
Cholesterol lowering drugs/statins: Because of the well-proven link between cholesterol and AD, statins are believed to be therapeutic for the disease. Simvastatin, via increased PI3K/AKT activity and endothelial NO synthase pathway, contributes towards inhibiting the learning and memory impairment in the Tg2576 mice [229]. However, simvastatin has no effect on the Aβ level in brain. Rather, administration of simvastatin to Aβ-immunized mice exacerbated the amyloid angiopathy [230]. The suppression of cholesterol metabolism with the help of HMG-CoA reductase inhibitor lovastatin, or its active metabolite lovastatin acid at 10-60 mg once-daily dose, caused dose-dependent Aβ reduction in human subjects [231]. Epidemiological studies have demonstrated that hypercholesterolemia was a risk factor for AD, and lovastatin caused a delayed AD onset and attenuated AD development [231,232]. Atorvastatin was reported to prevent the neuronal degeneration following Aβ induction. The effects of both atorvastatin and pitavastatin were mediated by attenuation of inflammation and oxidative stress and increase in the glutamatergic transporters [229,233]. Statins were also found to suppress inflammation and microglial iNOS synthesis and NO generation in AD via pleiotropic actions involving isoprenyl intermediates [234]. The neuroprotective pleiotropic effects of statin included an increase in SOD activity, activation of PKC, augmentation of endothelial nitric oxide synthase (eNOS) and reduction of CoQ10 levels [235,236]. The effects observed in clinical trials were analyzed for atorvastatin, cerivastatin, fluvastatin, pravastatin, rosuvastatin and simvastatin and were found to be independent of apoE genotype [237]. There were several controversies in regards to the statins’ beneficial role, however. It was later deduced that, despite lower reported AD incidence among the statin-treated users, there is no direct link between statins and the AD risk and development. Moreover, contradicting the phase II study, phase III randomized clinical trials showed a less beneficial role of statins [237]. Thus, it is suggested that clinical trials with a large patient population, different duration and different stages of disease are needed to find out the exact role of the cholesterol-reducing agents in AD. An up-regulation of the heme oxygenase/biliverdin reductase system, probably via inhibition of BACE1, is a likelypossible pathway for inhibiting the AD progression. It is hypothesized that the effects of simvastatin, lovastatin, atorvastatin and rosuvastatin are based on this mechanism [238]. Although still debatable, the use of statins may provide a useful strategy for suppressing the AD progression.
Future directions and conclusion
The present review gives an insight into the major pathophysiological risk factors promoting AD. Overall, it is observed that despite elaborate knowledge of the risk factors and mechanism of AD, only a modest choice of therapeutic tools is available for management, prevention, mitigation and treatment of the disease. Presently, symptomatic treatments are the most prevalent, and multiple studies are in progress to identify specific drugs targeting AD. With all the progress made so far, clinical trials have accepted the AChE inhibitors and a single NMDA receptor agonist as the probable AD therapeutics. AChE inhibitors, donepezil, ganthamine and rivastigmine are used in mild to moderate AD. In severe AD or in patients not responding to AChE inhibitors, memantine can be used as an alternative (Table 5). A combination of cholinesterase inhibitors, with memantine is also accepted as a treatment option, where the cholinergic drugs appeared more clinically beneficial even at the late stage of the disease [239]. Yet, it remains unclear at which stage of the disease the drugs should be started and which sequence of treatments should be used. A lack of knowledge on the AD-specific pharmacokinetics and bioavailability has led to a reasonably lowacceptance of the antioxidants, statins and anti-inflammatory agents as AD therapeutics. An understanding of their usage-dose, latency period, stage at which efficacy is at the peak and the genetic impact may help in proceeding with the agents reducing oxidative stress, inflammation and hyperlipidemia. Targeting BACE and presenilin, the components of the γ-secretase pathway, is envisaged as reasonable approach for inhibiting the amyloidogenic pathway of APP processing. Alternatively, stimulation of signaling pathways triggering the α-secretase-based non-amyloidogenic pathway appears as a reasonable strategy. A very optimistic approach is the targeting of APP 5’-UTR. Worldwide, the drug libraries are being screened for that purpose, with the aim of identifying both the inhibitors of amyloidogenic and the promoters of non-amyloidogenic pathways of Aβ. Vaccine-based approaches are also designed, bringing the attention to the usefulness of Aβ immunotherapy. The use of amyloid anti-aggregant strategies is also being investigated. Though clinical trials for the immunotherapies are in progress, detailed clinical studies on patients are yet awaited. However, we look forward to further research and clinical trials towards understanding and identifying novel independent and interdependent strategies that modulate amyloid metabolism in attenuating Aβ in AD.
Table 5.
FDA-approved drugs as AD therapeutics
| Drugs | Mode of action | AD symptoms |
|---|---|---|
| Donepezil | AChE inhibitor | Mild to severe AD |
| Memantine | NMDAR antagonist | Moderate to severe AD |
| Rivastigmine | AChE and BuChE inhibitor | Alzheimer-like pathology |
| Galantamine | Cholinergic inhibitor | Alzheimer-like pathology |
Disclosure of conflict of interest
None.
References
- 1.Coria F, Rubio I, Bayon C. Alzheimer’s disease, beta-amyloidosis, and aging. Rev Neurosci. 1994;5:275–292. doi: 10.1515/revneuro.1994.5.4.275. [DOI] [PubMed] [Google Scholar]
- 2.Mattila J, Soininen H, Koikkalainen J, Rueckert D, Wolz R, Waldemar G, Lotjonen J. Optimizing the diagnosis of early Alzheimer’s disease in mild cognitive impairment subjects. J Alzheimers Dis. 2012;32:969–979. doi: 10.3233/JAD-2012-120934. [DOI] [PubMed] [Google Scholar]
- 3.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M Alzheimer’s Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haan MN, Wallace R. Can dementia be prevented? Brain aging in a population-based context. Annu Rev Public Health. 2004;25:1–24. doi: 10.1146/annurev.publhealth.25.101802.122951. [DOI] [PubMed] [Google Scholar]
- 5.Dufouil C, Alperovitch A. [Epidemiology of Alzheimer’s disease] . Rev Prat. 2005;55:1869–1878. [PubMed] [Google Scholar]
