

Abstract
Background: Recent studies show inflammation-associated lymphangiogenesis (IAL) induced by vascular endothelial growth factor receptor 3 (VEGFR-3) pathway has a close relationship with chronic intestinal inflammation, and antilymphatic signaling pathways may repress IAL. However, whether the biologic function of lymphatic vessel is the same in severe acute intestinal inflammation still remain unknown. Methods: C57BL/6 mice were administered with 5% of dextran sodium sulfate (DSS) in drinking water for 7 days to establish severe acute colitis (SAC) model. Chronic colitis (CC) model was established by three cycles of 2% DSS for 5 days following water for 5 days. Mice were treated with VEGFR-3 antibody once daily in SAC group, or once every 3 days in CC group. The colon inflammation, submucosal edema, lymphatic vessel (LV) density, LV size, lymph flow, cytokines and immune cells infiltration were detected. Results: Both acute and chronic colitis resulted in a significant aggravation of colon inflammation in anti-VEGFR-3-treated mice, compared with PBS-treated colitis mice. Meanwhile, this was accompanied with decreased lymph drainage, increased submucosal edema, inflammatory cells infiltration and cytokines levels. In acute intestinal inflammation, significantly distorted and enlarged lymphatics were found but the LV number remained unchanged; not only significantly distorted and enlarged lymphatics but reduced LV number were found in chronic colitis. Conclusion: Blocking VEGFR-3 in acute and chronic colitis leads to deterioration of colon inflammation with impaired lymphatic function and different changes in LVs. In the therapy targeting VEGF-C/VEGFR-3 pathway for lymphangiogenesis, the phrase and severity of intestinal inflammation should be taken into account.
Keywords: Lymphangiogenesis, vascular endothelial growth factor receptor 3, acute colitis, chronic colitis
Introduction
Inflammatory bowel diseases (IBDs), such as ulcerative colitis (UC) and Crohn’s disease (CD), are the relapsing disorders with alteration of activation and remission of inflammation in the gastrointestinal tract. In recent years, the incidence of IBDs is increasing rapidly in Asia [1]. However, the etiology and pathogenesis of IBD are still poorly understood.
A large amount of studies on IBD has focused on the mechanisms of inflammation in the intestine, i.e. immune cells entry into the mucosa, cytokines and chemokines in the intestine, invasion of bacterial and foreign antigen, and angiogenesis. On the basis of these mechanisms, several effective therapies for IBD have been developed [2]. However, the mechanisms of inflammation out of the intestine are still unclear. Recently, increasing studies concentrate on the roles of lymphangiogenesis in the pathogenesis of inflammation.
Accumulating evidence has proven that inflammation associated lymphangiogenesis (IAL) is involved in the pathophysiology of various inflammatory disorders [3]. Acute and chronic inflammatory processes are associated with the growth and/or enlargement of blood and lymphatic vessels [4]. The lymphatic vessels mainly function to transport the extravasated fluid from tissues back to the blood, clear bacteria and inflammatory elements (infiltrated inflammatory cells, excess cytokines and chemokines), and modify the innate immunity. However, it is unclear whether lymphangiogenesisis able to amplify or protect against IBD development. Many lymphangiogenic signaling pathways are activated upon exposure to inflammatory stimuli, while the role of vascular endothelial growth factor (VEGF)-C/VEGF receptor (VEGFR)-3 signaling is the most important once in the process of IAL. A recent study targeting to this pathway shows that inhibition of VEGFR-3 signaling with specific neutralizing antibody significant decreases the numbers of lymphatic vessels (LVs) and lymphatic drainage, and at the same time increases the severity of inflammation in chronic inflammatory arthritis [5]. Similar results are also reported in a model of chronic airway inflammation [6] and IBD [7]. In contrast, stimulation of functional lymphangiogenesis via VEGFR-3 in transgenic mice expressing VEGF-C/-D significantly reduces chronic skin inflammation [8]. Similarly, systemic delivery of VEGF-C to simulate lymphatic function and enhance lymphangiogenesis markedly reduces chronic intestinal inflammation [9].
However, few studies have been conducted to investigate the role of VEGF-C/VEGFR-3 signaling in acute inflammation and available findings are still conflicting. The biologic function of lymphatic vessels in acute inflammation has remained less explored [10]. Transgenic delivery of lymphangiogenic factors VEGF-C and -D significantly limits acute skin inflammation in animal models, with enhanced lymphatic drainage and increased network of lymphatic vessels [10]. However, treatment with both VEGFR-2/-3 neutralizing antibodies leads to lymphangiogenesis independent extended edema in the mice tail and macrophages are excluded as contribution factors [11]. Stimulation of VEGF-C has been reported to protect against the development of acute intestinal inflammation induced by drinking 2.5% dextran sodium sulfate (DSS) for 10 days [9]. However, in our previous study, the colon inflammation was not improved or even deteriorated by the adenovirus mediated induction of lymphangiogenic factor VEGF-C or the administration of recombinant VEGF-C 156Ser protein in experimental acute colitis induced by drinking 5% DSS for 7 days (the data is being published). Therefore, roles of IAL and VEGF-C/VEGFR-3 pathway in development of acute and chronic intestinal inflammation are needed to be further investigated.