- 6.Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer's disease. Annu Rev Genomics Hum Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- 7.Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci. 2015;9:124. doi: 10.3389/fncel.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castellani RJ, Perry G. The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol. 2014;88:671–676. doi: 10.1016/j.bcp.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Jia Q, Deng Y, Qing H. Potential therapeutic strategies for Alzheimer's disease targeting or beyond beta-amyloid: insights from clinical trials. Biomed Res Int. 2014;2014:837157. doi: 10.1155/2014/837157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbogast CE, Welleford EA, Netting FE. State Dementia Plans and the Alzheimer’s Disease Movement: Framing Diagnosis, Prognosis, and Motivation. J Appl Gerontol. 2015 doi: 10.1177/0733464815602112. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.Iadecola C. Cerebrovascular effects of amyloidbeta peptides: mechanisms and implications for Alzheimer's dementia. Cell Mol Neurobiol. 2003;23:681–689. doi: 10.1023/A:1025092617651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thal DR, Griffin WS, Braak H. Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J Cell Mol Med. 2008;12:1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta. 2011;1812:507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canobbio I, Catricala S, Balduini C, Torti M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim Biophys Acta. 2011;1813:500–506. doi: 10.1016/j.bbamcr.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 18.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer’s disease. Am J Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17:676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orsucci D, Mancuso M, Ienco EC, Simoncini C, Siciliano G, Bonuccelli U. Vascular factors and mitochondrial dysfunction: a central role in the pathogenesis of Alzheimer’s disease. Curr Neurovasc Res. 2013;10:76–80. doi: 10.2174/156720213804805972. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y. Apolipoprotein E and Alzheimer disease. Neurology. 2006;66:S79–85. doi: 10.1212/01.wnl.0000192102.41141.9e. [DOI] [PubMed] [Google Scholar]
- 23.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33:1500–1513. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou Z, Liu C, Che C, Huang H. Clinical genetics of Alzheimer’s disease. Biomed Res Int. 2014;2014:291862. doi: 10.1155/2014/291862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zadori D, Veres G, Szalardy L, Klivenyi P, Toldi J, Vecsei L. Glutamatergic dysfunctioning in Alzheimer’s disease and related therapeutic targets. J Alzheimers Dis. 2014;42(Suppl 3):S177–187. doi: 10.3233/JAD-132621. [DOI] [PubMed] [Google Scholar]
- 26.Verhoeff NP. Acetylcholinergic neurotransmission and the beta-amyloid cascade: implications for Alzheimer’s disease. Expert Rev Neurother. 2005;5:277–284. doi: 10.1586/14737175.5.2.277. [DOI] [PubMed] [Google Scholar]
- 27.Luchetti S, Huitinga I, Swaab DF. Neurosteroid and GABA-A receptor alterations in Alzheimer's disease, Parkinson’s disease and multiple sclerosis. Neuroscience. 2011;191:6–21. doi: 10.1016/j.neuroscience.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Zlomuzica A, Dere D, Binder S, De Souza Silva MA, Huston JP, Dere E. Neuronal histamine and cognitive symptoms in Alzheimer’s disease. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.05.007. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer’s disease. Prog Neurobiol. 2012;99:15–41. doi: 10.1016/j.pneurobio.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 30.Vasto S, Candore G, Listi F, Balistreri CR, Colonna-Romano G, Malavolta M, Lio D, Nuzzo D, Mocchegiani E, Di Bona D, Caruso C. Inflammation, genes and zinc in Alzheimer’s disease. Brain Res Rev. 2008;58:96–105. doi: 10.1016/j.brainresrev.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Bush AI. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 32.Daneschvar HL, Aronson MD, Smetana GW. Do statins prevent Alzheimer’s disease? A narrative review. Eur J Intern Med. 2015;26:666–9. doi: 10.1016/j.ejim.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomsen T, Zendeh B, Fischer JP, Kewitz H. In vitro effects of various cholinesterase inhibitors on acetyl- and butyrylcholinesterase of healthy volunteers. Biochem Pharmacol. 1991;41:139–141. doi: 10.1016/0006-2952(91)90022-w. [DOI] [PubMed] [Google Scholar]
- 35.Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36:375–399. doi: 10.1007/s12272-013-0036-3. [DOI] [PubMed] [Google Scholar]
- 36.Nagabukuro H, Hashimoto T, Iwata M, Doi T. Effects of TAK-802, a novel acetylcholinesterase inhibitor, and tamsulosin, an alpha1-adrenoceptor antagonist, and their synergistic effects on the urodynamic characteristics in a guinea-pig model of functional bladder outlet obstruction. BJU Int. 2005;95:1071–1076. doi: 10.1111/j.1464-410X.2005.05469.x. [DOI] [PubMed] [Google Scholar]
- 37.Park SM, Ki SH, Han NR, Cho IJ, Ku SK, Kim SC, Zhao RJ, Kim YW. Tacrine, an oral acetylcholinesterase inhibitor, induced hepatic oxidative damage, which was blocked by liquiritigenin through GSK3-beta inhibition. Biol Pharm Bull. 2015;38:184–192. doi: 10.1248/bpb.b14-00430. [DOI] [PubMed] [Google Scholar]
- 38.Minarini A, Milelli A, Simoni E, Rosini M, Bolognesi ML, Marchetti C, Tumiatti V. Multifunctional tacrine derivatives in Alzheimer’s disease. Curr Top Med Chem. 2013;13:1771–1786. doi: 10.2174/15680266113139990136. [DOI] [PubMed] [Google Scholar]
- 39.Li C, Carlier PR, Ren H, Kan KK, Hui K, Wang H, Li W, Li Z, Xiong K, Clement EC, Xue H, Liu X, Li M, Pang Y, Han Y. Alkylene tether-length dependent gamma-aminobutyric acid type A receptor competitive antagonism by tacrine dimers. Neuropharmacology. 2007;52:436–443. doi: 10.1016/j.neuropharm.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 40.Colovic MB, Krstic DZ, Lazarevic-Pasti TD, Bondzic AM, Vasic VM. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol. 2013;11:315–335. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero A, Cacabelos R, Oset-Gasque MJ, Samadi A, Marco-Contelles J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett. 2013;23:1916–1922. doi: 10.1016/j.bmcl.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 42.Alonso D, Dorronsoro I, Rubio L, Munoz P, Garcia-Palomero E, Del Monte M, Bidon-Chanal A, Orozco M, Luque FJ, Castro A, Medina M, Martinez A. Donepezil-tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg Med Chem. 2005;13:6588–6597. doi: 10.1016/j.bmc.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 43.Guzior N, Wieckowska A, Panek D, Malawska B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem. 2015;22:373–404. doi: 10.2174/0929867321666141106122628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lupp A, Appenroth D, Fang L, Decker M, Lehmann J, Fleck C. Tacrine-NO donor and tacrine-ferulic acid hybrid molecules as new anti-Alzheimer agents: hepatotoxicity and influence on the cytochrome P450 system in comparison to tacrine. Arzneimittelforschung. 2010;60:229–237. doi: 10.1055/s-0031-1296278. [DOI] [PubMed] [Google Scholar]
- 45.Marco-Contelles J, Leon R, de Los Rios C, Guglietta A, Terencio J, Lopez MG, Garcia AG, Villarroya M. Novel multipotent tacrine-dihydropyridine hybrids with improved acetylcholinesterase inhibitory and neuroprotective activities as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2006;49:7607–7610. doi: 10.1021/jm061047j. [DOI] [PubMed] [Google Scholar]
- 46.Huang L, Su T, Shan W, Luo Z, Sun Y, He F, Li X. Inhibition of cholinesterase activity and amyloid aggregation by berberine-phenyl-benzoheterocyclic and tacrine-phenyl-benzoheterocyclic hybrids. Bioorg Med Chem. 2012;20:3038–3048. doi: 10.1016/j.bmc.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 47.Cacabelos R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat. 2007;3:303–333. [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto H, Iimura Y, Yamanishi Y, Yamatsu K. Synthesis and structure-activity relationships of acetylcholinesterase inhibitors: 1-benzyl-4-[(5,6-dimethoxy-1-oxoindan-2-yl)methyl] piperidine hydrochloride and related compounds. J Med Chem. 1995;38:4821–4829. doi: 10.1021/jm00024a009. [DOI] [PubMed] [Google Scholar]
- 49.Pang YP, Kozikowski AP. Prediction of the binding site of 1-benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl] piperidine in acetylcholinesterase by docking studies with the SYSDOC program. J Comput Aided Mol Des. 1994;8:683–693. doi: 10.1007/BF00124015. [DOI] [PubMed] [Google Scholar]
- 50.Hopkins TJ, Rupprecht LE, Hayes MR, Blendy JA, Schmidt HD. Galantamine, an acetylcholinesterase inhibitor and positive allosteric modulator of nicotinic acetylcholine receptors, attenuates nicotine taking and seeking in rats. Neuropsychopharmacology. 2012;37:2310–2321. doi: 10.1038/npp.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Albuquerque EX, Santos MD, Alkondon M, Pereira EF, Maelicke A. Modulation of nicotinic receptor activity in the central nervous system: a novel approach to the treatment of Alzheimer disease. Alzheimer Dis Assoc Disord. 2001;15(Suppl 1):S19–25. doi: 10.1097/00002093-200108001-00004. [DOI] [PubMed] [Google Scholar]
- 52.Greenblatt HM, Kryger G, Lewis T, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 A resolution. FEBS Lett. 1999;463:321–326. doi: 10.1016/s0014-5793(99)01637-3. [DOI] [PubMed] [Google Scholar]
- 53.Bolognesi ML, Bartolini M, Cavalli A, Andrisano V, Rosini M, Minarini A, Melchiorre C. Design, synthesis, and biological evaluation of conformationally restricted rivastigmine analogues. J Med Chem. 2004;47:5945–5952. doi: 10.1021/jm049782n. [DOI] [PubMed] [Google Scholar]
- 54.Rodrigues Simoes MC, Dias Viegas FP, Moreira MS, de Freitas Silva M, Riquiel MM, da Rosa PM, Castelli MR, dos Santos MH, Soares MG, Viegas C Jr. Donepezil: an important prototype to the design of new drug candidates for Alzheimer’s disease. Mini Rev Med Chem. 2014;14:2–19. doi: 10.2174/1389557513666131119201353. [DOI] [PubMed] [Google Scholar]
- 55.Toda N, Tago K, Marumoto S, Takami K, Ori M, Yamada N, Koyama K, Naruto S, Abe K, Yamazaki R, Hara T, Aoyagi A, Abe Y, Kaneko T, Kogen H. Design, synthesis and structureactivity relationships of dual inhibitors of acetylcholinesterase and serotonin transporter as potential agents for Alzheimer’s disease. Bioorg Med Chem. 2003;11:1935–1955. doi: 10.1016/s0968-0896(03)00091-9. [DOI] [PubMed] [Google Scholar]
- 56.Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P. Cholinesterase inhibitors: xanthostigmine derivatives blocking the acetylcholinesterase-induced beta-amyloid aggregation. J Med Chem. 2005;48:4444–4456. doi: 10.1021/jm049515h. [DOI] [PubMed] [Google Scholar]
- 57.Anand P, Singh B. Synthesis and evaluation of novel 4-[(3H,3aH,6aH)-3-phenyl)-4,6-dioxo-2-phenyldihydro-2H-pyrrolo[3,4-d] isoxazol-5(3H,6H,6aH)-yl] benzoic acid derivatives as potent acetylcholinesterase inhibitors and anti-amnestic agents. Bioorg Med Chem. 2012;20:521–530. doi: 10.1016/j.bmc.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 58.Razavi SF, Khoobi M, Nadri H, Sakhteman A, Moradi A, Emami S, Foroumadi A, Shafiee A. Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem. 2013;64:252–259. doi: 10.1016/j.ejmech.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 59.Rizzo S, Bartolini M, Ceccarini L, Piazzi L, Gobbi S, Cavalli A, Recanatini M, Andrisano V, Rampa A. Targeting Alzheimer’s disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorg Med Chem. 2010;18:1749–1760. doi: 10.1016/j.bmc.2010.01.071. [DOI] [PubMed] [Google Scholar]
- 60.Shen Y, Zhang J, Sheng R, Dong X, He Q, Yang B, Hu Y. Synthesis and biological evaluation of novel flavonoid derivatives as dual binding acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem. 2009;24:372–380. doi: 10.1080/14756360802187885. [DOI] [PubMed] [Google Scholar]
- 61.Sawasdee P, Sabphon C, Sitthiwongwanit D, Kokpol U. Anticholinesterase activity of 7-methoxyflavones isolated from Kaempferia parviflora. Phytother Res. 2009;23:1792–1794. doi: 10.1002/ptr.2858. [DOI] [PubMed] [Google Scholar]
- 62.Walton HS, Dodd PR. Glutamate-glutamine cycling in Alzheimer’s disease. Neurochem Int. 2007;50:1052–1066. doi: 10.1016/j.neuint.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 63.Innocent N, Cousins SL, Stephenson FA. NMDA receptor/amyloid precursor protein interactions: a comparison between wild-type and amyloid precursor protein mutations associated with familial Alzheimer’s disease. Neurosci Lett. 2012;515:131–136. doi: 10.1016/j.neulet.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 64.Golde TE. Disease modifying therapy for AD? J Neurochem. 2006;99:689–707. doi: 10.1111/j.1471-4159.2006.04211.x. [DOI] [PubMed] [Google Scholar]
- 65.Farlow MR, Graham SM, Alva G. Memantine for the treatment of Alzheimer’s disease: tolerability and safety data from clinical trials. Drug Saf. 2008;31:577–585. doi: 10.2165/00002018-200831070-00003. [DOI] [PubMed] [Google Scholar]
- 66.Cummings JL. Treatment of Alzheimer’s disease: the role of symptomatic agents in an era of disease-modifying therapies. Rev Neurol Dis. 2007;4:57–62. [PubMed] [Google Scholar]
- 67.Gilling KE, Jatzke C, Hechenberger M, Parsons CG. Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology. 2009;56:866–875. doi: 10.1016/j.neuropharm.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 68.Danysz W, Parsons CG. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br J Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miguel-Hidalgo JJ, Paul IA, Wanzo V, Banerjee PK. Memantine prevents cognitive impairment and reduces Bcl-2 and caspase 8 immunoreactivity in rats injected with amyloid beta1-40. Eur J Pharmacol. 2012;692:38–45. doi: 10.1016/j.ejphar.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 70.Pohanka M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int J Mol Sci. 2012;13:2219–2238. doi: 10.3390/ijms13022219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dominguez E, Chin TY, Chen CP, Wu TY. Management of moderate to severe Alzheimer’s disease: focus on memantine. Taiwan J Obstet Gynecol. 2011;50:415–423. doi: 10.1016/j.tjog.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Prokselj T, Jerin A, Kogoj A. Memantine may affect pseudobulbar affect in patients with Alzheimer’s disease. Acta Neuropsychiatr. 2013;25:361–366. doi: 10.1017/neu.2013.14. [DOI] [PubMed] [Google Scholar]
- 73.Atri A, Molinuevo JL, Lemming O, Wirth Y, Pulte I, Wilkinson D. Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther. 2013;5:6. doi: 10.1186/alzrt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gauthier S, Molinuevo JL. Benefits of combined cholinesterase inhibitor and memantine treatment in moderate-severe Alzheimer’s disease. Alzheimers Dement. 2013;9:326–331. doi: 10.1016/j.jalz.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 75.Bell KF, de Kort GJ, Steggerda S, Shigemoto R, Ribeiro-da-Silva A, Cuello AC. Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci Lett. 2003;353:143–147. doi: 10.1016/j.neulet.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 76.Ulrich D. Amyloid-beta Impairs Synaptic Inhibition via GABA(A) Receptor Endocytosis. J Neurosci. 2015;35:9205–9210. doi: 10.1523/JNEUROSCI.0950-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jansen KL, Faull RL, Dragunow M, Synek BL. Alzheimer's disease: changes in hippocampal N-methyl-D-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors--an autoradiographic study. Neuroscience. 1990;39:613–627. doi: 10.1016/0306-4522(90)90246-z. [DOI] [PubMed] [Google Scholar]