In the present study, the role of VEGFR-3 signaling was explored in acute and chronic intestinal inflammation. Through blocking VEGFR-3 signaling with specific neutralizing antibody mice, the lymphatic vessel density (LVD), LV size, the severity of colon inflammation, and lymphatic drainage were evaluated by examining the changes in lymphatics and lymph flow, infiltrated inflammatory cells, and clearance of inflammatory edema and cells. Our findings may provide a complete understanding about the role of IAL and VEGFR-3 signaling in acute and chronic colitis.
Materials and methods
Animals
Female C57BL/6 mice aged 8-12 weeks were purchased from the Shanghai Laboratory Animal Center (Shanghai, China). The mice were housed under a specific pathogen-free (SPF) condition in the Animal Experimental Center of Tongji Hospital. All animals were given ad libitum access to food and water according to experimental requirements. All animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Tongji University. Mice were euthanized by CO2 inhalation, followed by cervical dislocation.
Mouse colitis model and study design
For the establishment of acute colitis model, C57BL/6 mice given ad libitum access to 5% DSS (MW 36,000-50,000; MP Biomedical, Solon, OH, USA) in water for 7 days (d0-d6). Mice were grouped according to their body weight. DSS water was freshly prepared every day. The mean DSS-water consumption was recorded daily. Mice were treated by intraperitoneal (i.p.) injection once daily with 500 μg of anti-mouse VEGFR-3 (Catalog: #140904, BioLegend Inc., San Diego, CA.) in 100 μl of phosphate-buffered saline (PBS) or 100 μl of PBS as a control (n = 5 per group) from day 0 to day 6 [12]. Animals were killed on day 8.
C57BL/6 mice were given ad libitum access to 2% DSS in water for 5 days and then to DSS free water for another 5 days for 2 cycles to induce chronic colitis. The study design is shown in Figure 1. Mice were intraperitoneally treated with 500 μg of anti-mouse VEGFR-3 (clone AFL4) in 100 μl PBS [13] or 100 μl of PBS as a control (n = 5 per group) once every 3 days starting from the first day of DSS administration [7] (a total of 9 injections). Three days after the last injection, mice were euthanized.
Figure 1.
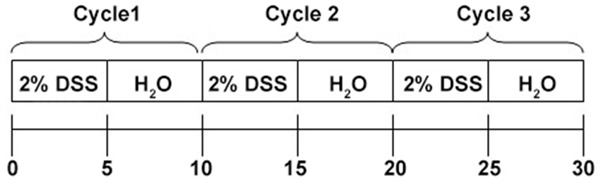
Schematic chart of study design.
All animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Tongji University. The entire colons were removed, fixed in 10% buffered formalin solution and then embedded in paraffin for histological study.
Disease activity index
The disease activity index (DAI) was evaluated daily during the DSS treatment. DAI was determined by using the combined score of weight loss as compared to initial weight, stool consistency, and hemafecia (Table 1) [14,15]. The hemafecia was tested using the Hemoccult test kit (Nanjing Jiancheng Technology Co., LTD, Nanjing, China). The final DAI score of each animal was calculated blindly.
Table 1.
Scores of disease activity index
| Score | Weight loss | Stool consistency∆ | Hemafecia |
|---|---|---|---|
| 0 | No loss | Normal | No blood |
| 1 | 1-5% | ||
| 2 | 5-10% | Loose stools | Hemoccult positive |
| 3 | 10-20% | ||
| 4 | > 20% | Diarrhea | Gross bleeding |
Normal stools = well formed pellets, loose stools = pasty and semi-formed stools which do not stick to the anus, diarrhea = liquid stools that stick to the anus [15].
Histological examination of colitis
To evaluate the severity of colitis, the colon was divided into three equal segments (proximal, middle, and distal), and each was fixed in 10% buffered formalin solution. Paraffin-embedded sections (5 μm in thickness) were stained with hematoxylin/eosin (H&E). In brief, the sections were scored according to the crypt, hyperplastic epithelium, crypt distortion (distorted epithelium), and severity of inflammation (acute and chronic). Each was scored from 0 to 3 and percentage area involvement with the product of the two. This was done by each piece of tissue [15] and performed by two pathologists independently.
Myeloperoxidase (MPO) measurement
MPO activity is detected with a modified procedure for the quantification of neutrophil accumulation in tissues. In brief, a portion of fresh colon tissue was homogenized in 1 ml of 50 mmol/L PBS (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide (Sigma-Aldrich, ST. Louis, Germany) on ice using a tissue homogenizer for 4 min at 30 Hz. The homogenate was sonicated three times and then centrifuged at 14,000 rpm for 15 min. Supernatant was collected and transferred into PBS (pH 6.0) containing 0.17 mg/ml 3.3’-dimethoxybenzidine (Sigma-Aldrich, ST. Louis, Germany) and 0.0005% H2O2, and the absorbance was measured at 460 nm. MPO activity of the supernatant was expressed as units per mg of total protein. Total protein content was determined with a bicinchoninic acid protein assay kit (Sigma-Aldrich, ST. Louis, Germany).