- 78.Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, Schweighoffer F, Desire L. Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J Neurochem. 2008;106:392–404. doi: 10.1111/j.1471-4159.2008.05396.x. [DOI] [PubMed] [Google Scholar]
- 79.Cooney C, Murphy S, Tessema H, Freyne A. Use of low-dose gabapentin for aggressive behavior in vascular and Mixed Vascular/Alzheimer Dementia. J Neuropsychiatry Clin Neurosci. 2013;25:120–125. doi: 10.1176/appi.neuropsych.12050115. [DOI] [PubMed] [Google Scholar]
- 80.Adam F, Bordenave L, Sessler DI, Chauvin M. Effects of a single 1200-mg preoperative dose of gabapentin on anxiety and memory. Ann Fr Anesth Reanim. 2012;31:e223–227. doi: 10.1016/j.annfar.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 81.Lin TW, Kuo YM. Exercise benefits brain function: the monoamine connection. Brain Sci. 2013;3:39–53. doi: 10.3390/brainsci3010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen CP, Eastwood SL, Hope T, McDonald B, Francis PT, Esiri MM. Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioural changes. Neuropathol Appl Neurobiol. 2000;26:347–355. doi: 10.1046/j.1365-2990.2000.00254.x. [DOI] [PubMed] [Google Scholar]
- 83.Cochet M, Donneger R, Cassier E, Gaven F, Lichtenthaler SF, Marin P, Bockaert J, Dumuis A, Claeysen S. 5-HT4 receptors constitutively promote the non-amyloidogenic pathway of APP cleavage and interact with ADAM10. ACS Chem Neurosci. 2013;4:130–140. doi: 10.1021/cn300095t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hedlund PB, Sutcliffe JG. Functional, molecular and pharmacological advances in 5-HT7 receptor research. Trends Pharmacol Sci. 2004;25:481–486. doi: 10.1016/j.tips.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 85.Ramirez MJ. 5-HT6 receptors and Alzheimer’s disease. Alzheimers Res Ther. 2013;5:15. doi: 10.1186/alzrt169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Madjid N, Tottie EE, Luttgen M, Meister B, Sandin J, Kuzmin A, Stiedl O, Ogren SO. 5-Hydroxytryptamine 1A receptor blockade facilitates aversive learning in mice: interactions with cholinergic and glutamatergic mechanisms. J Pharmacol Exp Ther. 2006;316:581–591. doi: 10.1124/jpet.105.092262. [DOI] [PubMed] [Google Scholar]
- 87.Schechter LE, Smith DL, Rosenzweig-Lipson S, Sukoff SJ, Dawson LA, Marquis K, Jones D, Piesla M, Andree T, Nawoschik S, Harder JA, Womack MD, Buccafusco J, Terry AV, Hoebel B, Rada P, Kelly M, Abou-Gharbia M, Barrett JE, Childers W. Lecozotan (SRA-333): a selective serotonin 1A receptor antagonist that enhances the stimulated release of glutamate and acetylcholine in the hippocampus and possesses cognitive-enhancing properties. J Pharmacol Exp Ther. 2005;314:1274–1289. doi: 10.1124/jpet.105.086363. [DOI] [PubMed] [Google Scholar]
- 88.Sumiyoshi T, Park S, Jayathilake K, Roy A, Ertugrul A, Meltzer HY. Effect of buspirone, a serotonin1A partial agonist, on cognitive function in schizophrenia: a randomized, double-blind, placebo-controlled study. Schizophr Res. 2007;95:158–168. doi: 10.1016/j.schres.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 89.Miller LG, Thompson ML, Byrnes JJ, Greenblatt DJ, Shemer A. Kinetics, brain uptake, and receptor binding of tandospirone and its metabolite 1-(2-pyrimidinyl)-piperazine. J Clin Psychopharmacol. 1992;12:341–345. [PubMed] [Google Scholar]
- 90.Ramirez MJ, Lai MK, Tordera RM, Francis PT. Serotonergic therapies for cognitive symptoms in Alzheimer’s disease: rationale and current status. Drugs. 2014;74:729–736. doi: 10.1007/s40265-014-0217-5. [DOI] [PubMed] [Google Scholar]
- 91.Maher-Edwards G, Zvartau-Hind M, Hunter AJ, Gold M, Hopton G, Jacobs G, Davy M, Williams P. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res. 2010;7:374–385. doi: 10.2174/156720510791383831. [DOI] [PubMed] [Google Scholar]
- 92.Maher-Edwards G, Dixon R, Hunter J, Gold M, Hopton G, Jacobs G, Hunter J, Williams P. SB-742457 and donepezil in Alzheimer disease: a randomized, placebo-controlled study. Int J Geriatr Psychiatry. 2011;26:536–544. doi: 10.1002/gps.2562. [DOI] [PubMed] [Google Scholar]
- 93.Bezprozvanny I. The rise and fall of Dimebon. Drug News Perspect. 2010;23:518–523. doi: 10.1358/dnp.2010.23.8.1500435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, McKnight SL, Pieper AA, De Brabander JK, Ready JM. Development of proneurogenic, neuroprotective small molecules. J Am Chem Soc. 2011;133:1428–1437. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kendall I, Slotten HA, Codony X, Burgueno J, Pauwels PJ, Vela JM, Fone KC. E-6801, a 5-HT6 receptor agonist, improves recognition memory by combined modulation of cholinergic and glutamatergic neurotransmission in the rat. Psychopharmacology (Berl) 2011;213:413–430. doi: 10.1007/s00213-010-1854-3. [DOI] [PubMed] [Google Scholar]
- 96.Nagy CF, Kumar D, Perdomo CA, Wason S, Cullen EI, Pratt RD. Concurrent administration of donepezil HCl and sertraline HCl in healthy volunteers: assessment of pharmacokinetic changes and safety following single and multiple oral doses. Br J Clin Pharmacol. 2004;58(Suppl 1):25–33. doi: 10.1111/j.1365-2125.2004.01801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keowkase R, Aboukhatwa M, Luo Y. Fluoxetine protects against amyloid-beta toxicity, in part via daf-16 mediated cell signaling pathway, in Caenorhabditis elegans. Neuropharmacology. 2010;59:358–365. doi: 10.1016/j.neuropharm.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kohler CA, da Silva WC, Benetti F, Bonini JS. Histaminergic mechanisms for modulation of memory systems. Neural Plast. 2011;2011:328602. doi: 10.1155/2011/328602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dai H, Kaneko K, Kato H, Fujii S, Jing Y, Xu A, Sakurai E, Kato M, Okamura N, Kuramasu A, Yanai K. Selective cognitive dysfunction in mice lacking histamine H1 and H2 receptors. Neurosci Res. 2007;57:306–313. doi: 10.1016/j.neures.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 100.Threlfell S, Cragg SJ, Kallo I, Turi GF, Coen CW, Greenfield SA. Histamine H3 receptors inhibit serotonin release in substantia nigra pars reticulata. J Neurosci. 2004;24:8704–8710. doi: 10.1523/JNEUROSCI.2690-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Philippu A, Prast H. Importance of histamine in modulatory processes, locomotion and memory. Behav Brain Res. 2001;124:151–159. doi: 10.1016/s0166-4328(01)00226-1. [DOI] [PubMed] [Google Scholar]
- 102.Naddafi F, Mirshafiey A. The neglected role of histamine in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2013;28:327–336. doi: 10.1177/1533317513488925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Geng S, Gao YD, Yang J, Zou JJ, Guo W. Potential role of store-operated Ca2+ entry in Th2 response induced by histamine in human monocyte-derived dendritic cells. Int Immunopharmacol. 2012;12:358–367. doi: 10.1016/j.intimp.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 104.Mizuguchi H, Ono S, Hattori M, Fukui H. Inverse agonistic activity of antihistamines and suppression of histamine H1 receptor gene expression. J Pharmacol Sci. 2012;118:117–121. doi: 10.1254/jphs.11177SC. [DOI] [PubMed] [Google Scholar]
- 105.Motawaj M, Peoc’h K, Callebert J, Arrang JM. CSF levels of the histamine metabolite tele-methylhistamine are only slightly decreased in Alzheimer’s disease. J Alzheimers Dis. 2010;22:861–871. doi: 10.3233/JAD-2010-100381. [DOI] [PubMed] [Google Scholar]
- 106.Kharmate G, Liu Z, Patterson E, Khan MM. Histamine affects STAT6 phosphorylation via its effects on IL-4 secretion: role of H1 receptors in the regulation of IL-4 production. Int Immunopharmacol. 2007;7:277–286. doi: 10.1016/j.intimp.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baronio D, Gonchoroski T, Castro K, Zanatta G, Gottfried C, Riesgo R. Histaminergic system in brain disorders: lessons from the translational approach and future perspectives. Ann Gen Psychiatry. 2014;13:34. doi: 10.1186/s12991-014-0034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Passani MB, Blandina P. Cognitive implications for H3 and 5-HT3 receptor modulation of cortical cholinergic function: a parallel story. Methods Find Exp Clin Pharmacol. 1998;20:725–733. doi: 10.1358/mf.1998.20.8.487510. [DOI] [PubMed] [Google Scholar]
- 109.Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol Lett. 2008;117:198–202. doi: 10.1016/j.imlet.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 110.Bonotis K, Krikki E, Holeva V, Aggouridaki C, Costa V, Baloyannis S. Systemic immune aberrations in Alzheimer’s disease patients. J Neuroimmunol. 2008;193:183–187. doi: 10.1016/j.jneuroim.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 111.Rivera-Oliver M, Diaz-Rios M. Using caffeine and other adenosine receptor antagonists and agonists as therapeutic tools against neurodegenerative diseases: a review. Life Sci. 2014;101:1–9. doi: 10.1016/j.lfs.2014.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gahr M, Nowak DA, Connemann BJ, Schonfeldt-Lecuona C. Cerebral Amyloidal Angiopathy--a disease with implications for neurology and psychiatry. Brain Res. 2013;1519:19–30. doi: 10.1016/j.brainres.2013.04.052. [DOI] [PubMed] [Google Scholar]
- 113.Cupino TL, Zabel MK. Alzheimer’s silent partner: cerebral amyloid angiopathy. Transl Stroke Res. 2014;5:330–337. doi: 10.1007/s12975-013-0309-7. [DOI] [PubMed] [Google Scholar]
- 114.Zeitlin R, Patel S, Burgess S, Arendash GW, Echeverria V. Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res. 2011;1417:127–136. doi: 10.1016/j.brainres.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 115.Gelber RP, Petrovitch H, Masaki KH, Ross GW, White LR. Coffee intake in midlife and risk of dementia and its neuropathologic correlates. J Alzheimers Dis. 2011;23:607–615. doi: 10.3233/JAD-2010-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Melani A, Pantoni L, Bordoni F, Gianfriddo M, Bianchi L, Vannucchi MG, Bertorelli R, Monopoli A, Pedata F. The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res. 2003;959:243–250. doi: 10.1016/s0006-8993(02)03753-8. [DOI] [PubMed] [Google Scholar]
- 117.Azizi G, Navabi SS, Al-Shukaili A, Seyedzadeh MH, Yazdani R, Mirshafiey A. The Role of Inflammatory Mediators in the Pathogenesis of Alzheimer’s Disease. Sultan Qaboos Univ Med J. 2015;15:e305–316. doi: 10.18295/squmj.2015.15.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Meraz-Rios MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernandez J, Campos-Pena V. Inflammatory process in Alzheimer’s Disease. Front Integr Neurosci. 2013;7:59. doi: 10.3389/fnint.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ribizzi G, Fiordoro S, Barocci S, Ferrari E, Megna M. Cytokine polymorphisms and Alzheimer disease: possible associations. Neurol Sci. 2010;31:321–325. doi: 10.1007/s10072-010-0221-9. [DOI] [PubMed] [Google Scholar]
- 121.Lee SJ, Drabik K, Van Wagoner NJ, Lee S, Choi C, Dong Y, Benveniste EN. ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signalregulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol. 2000;165:4658–4666. doi: 10.4049/jimmunol.165.8.4658. [DOI] [PubMed] [Google Scholar]
- 122.Greer MM. Inflammation and the pathophysiology of Alzheimer’s disease. Dialogues Clin Neurosci. 2000;2:233–239. doi: 10.31887/DCNS.2000.2.3/mgreer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yeh ET. A new perspective on the biology of C-reactive protein. Circ Res. 2005;97:609–611. doi: 10.1161/01.RES.0000186188.38344.13. [DOI] [PubMed] [Google Scholar]
- 124.Guo JT, Yu J, Grass D, de Beer FC, Kindy MS. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J Neurosci. 2002;22:5900–5909. doi: 10.1523/JNEUROSCI.22-14-05900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Richardson C, Gard PR, Klugman A, Isaac M, Tabet N. Blood pro-inflammatory cytokines in Alzheimer’s disease in relation to the use of acetylcholinesterase inhibitors. Int J Geriatr Psychiatry. 2013;28:1312–1317. doi: 10.1002/gps.3966. [DOI] [PubMed] [Google Scholar]
- 127.Colton CA, Chernyshev ON, Gilbert DL, Vitek MP. Microglial contribution to oxidative stress in Alzheimer’s disease. Ann N Y Acad Sci. 2000;899:292–307. doi: 10.1111/j.1749-6632.2000.tb06195.x. [DOI] [PubMed] [Google Scholar]
- 128.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J Biol Chem. 1998;273:29719–29726. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 129.Koistinaho M, Kettunen MI, Goldsteins G, Keinanen R, Salminen A, Ort M, Bures J, Liu D, Kauppinen RA, Higgins LS, Koistinaho J. Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc Natl Acad Sci U S A. 2002;99:1610–1615. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shen Y, Yang L, Li R. What does complement do in Alzheimer’s disease? Old molecules with new insights. Transl Neurodegener. 2013;2:21. doi: 10.1186/2047-9158-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lue LF, Walker DG. Modeling Alzheimer’s disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- 132.Szekely CA, Zandi PP. Non-steroidal antiinflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol Disord Drug Targets. 2010;9:132–139. doi: 10.2174/187152710791012026. [DOI] [PubMed] [Google Scholar]
- 133.Hoozemans JJ, Veerhuis R, Janssen I, Rozemuller AJ, Eikelenboom P. Interleukin-1beta induced cyclooxygenase 2 expression and prostaglandin E2 secretion by human neuroblastoma cells: implications for Alzheimer’s disease. Exp Gerontol. 2001;36:559–570. doi: 10.1016/s0531-5565(00)00226-6. [DOI] [PubMed] [Google Scholar]
- 134.Blom MA, van Twillert MG, de Vries SC, Engels F, Finch CE, Veerhuis R, Eikelenboom P. NSAIDS inhibit the IL-1 beta-induced IL-6 release from human post-mortem astrocytes: the involvement of prostaglandin E2. Brain Res. 1997;777:210–218. doi: 10.1016/s0006-8993(97)01204-3. [DOI] [PubMed] [Google Scholar]
- 135.Choi SH, Aid S, Caracciolo L, Minami SS, Niikura T, Matsuoka Y, Turner RS, Mattson MP, Bosetti F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J Neurochem. 2013;124:59–68. doi: 10.1111/jnc.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA. NSAID and antioxidant prevention of Alzheimer’s disease: lessons from in vitro and animal models. Ann N Y Acad Sci. 2004;1035:68–84. doi: 10.1196/annals.1332.005. [DOI] [PubMed] [Google Scholar]