Immunofluorescence staining
Three colon rings were obtained from each mouse. Tissues were embedded in optimal cutting temperature (OCT) compound, frozen in liquid nitrogen, and cut into 6-μm sections. Sections were placed on glass slides, air dried, and fixed with acetone for 2 min at -20°C. After rehydration with 80% methanol at 4°C, and PBS with 12% bovine serum albumin (BSA), the sections were incubated with primary antibodies. Standard H&E staining and immunofluorescence staining were performed as described previously [8]. Following primary antibodies were used: anti-mouse LYVE-1 polyclonal antibody (ab14917, 1:200, Abcam, Cambridge, UK), anti-mouse MECA-32+ monoclonal antibody (ab27853, 1:50, Abcam), anti-mouse CD11c (BD Biosciences PharMingen), anti-mouse CD31 (1:50; BD Pharmingen), anti-mouse CD68 (1:200; BD Pharmingen), anti-mouse MPO (1:10; Abcam), and CD45/B220 (1:50; BD Pharmingen). Alexa488- or Alexa594-coupled secondary antibodies and DAPI were purchased from Invitrogen.
Sections were mounted with FluorSave reagent (Calbiochem, San Diego, California, USA) and observed under an epifluorescence microscope (Nikon Corporation, JAPAN). Images were obtained with an oil immersion objective. Infiltrating leukocytes were counted in five randomly selected fields within inflammatory mucosa under a light microscope at a magnification of 400×. After H&E staining, the width of colon submucosa (inflammatory edema) was measured in five randomly selected fields per section under a light microscope at 100× [7]. The colon submucosa refers to the tissues surrounding the lamina propria at the base of epithelial crypts, the muscularis mucosa, submucosa, and the muscle layers using NIS Elements 4.0 Microscope Imaging Software (Nikon) [7].
Morphometric and immunofluorescence intensity measurements
Double immunofluorescence staining of the colon for LYVE-1 and CD31 was performed for morphometric analysis. Vascular structures with a lumen were analyzed. To exclude non-specific staining, vascular structures less than 8 μm (1 μm = 6.8 pixels) in diameter were excluded. The CD31+LYVE-1+ (green) vessels were lymphatic vessels and CD31+LYVE-1- (red) vessels were blood vessels [16]. For each colon analyzed, LVD, LV size, and blood vessels density (BVD) were measured in 5 randomly selected fields of “hot spots” view (with the highest density of lymphatic/blood vessels) per section (mucosal, submucosal and serosal layers) using Photoshop extended CS4 software. Computer-assisted analyses of digital images were performed using the NIS Elements 4.0 Microscope Imaging Software (Nikon) as describe dpreviously [10]. The average size of vessels per area was calculated. Scoring and counting of vessels were performed independently by two investigators who were blind to this study. Results are expressed as vessel number per millimeter.
Lymph flow assessment after evans blue staining [9, 10]
Ten micrograms of Evan blue dye (Sigma-Aldrich) in 10 μl of PBS was injected into the rectal mucosa of anesthetized, anti-VEGFR-3 antibody-treated mice and control mice (n = 5/timepoint) using a Hamilton syringe. After 16 h,mice were killed. Evans blue dye was extracted from the distal colon tissues of comparable weight by incubating them at 55°C overnight in formamide (Sigma-Aldrich). The background-subtracted absorbance was measured with an infinite M200 microplate reader at the wavelength ranging from 620 nm to 740 nm. The concentration of Evan blue dye in the extract was calculated from a standard curve and is presented as absolute amount of dye in the colon tissues.
Enzyme-linked immunosorbent assay (ELISA)
Colon tissues (n = 5 per group) were homogenized in lysis buffer containing a protease inhibitor cocktail for radioimmunoprecipitation assay (150 mM NaCl, 50 mM Tris, pH 7.5, Sigma Chemicals). Homogenates were centrifuged for 10 min at 14000 g, and the supernatant was collected for the detection of TNF-α, IL-1β, IL-6, IL-4 and IL-12 with commercially available ELISA kits according to the manufacturer’s protocols. The absorbance was measured with an Infinite M200 microplate reader (Tecan Group Ltd., Männedorf, Switzerland). Protein contents were normalized by per milligram of tissue.
Statistical analysis
Statistical analyses were performed with IBM SPSS statistical software version 22 for Windows (IBM Corp., New York, USA). Data are presented as means ± standard error (SEM). Comparisons of means between two groups were conducted with 2-tailed, unpaired Student’s t test. Figures were generated by GraphPad Prism 6 (GraphPad Software, Inc. La Jolla, CA, USA). A value of two-tailed P < 0.05 was considered statistically significant.
Results
VEGFR-3 blocking deteriorates disease activity in acute and chronic colitis
To investigate the effects of VEGFR-3 blockingon acute colonic inflammation, mice were treated with 500 μg of anti-mouse VEGFR-3 antibodyor PBS as a control in acute colitis model. The DAI scores significantly increased in anti-VEGFR-3-treated mice from d2 to d6 as compared to control group (P = 0.0092, Figure 2A). Mice treated with anti-VEGFR-3 antibody were Hemoccult positive on day 2, and displayed more severe diarrhea. From day 4, anti-VEGFR-3-treated mice had more visible blood in their stool and evident macroscopic rectal bleeding. In chronic colitis model, mice were treated with anti-VEGFR-3 antibody once every 3 day during the chronic colitis. Anti-VEGFR-3-treated mice also showed higher DAI scores as compared to PBS-treated mice, particularly during the DSS treatment. In the first cycle of acute colitis phrase in which 2% DSS was administered for 5 days, the DAI score was lower than that in acute colitis induced by 5% DSS for 7 days (3.44±1.53 vs 7.03±3.05, Figure 2A and 2B). From the third cycle, DAIs in anti-VEGFR-3-treated mice was significantly higher than that in PBS-treated mice (Figure 2B, P < 0.001).
Figure 2.
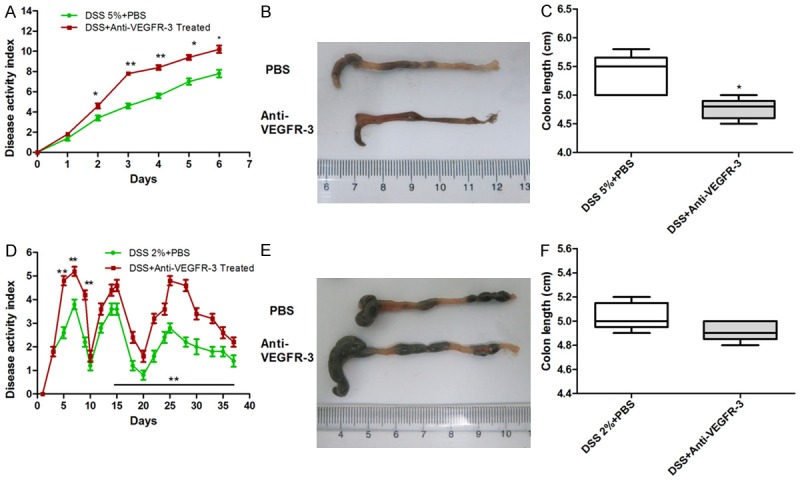
Blocking VEGFR-3 exacerbates colon inflammation in acute and chronic colitis. C57BL/6 mice fed with 5% DSS for 7 days to induce acute colitis were treated intraperitoneally with anti-VEGFR-3 antibody once daily (n = 5/group). Anti-VEGFR-3 antibody treatment significantly increases intestinal inflammation with higher DAI scores (A) and shorter colon length (B, C) (t test, P < 0.05). Mice fed with 3 cycles of 2% DSS to induce chronic colitis were treated intraperitoneally with anti-VEGFR-3 antibody once every 3 days. Anti-VEGFR-3 antibody treatment leads to significant exacerbation of chronic colitis with higher DAI score (D, n = 5/time point) (t test, P < 0.01), but the colon length remains unchanged (E, F) (t test, P > 0.05).
Colon length was measured to determine the severity of colitis. In acute colitis mice, high concentration of DSS could lead to significant reduction in colon length in the presence of VEGFR-3 blocking treatment as described in Figures 2C and 2E (P = 0.0196), but the colon length remained unchanged in chronic colitis mice (Figure 2D and 2F, P > 0.05). MPO activity markedly increased in anti-VEGFR-3-treated acute colitis mice (P = 0.0249, Figure 3). The amount of DSS intake was comparable between groups (data not shown).
Figure 3.
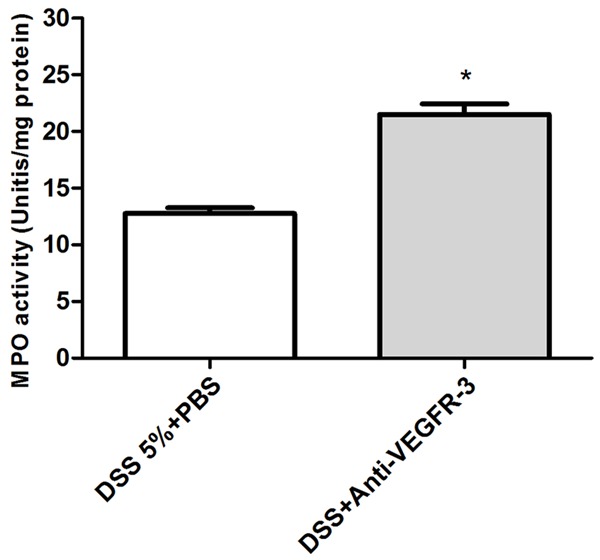
MPO activity as an index of neutrophils infiltration incolon tissues of acute colitis mice. Anti-VEGFR-3 antibody treatment significantly increases the MPO activity as compared to PBS treated mice. (t test, P < 0.05).