- 139.Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-beta and cerebrovascular pathology. PLoS One. 2013;8:e68612. doi: 10.1371/journal.pone.0068612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nenov MN, Laezza F, Haidacher SJ, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon BA, Dineley KT, Denner L. Cognitive enhancing treatment with a PPARgamma agonist normalizes dentate granule cell presynaptic function in Tg2576 APP mice. J Neurosci. 2014;34:1028–1036. doi: 10.1523/JNEUROSCI.3413-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kubo T, Yamaguchi A, Iwata N, Yamashita T. The therapeutic effects of Rho-ROCK inhibitors on CNS disorders. Ther Clin Risk Manag. 2008;4:605–615. doi: 10.2147/tcrm.s2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rogers JT, Lahiri DK. Metal and inflammatory targets for Alzheimer’s disease. Curr Drug Targets. 2004;5:535–551. doi: 10.2174/1389450043345272. [DOI] [PubMed] [Google Scholar]
- 144.Bleumink GS, Feenstra J, Sturkenboom MC, Stricker BH. Nonsteroidal anti-inflammatory drugs and heart failure. Drugs. 2003;63:525–534. doi: 10.2165/00003495-200363060-00001. [DOI] [PubMed] [Google Scholar]
- 145.Whelton A, Hamilton CW. Nonsteroidal antiinflammatory drugs: effects on kidney function. J Clin Pharmacol. 1991;31:588–598. doi: 10.1002/j.1552-4604.1991.tb03743.x. [DOI] [PubMed] [Google Scholar]
- 146.Satoh H, Takeuchi K. Management of NSAID/aspirin-induced small intestinal damage by GI-sparing NSAIDs, anti-ulcer drugs and food constituents. Curr Med Chem. 2012;19:82–89. doi: 10.2174/092986712803413980. [DOI] [PubMed] [Google Scholar]
- 147.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci. 2010;2 doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stoltenberg M, Bruhn M, Sondergaard C, Doering P, West MJ, Larsen A, Troncoso JC, Danscher G. Immersion autometallographic tracing of zinc ions in Alzheimer betaamyloid plaques. Histochem Cell Biol. 2005;123:605–611. doi: 10.1007/s00418-005-0787-0. [DOI] [PubMed] [Google Scholar]
- 149.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 150.Danscher G, Jensen KB, Frederickson CJ, Kemp K, Andreasen A, Juhl S, Stoltenberg M, Ravid R. Increased amount of zinc in the hippocampus and amygdala of Alzheimer’s diseased brains: a proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J Neurosci Methods. 1997;76:53–59. doi: 10.1016/s0165-0270(97)00079-4. [DOI] [PubMed] [Google Scholar]
- 151.Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol. 2006;155:30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 152.Yumoto S, Kakimi S, Ohsaki A, Ishikawa A. Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. J Inorg Biochem. 2009;103:1579–1584. doi: 10.1016/j.jinorgbio.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 153.Yang X, Yang Y, Li G, Wang J, Yang ES. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J Mol Neurosci. 2008;34:165–171. doi: 10.1007/s12031-007-9033-7. [DOI] [PubMed] [Google Scholar]
- 154.Perry G, Taddeo MA, Petersen RB, Castellani RJ, Harris PL, Siedlak SL, Cash AD, Liu Q, Nunomura A, Atwood CS, Smith MA. Adventiously-bound redox active iron and copper are at the center of oxidative damage in Alzheimer disease. Biometals. 2003;16:77–81. doi: 10.1023/a:1020731021276. [DOI] [PubMed] [Google Scholar]
- 155.Velez-Pardo C, Ospina GG, Jimenez del Rio M. Abeta[25-35] peptide and iron promote apoptosis in lymphocytes by an oxidative stress mechanism: involvement of H2O2, caspase-3, NF-kappaB, p53 and c-Jun. Neurotoxicology. 2002;23:351–365. doi: 10.1016/s0161-813x(02)00081-5. [DOI] [PubMed] [Google Scholar]
- 156.Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99:438–449. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- 157.Lu PH, Lee GJ, Raven EP, Tingus K, Khoo T, Thompson PM, Bartzokis G. Age-related slowing in cognitive processing speed is associated with myelin integrity in a very healthy elderly sample. J Clin Exp Neuropsychol. 2011;33:1059–1068. doi: 10.1080/13803395.2011.595397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thomson AM, Rogers JT, Leedman PJ. Ironregulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol. 1999;31:1139–1152. doi: 10.1016/s1357-2725(99)00080-1. [DOI] [PubMed] [Google Scholar]
- 159.Pinero DJ, Hu J, Connor JR. Alterations in the interaction between iron regulatory proteins and their iron responsive element in normal and Alzheimer’s diseased brains. Cell Mol Biol (Noisy-le-grand) 2000;46:761–776. [PubMed] [Google Scholar]
- 160.Silvestri L, Camaschella C. A potential pathogenetic role of iron in Alzheimer’s disease. J Cell Mol Med. 2008;12:1548–1550. doi: 10.1111/j.1582-4934.2008.00356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Atwood CS, Huang X, Khatri A, Scarpa RC, Kim YS, Moir RD, Tanzi RE, Roher AE, Bush AI. Copper catalyzed oxidation of Alzheimer Abeta. Cell Mol Biol (Noisy-le-grand) 2000;46:777–783. [PubMed] [Google Scholar]
- 162.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 163.Hayashi T, Shishido N, Nakayama K, Nunomura A, Smith MA, Perry G, Nakamura M. Lipid peroxidation and 4-hydroxy-2-nonenal formation by copper ion bound to amyloid-beta peptide. Free Radic Biol Med. 2007;43:1552–1559. doi: 10.1016/j.freeradbiomed.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 164.Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G, Frederickson CJ. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000;852:274–278. doi: 10.1016/s0006-8993(99)02096-x. [DOI] [PubMed] [Google Scholar]
- 165.Bush AI, Multhaup G, Moir RD, Williamson TG, Small DH, Rumble B, Pollwein P, Beyreuther K, Masters CL. A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J Biol Chem. 1993;268:16109–16112. [PubMed] [Google Scholar]
- 166.Kambe T, Yamaguchi-Iwai Y, Sasaki R, Nagao M. Overview of mammalian zinc transporters. Cell Mol Life Sci. 2004;61:49–68. doi: 10.1007/s00018-003-3148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Multhaup G, Mechler H, Masters CL. Characterization of the high affinity heparin binding site of the Alzheimer’s disease beta A4 amyloid precursor protein (APP) and its enhancement by zinc(II) J Mol Recognit. 1995;8:247–257. doi: 10.1002/jmr.300080403. [DOI] [PubMed] [Google Scholar]
- 168.Cuajungco MP, Lees GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol Dis. 1997;4:137–169. doi: 10.1006/nbdi.1997.0163. [DOI] [PubMed] [Google Scholar]
- 169.Pratico D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VM. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002;16:1138–1140. doi: 10.1096/fj.02-0012fje. [DOI] [PubMed] [Google Scholar]
- 170.Polizzi S, Pira E, Ferrara M, Bugiani M, Papaleo A, Albera R, Palmi S. Neurotoxic effects of aluminium among foundry workers and Alzheimer’s disease. Neurotoxicology. 2002;23:761–774. doi: 10.1016/S0161-813X(02)00097-9. [DOI] [PubMed] [Google Scholar]
- 171.Hegde ML, Anitha S, Latha KS, Mustak MS, Stein R, Ravid R, Rao KS. First evidence for helical transitions in supercoiled DNA by amyloid Beta Peptide (1-42) and aluminum: a new insight in understanding Alzheimer’s disease. J Mol Neurosci. 2004;22:19–31. doi: 10.1385/jmn:22:1-2:19. [DOI] [PubMed] [Google Scholar]
- 172.Amit T, Avramovich-Tirosh Y, Youdim MB, Mandel S. Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008;22:1296–1305. doi: 10.1096/fj.07-8627rev. [DOI] [PubMed] [Google Scholar]