Systemic blocking of VEGFR-3 increases histological score and inflammatory edemain acute and chronic colitis
The normal mouse colon showed intact epithelium, well defined crypt length, no edema,no neutrophil infiltration in the mucosa and submucosa, and no ulcers or erosions (Figure 4A). In PBS-treated acute colitis mice, shortening and loss of crypts were observed, with more extensive mucosal involvement, and finally the whole colon was involved, and mucin, goblet cell depletion, epithelial erosion and sometimes ulceration were also noted (Figure 4B). Anti-VEGFR-3 treated mice had a marked increase in histologic damage (Figure 4C and 4D, P = 0.0039) and inflammatory edema (Figure 4E, P = 0.0257) as compared to PBS treated mice. Infiltration of immune cells including neutrophils and lymphocytes in the lamina propria and submucosa was more obvious inanti-VEGFR-3 treated mice (Figure 4A and 4B).
Figure 4.
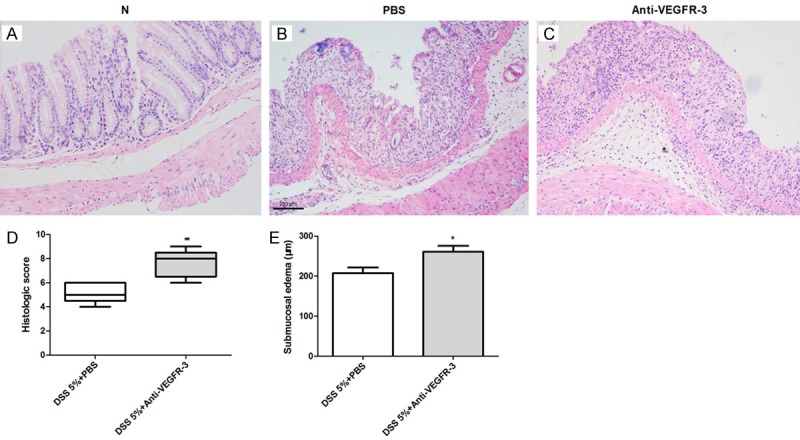
Histological changes in mice treated with anti-VEGFR-3 antibodyin acute colitis. (A) Normal histology of mouse colon. Anti-VEGFR-3 antibody in mice worsens histologic scores (B, D) (t test, P < 0.05) and increases submucosal edema (C, E) (t test, P < 0.01) as compared to PBS-treated mice.
In chronic colitis model, mice were killed on day 33. Histological examination showed mononuclear leukocyte infiltration, disruption of crypt architecture and widening of the gap between crypt baseand muscularis with deep mucosal lymphocytosis (Figure 5A and 5B). Systemic delivery of anti-VEGFR-3 antibody worsened the chronic intestinal inflammation as shown by histological scores (Figure 5C, P = 0.021) and inflammatory edema (Figure 5D, P = 0.0005).
Figure 5.
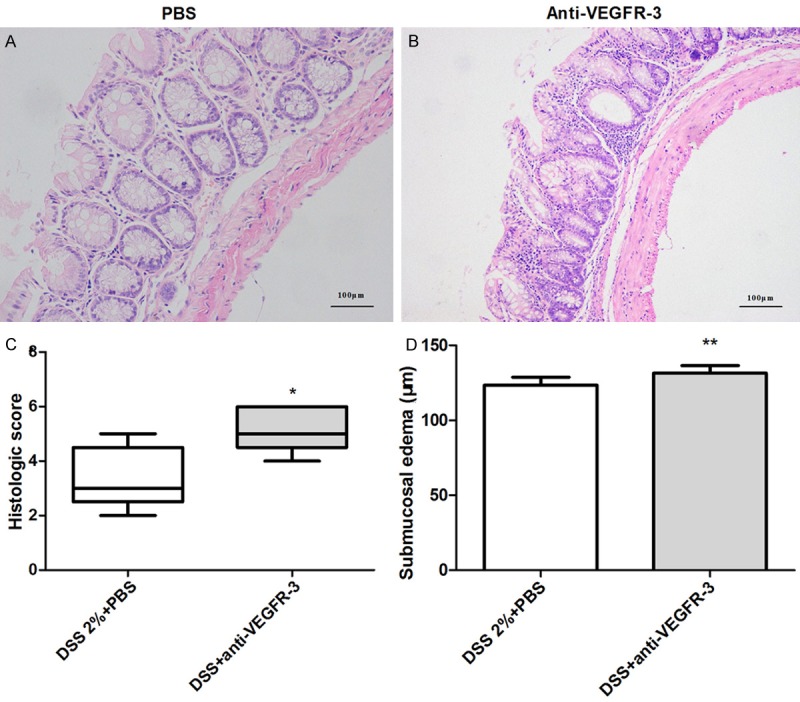
Histological changes in mice treated with anti-VEGFR-3 antibody in chronic colitis. Compared with PBS-treated mice (A), mice with anti-VEGFR-3 antibody treatment had a higher histologic score (B, C, t test, P < 0.05) and more severe submucosal edema (D) (t test, P < 0.01).
Inflammatory lymphangiogenesis is inhibited in chronic colitis but not in acute colitis after VEGFR-3 blocking
The proximal, middle, and distal colonic segments from PBS-treated and anti-VEGFR-3-treated mice were processed for staining with anti-LYVE-1 and anti-CD31 antibodies (Abs), and then the area density and dimension of LVs were determined during acute and chronic colitis. Significant difference was not observed in LVD (Figure 6A-C, P > 0.05), but LV size (with enlarged lumen) increased significantly (Figure 6D, P = 0.0437) in anti-VEGFR-3-treated mice with acute colitis, as compared to control mice. However, in chronic colitis mice, anti-VEGFR-3 treatment significantly reduced the LVD in mice, as compared to PBS-treated mice (Figure 6E-G, P = 0.012). Meanwhile, markedly enlarged and tortuous LVs were also noted in anti-VEGFR-3-treated mice (Figure 6F, P = 0.0328).
Figure 6.

Changes in lymphatic vessels after VEGFR-3 blocking in acute and chronic colitis. Double immunofluorescence staining was performed for differentiating lymphatic vessels (green, CD31+LYVE-1+) from blood vessels (red, CD31+LYVE-1-), with DAPI staining for nucleus (blue). Compared with PBS-treated mice (A), mice with anti-VEGFR-3 antibody treatment had no significant change in LVD (B, C, P > 0.05), but significantly enlarged lymphatics were observed (D, t test, P < 0.05) in acute colitis mice. In chronic colitis mice, LVD was significantly decreased in anti-VEGFR-3-treated mice, as compared to PBS-treated mice (E-G, t test, P< 0.05), accompanied by dramatically enlarged and distorted lymphatics (F, H, t test, P < 0.05).
Computer-based morphometric analyses after double immunofluorescence staining for CD31+ LYVE-1- blood vessels revealed high density of blood vessels in acute or chronic colitis mice, particularly in acute inflammatory colons with evident submucosal edema (Red, Figure 6). However, no significant difference in BVD was observed between PBS-treated mice and anti-VEGFR-3-treated mice in both acute (19.8±3.2 vs 21.8±1.74, P > 0.05) and chronic colitis (12.04±0.84 vs 11.64±1.11, P > 0.05) mice as compared to PBS-treated mice.
VEGFR-3 blocking decreases inflammatory cells infiltration in acute and chronic colitis mice
Different immune cells were detected in acute and chronic colitis mice with anti-VEGFR-3 treatment by immunofluorescent staining. Quantification of infiltrating leukocytes showed a significantly larger number of MPO+ neutrophils (Figure 7A1-A3, P = 0.0249), CD11c+ dendritic cells (Figure 7B1-B3, P = 0.0007) and CD68+ macrophages (Figure 7C1-C3, P = 0.034) in anti-VEGFR-3-treated mice as compared to PBS-treated mice in acute colitis group. However, there were no significant differences in the number of infiltrated CD3+ T cells and CD45/B220+ B cells between two groups (Figure 7, P > 0.05).
Figure 7.