- 173.Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P. Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis. 2009;17:457–468. doi: 10.3233/JAD-2009-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mandel S, Amit T, Bar-Am O, Youdim MB. Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol. 2007;82:348–360. doi: 10.1016/j.pneurobio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 175.Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822:631–638. doi: 10.1016/j.bbadis.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 176.Yatin SM, Varadarajan S, Butterfield DA. Vitamin E Prevents Alzheimer’s Amyloid beta-Peptide (1-42)-Induced Neuronal Protein Oxidation and Reactive Oxygen Species Production. J Alzheimers Dis. 2000;2:123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- 177.Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 178.Yao Y, Chinnici C, Tang H, Trojanowski JQ, Lee VM, Pratico D. Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J Neuroinflammation. 2004;1:21. doi: 10.1186/1742-2094-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alphatocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 180.Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JC Cache County Study Group. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 181.Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, Prieto S, Chen P, Loreck DJ, Trapp G, Bakshi RS, Mintzer JE, Heidebrink JL, Vidal-Cardona A, Arroyo LM, Cruz AR, Zachariah S, Kowall NW, Chopra MP, Craft S, Thielke S, Turvey CL, Woodman C, Monnell KA, Gordon K, Tomaska J, Segal Y, Peduzzi PN, Guarino PD. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311:33–44. doi: 10.1001/jama.2013.282834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ Alzheimer’s Disease Cooperative Study Group. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 183.Lee J, Boo JH, Ryu H. The failure of mitochondria leads to neurodegeneration: Do mitochondria need a jump start? Adv Drug Deliv Rev. 2009;61:1316–1323. doi: 10.1016/j.addr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gutzmann H, Hadler D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: update on a 2-year double-blind multicentre study. J Neural Transm Suppl. 1998;54:301–310. doi: 10.1007/978-3-7091-7508-8_30. [DOI] [PubMed] [Google Scholar]
- 185.Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, Weiner MF, Zamrini E, Thomas RG. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology. 2003;61:1498–1502. doi: 10.1212/01.wnl.0000096376.03678.c1. [DOI] [PubMed] [Google Scholar]
- 186.Siedlak SL, Casadesus G, Webber KM, Pappolla MA, Atwood CS, Smith MA, Perry G. Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer’s disease. Free Radic Res. 2009;43:156–164. doi: 10.1080/10715760802644694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pocernich CB, Lange ML, Sultana R, Butterfield DA. Nutritional approaches to modulate oxidative stress in Alzheimer’s disease. Curr Alzheimer Res. 2011;8:452–469. doi: 10.2174/156720511796391908. [DOI] [PubMed] [Google Scholar]
- 188.Shinto L, Quinn J, Montine T, Dodge HH, Woodward W, Baldauf-Wagner S, Waichunas D, Bumgarner L, Bourdette D, Silbert L, Kaye J. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J Alzheimers Dis. 2014;38:111–120. doi: 10.3233/JAD-130722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ono K, Hirohata M, Yamada M. Alpha-lipoic acid exhibits anti-amyloidogenicity for betaamyloid fibrils in vitro. Biochem Biophys Res Commun. 2006;341:1046–1052. doi: 10.1016/j.bbrc.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 190.McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kapay NA, Isaev NK, Stelmashook EV, Popova OV, Zorov DB, Skrebitsky VG, Skulachev VP. In vivo injected mitochondria-targeted plastoquinone antioxidant SkQR1 prevents beta-amyloid-induced decay of long-term potentiation in rat hippocampal slices. Biochemistry (Mosc) 2011;76:1367–1370. doi: 10.1134/S0006297911120108. [DOI] [PubMed] [Google Scholar]
- 192.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chernyak BV, Antonenko YN, Domnina LV, Ivanova OY, Lyamzaev KG, Pustovidko AV, Rokitskaya TI, Severina II, Simonyan RA, Trendeleva TA, Zvyagilskaya RA. Novel penetrating cations for targeting mitochondria. Curr Pharm Des. 2013;19:2795–2806. doi: 10.2174/1381612811319150015. [DOI] [PubMed] [Google Scholar]
- 194.Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology. 2014;76 Pt A:27–50. doi: 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 195.Robinson RA, Joshi G, Huang Q, Sultana R, Baker AS, Cai J, Pierce W, St Clair DK, Markesbery WR, Butterfield DA. Proteomic analysis of brain proteins in APP/PS-1 human double mutant knock-in mice with increasing amyloid beta-peptide deposition: insights into the effects of in vivo treatment with N-acetylcysteine as a potential therapeutic intervention in mild cognitive impairment and Alzheimer’s disease. Proteomics. 2011;11:4243–4256. doi: 10.1002/pmic.201000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Butterfield DA, Poon HF. The senescenceaccelerated prone mouse (SAMP8): a model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer’s disease. Exp Gerontol. 2005;40:774–783. doi: 10.1016/j.exger.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 197.Parachikova A, Green KN, Hendrix C, LaFerla FM. Formulation of a medical food cocktail for Alzheimer’s disease: beneficial effects on cognition and neuropathology in a mouse model of the disease. PLoS One. 2010;5:e14015. doi: 10.1371/journal.pone.0014015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Javed H, Khan MM, Ahmad A, Vaibhav K, Ahmad ME, Khan A, Ashafaq M, Islam F, Siddiqui MS, Safhi MM, Islam F. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience. 2012;210:340–352. doi: 10.1016/j.neuroscience.2012.02.046. [DOI] [PubMed] [Google Scholar]
- 199.Vijayapadma V, Ramyaa P, Pavithra D, Krishnasamy R. Protective effect of lutein against benzo(a)pyrene-induced oxidative stress in human erythrocytes. Toxicol Ind Health. 2014;30:284–293. doi: 10.1177/0748233712457439. [DOI] [PubMed] [Google Scholar]
- 200.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 201.Ringman JM, Frautschy SA, Teng E, Begum AN, Bardens J, Beigi M, Gylys KH, Badmaev V, Heath DD, Apostolova LG, Porter V, Vanek Z, Marshall GA, Hellemann G, Sugar C, Masterman DL, Montine TJ, Cummings JL, Cole GM. Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther. 2012;4:43. doi: 10.1186/alzrt146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 203.Ho L, Chen LH, Wang J, Zhao W, Talcott ST, Ono K, Teplow D, Humala N, Cheng A, Percival SS, Ferruzzi M, Janle E, Dickstein DL, Pasinetti GM. Heterogeneity in red wine polyphenolic contents differentially influences Alzheimer’s disease-type neuropathology and cognitive deterioration. J Alzheimers Dis. 2009;16:59–72. doi: 10.3233/JAD-2009-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, Renaud S, Breteler MB. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997;153:185–192. [PubMed] [Google Scholar]
- 205.Zhou J, Zhang S, Zhao X, Wei T. Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J Pineal Res. 2008;45:157–165. doi: 10.1111/j.1600-079X.2008.00570.x. [DOI] [PubMed] [Google Scholar]
- 206.He P, Ouyang X, Zhou S, Yin W, Tang C, Laudon M, Tian S. A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’ disease. Horm Behav. 2013;64:1–7. doi: 10.1016/j.yhbeh.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 207.Lu P, Mamiya T, Lu LL, Mouri A, Zou L, Nagai T, Hiramatsu M, Ikejima T, Nabeshima T. Silibinin prevents amyloid beta peptide-induced memory impairment and oxidative stress in mice. Br J Pharmacol. 2009;157:1270–1277. doi: 10.1111/j.1476-5381.2009.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Stackman RW, Eckenstein F, Frei B, Kulhanek D, Nowlin J, Quinn JF. Prevention of age-related spatial memory deficits in a transgenic mouse model of Alzheimer’s disease by chronic Ginkgo biloba treatment. Exp Neurol. 2003;184:510–520. doi: 10.1016/s0014-4886(03)00399-6. [DOI] [PubMed] [Google Scholar]