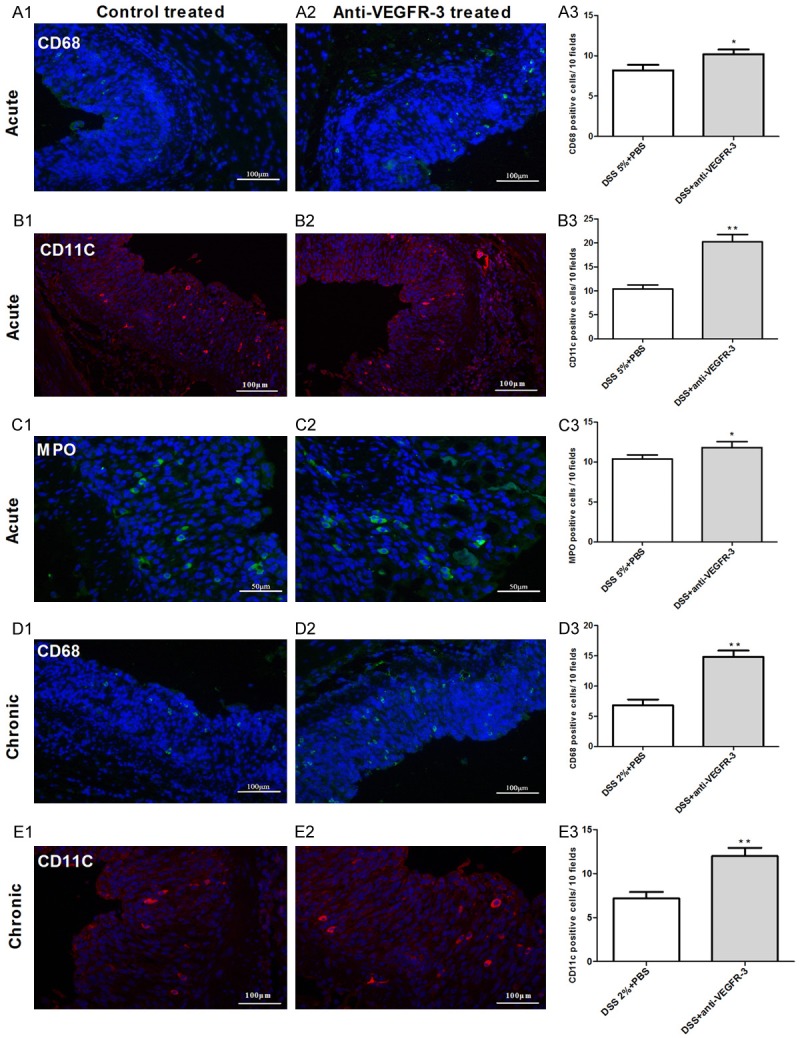
Inflammatory cells infiltration increases after blocking VEGFR-3 signaling in acute and chronic colitis. Quantification of infiltrating leukocytes shows a significantly larger number of CD68+ macrophages (A1-3, D1-3) and CD11c+ dendritic cells (B1-3, E1-3) in acute and chronic colitis mice treated with anti-VEGFR-3 antibody as compared to PBS-treated mice. MPO+ neutrophils increase in acute colitis mice with anti-VEGFR-3 treatment as compared to control mice (C1-3). Magnification: 200× (t test, *P < 0.05; **P < 0.01).
In chronic colitis, VEGFR-3 blocking was also hypothesized to inhibit the migration of immune cells toward draining lymph nodes (DLNs). Thus, the inflammatory cells were further investigated in the colon of PBS- and anti-VEGFR-3-treated mice. The number of CD68+ macrophages (Figure 7D1-D3, P = 0.004) and CD11c+ dendritic cells (Figure 7E1-E3, P = 0.039) increased dramatically in the colon of mice treated with anti-VEGFR-3 antibody as compared to control mice. These results indicate that the bio-behaviors of some inflammatory cells aremodulated by anti-VEGFR-3 antibody in chronic colitis. Significant difference was not observed in other types of inflammatory cells after blocking VEGFR-3 signaling (P > 0.05).
Cytokines in the colon of acute and chronic colitis mice after VEGFR-3 blocking
To further analyze some of the cytokine expression involved in colonic inflammation treated by anti-VEGFR-3 antibodies in acute and chronic DSS-induced colitis, fresh colons were removed at the end of models and further processed for cytokine production by ELISA assay, as described in the Materials and Methods section.Our results showed that the contents of TNF-α (Figure 8A, P = 0.0166), IL-1β (Figure 8B, P = 0.0433), IL-6 (Figure 8D, P = 0.0279), and IL-12 (Figure 8E, P = 0.0068) in the colon significantly increased in acute colitis mice with anti-VEGFR-3 treatment as compared to PBS-treatedmice. In chronic colitis mice, TNF-α (Figure 8F, P = 0.0009), IL-1β (Figure 8G, P = 0.024), IL-4 (Figure 8H, P = 0.0497), IL-6 (Figure 8I, P = 0.0153), and IL-12 (Figure 8J, P = 0.0308) contents increased dramatically after anti-VEGFR-3 antibody treatment as compared to control group. These indicate a mixed immune reaction of Th1/Th17 and Th2 cells in theacute and chronic colitis after anti-VEGFR-3 antibody treatment.
Figure 8.

Cytokine contents increase in colon tissues after blocking VEGFR-3 signaling in acute and chronic colitis. TNF-α, IL-1β, IL-6, and IL-12 contents increased in C57BL/6 mice treated with anti-VEGFR-3 antibody as compared to control mice in acute colitis. TNF-α, IL-1β, IL-4, IL-6 and IL-12 contents increased in colon tissues of anti-VEGFR-3-treated mice as compared to control mice (t test, *P < 0.05; **P < 0.01).
Lymph flow decreases in acute and chronic colitisafter VEGFR-3 blocking
To investigate whether VEGFR-3 blocking also decreased lymphatic drainagein acute colitis, as reported in chronic colitis, Evans blue dye was injected into the distant colonic mucosa of anti-VEGFR-3 treated mice. The Evans blue dye was extracted from distal colons and its amount was measured. Results revealed that lymphatic drainage was decreased in both acute and chronic colitis. Anti-VEGFR-3-treated mice had significantly more dye in the inflammatory tissues of acute (Figure 9A, P = 0.0344) and chronic colitis (Figure 9B, P = 0.025) mice as compared to PBS-treated mice, suggestive of reduced lymphatic drainage.