- 209.Prasanthi JR, Dasari B, Marwarha G, Larson T, Chen X, Geiger JD, Ghribi O. Caffeine protects against oxidative stress and Alzheimer’s disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic Biol Med. 2010;49:1212–1220. doi: 10.1016/j.freeradbiomed.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cao C, Cirrito JR, Lin X, Wang L, Verges DK, Dickson A, Mamcarz M, Zhang C, Mori T, Arendash GW, Holtzman DM, Potter H. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer’s disease transgenic mice. J Alzheimers Dis. 2009;17:681–697. doi: 10.3233/JAD-2009-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kim TI, Lee YK, Park SG, Choi IS, Ban JO, Park HK, Nam SY, Yun YW, Han SB, Oh KW, Hong JT. l-Theanine, an amino acid in green tea, attenuates beta-amyloid-induced cognitive dysfunction and neurotoxicity: reduction in oxidative damage and inactivation of ERK/p38 kinase and NF-kappaB pathways. Free Radic Biol Med. 2009;47:1601–1610. doi: 10.1016/j.freeradbiomed.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 212.Iuvone T, De Filippis D, Esposito G, D’Amico A, Izzo AA. The spice sage and its active ingredient rosmarinic acid protect PC12 cells from amyloid-beta peptide-induced neurotoxicity. J Pharmacol Exp Ther. 2006;317:1143–1149. doi: 10.1124/jpet.105.099317. [DOI] [PubMed] [Google Scholar]
- 213.Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989;14:175–185. doi: 10.1007/BF02797132. [DOI] [PubMed] [Google Scholar]
- 214.Goodman Y, Steiner MR, Steiner SM, Mattson MP. Nordihydroguaiaretic acid protects hippocampal neurons against amyloid beta-peptide toxicity, and attenuates free radical and calcium accumulation. Brain Res. 1994;654:171–176. doi: 10.1016/0006-8993(94)91586-5. [DOI] [PubMed] [Google Scholar]
- 215.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova N, Calingasan NY, Yang L, Tampellini D, Starkov AA, Chan RB, Di Paolo G, Pujol A, Beal MF. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum Mol Genet. 2012;21:5091–5105. doi: 10.1093/hmg/dds355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S, Levonen AL, Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 218.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wong BX, Hung YH, Bush AI, Duce JA. Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology. Front Aging Neurosci. 2014;6:91. doi: 10.3389/fnagi.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Pierrot N, Tyteca D, D’Auria L, Dewachter I, Gailly P, Hendrickx A, Tasiaux B, Haylani LE, Muls N, N’Kuli F, Laquerriere A, Demoulin JB, Campion D, Brion JP, Courtoy PJ, Kienlen-Campard P, Octave JN. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med. 2013;5:608–625. doi: 10.1002/emmm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mukherjee P, Faber AC, Shelton LM, Baek RC, Chiles TC, Seyfried TN. Thematic review series: sphingolipids. Ganglioside GM3 suppresses the proangiogenic effects of vascular endothelial growth factor and ganglioside GD1a. J Lipid Res. 2008;49:929–938. doi: 10.1194/jlr.R800006-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hicks DA, Nalivaeva NN, Turner AJ. Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol. 2012;3:189. doi: 10.3389/fphys.2012.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang H, Wu LM, Wu J. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. 2011;2011:949072. doi: 10.1155/2011/949072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Takeda S, Sato N, Morishita R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front Aging Neurosci. 2014;6:171. doi: 10.3389/fnagi.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Phan HT, Hata T, Morita M, Yoda T, Hamada T, Vestergaard MC, Takagi M. The effect of oxysterols on the interaction of Alzheimer’s amyloid beta with model membranes. Biochim Biophys Acta. 2013;1828:2487–2495. doi: 10.1016/j.bbamem.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 227.Vatassery GT, Smith WE, Quach HT, Lai JC. In vitro oxidation of vitamin E, vitamin C, thiols and cholesterol in rat brain mitochondria incubated with free radicals. Neurochem Int. 1995;26:527–535. doi: 10.1016/0197-0186(94)00147-m. [DOI] [PubMed] [Google Scholar]
- 228.Arciello M, Gori M, Balsano C. Mitochondrial dysfunctions and altered metals homeostasis: new weapons to counteract HCV-related oxidative stress. Oxid Med Cell Longev. 2013;2013:971024. doi: 10.1155/2013/971024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Shinohara M, Sato N, Shimamura M, Kurinami H, Hamasaki T, Chatterjee A, Rakugi H, Morishita R. Possible modification of Alzheimer’s disease by statins in midlife: interactions with genetic and non-genetic risk factors. Front Aging Neurosci. 2014;6:71. doi: 10.3389/fnagi.2014.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kou J, Song M, Pattanayak A, Lim JE, Yang J, Cao D, Li L, Fukuchi K. Combined treatment of Abeta immunization with statin in a mouse model of Alzheimer’s disease. J Neuroimmunol. 2012;244:70–83. doi: 10.1016/j.jneuroim.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Buxbaum JD, Cullen EI, Friedhoff LT. Pharmacological concentrations of the HMGCoA reductase inhibitor lovastatin decrease the formation of the Alzheimer beta-amyloid peptide in vitro and in patients. Front Biosci. 2002;7:a50–59. doi: 10.2741/A739. [DOI] [PubMed] [Google Scholar]
- 232.Friedhoff LT, Cullen EI, Geoghagen NS, Buxbaum JD. Treatment with controlled-release lovastatin decreases serum concentrations of human beta-amyloid (A beta) peptide. Int J Neuropsychopharmacol. 2001;4:127–130. doi: 10.1017/S1461145701002310. [DOI] [PubMed] [Google Scholar]
- 233.Zhang LL, Sui HJ, Liang B, Wang HM, Qu WH, Yu SX, Jin Y. Atorvastatin prevents amyloidbeta peptide oligomer-induced synaptotoxicity and memory dysfunction in rats through a p38 MAPK-dependent pathway. Acta Pharmacol Sin. 2014;35:716–726. doi: 10.1038/aps.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci. 2005;25:299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999;30:1969–1973. doi: 10.1161/01.str.30.9.1969. [DOI] [PubMed] [Google Scholar]
- 236.Butterfield DA, Barone E, Mancuso C. Cholesterol-independent neuroprotective and neurotoxic activities of statins: perspectives for statin use in Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res. 2011;64:180–186. doi: 10.1016/j.phrs.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Barone E, Di Domenico F, Butterfield DA. Statins more than cholesterol lowering agents in Alzheimer disease: their pleiotropic functions as potential therapeutic targets. Biochem Pharmacol. 2014;88:605–616. doi: 10.1016/j.bcp.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mancuso C, Santangelo R, Calabrese V. The heme oxygenase/biliverdin reductase system: a potential drug target in Alzheimers disease. J Biol Regul Homeost Agents. 2013;27:75–87. [PubMed] [Google Scholar]
- 239.Ehret MJ, Chamberlin KW. Current Practices in the Treatment of Alzheimer Disease: Where is the Evidence After the Phase III Trials? Clin Ther. 2015;37:1604–1616. doi: 10.1016/j.clinthera.2015.05.510. [DOI] [PubMed] [Google Scholar]