Figure 9.
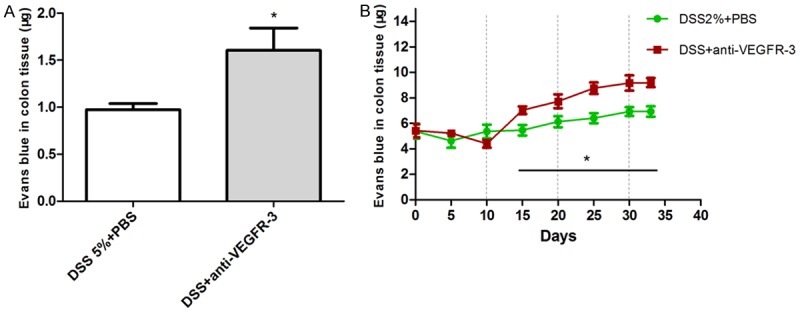
Lymph flowdecreases in colon tissuesafter blocking VEGFR-3 signaling in acute and chronic colitis. A. Anti-VEGFR-3-treated mice had a decreased lymph flow as compared to PBS-treated mice in acute colitis; B. In chronic colitis, anti-VEGFR-3-treated mice undergoing 3 cycles of 2% DSS treatment had more Evens Blue dyein the distal colons, especially from the second cycle, as compared to mice treated with PBS (t test, *P < 0.05).
Discussion
Although the lymphangiogenesis with lymphatic vessel obstruction and dysfunction has been recognized as features observed in human IBD, the role of lymphatics in the pathophysiology of IBD is poorly understood. IAL has been reported to contribute to the pathogenesis of intestinal inflammation in recent years. Studies also show that to control the growth and function of lymphatic vessels through modulating key signaling pathwaysis able to improve the intestinal inflammation in IBD patients.
In the present study,our results demonstratedthat blocking VEGFR-3 signaling aggravatedthe intestinal inflammation in both acute and chronic colitis. Mice treated with anti-VEGFR-3 antibody had more severe colonic inflammation, increased submucosa edema and inflammatory cells infiltration, accompanied by distorted and enlarged lymphatic vessels and decreased lymphatic function. However, systemic inhibition of VEGFR-3 reduced the LVD only in chronic colitis, but not in acute colitis. These results show blocking VEGFR-3 signaling has distinct effects indifferent phrases (acute or chronic) and/or severities (mild or severe) of colitis. Our finding may help the comprehensive understanding of biological role of VEGFR-3 signaling in intestinal inflammation, and provide a potential target for the therapy of IBD in different phrases.
The VEGF-C/VEGFR-3 signaling is the best characterized in the process of IAL [3,9], and involved in the lymphangiogenesis and inflammation controlling in many chronic inflammatory diseases [5-9]. Stimulating VEGFR-3 tyrosine kinase by VEGF-C may regenerate functional collecting LVs in mice after lymph node dissection. VEGFR-3 activation has an important role in the maintenance and survival of lymphatic endothelial cells (LECs) and vascular integrity, and thus inhibition of VEGFR-3 signaling might alter the lymphatic function,leading to the intestinal injury [17]. Blocking VEGFR-3 with specific antibody aggravated intestinal inflammation, deteriorated submucosal edema, and increased large and tortuous LVs, LVD and size, indicating a decreased lymphatic vessel function [7]. Similar findings were noted in previous studies on chronic colitis after anti-VEGFR-3 antibody-treatment. This indicates that inhibition of VEGFR-3 signaling suppresses the growth of lymphatic vessels and the survival of LECs, and damages the vascular integrity, leading to the LVs dysfunction during the chronic colonic inflammation. A study of Danese et al., showed that the exacerbation of experimental chronic colitis induced by VEGFR-3 blocking was associated with decreased afferent lymph flow and inflammatory cell migration to draining lymph nodes (DLN) [9]. Similarly, our findings also indicated that the decreased LVs function after blocking VEGFR-3 signaling was found in not only chronic DSS colitis, but acute colitis. Therefore, these confirm that controlling VEGFR-3 signaling can alter the intestinal inflammation through affecting the LVs function.
To date, lots of studies focused on chronic colitis, but few studies have been conducted to investigate the effects of VEGFR-3 signaling on acute colitis. To investigate the biologic role of IAL in acute inflammation, Helgenberger et al. found increased lymphatic vessels significantly enhanced lymphatic drainage in the inflamed ear skin of VEGF-C or VEGF-D transgenic mice [10]. Although DSS-induced experimental colitis in mice provides a model of acute intestinal injury and is has been widely used because of its simplicity and many similarities with to human UC, few studyies was reported the role of VEGFR-3 signaling in acute colitis in the past.
The severity of acute colon inflammation is dependent on the dose of DSS in drinking water. Usually, the dose of DSS ranges from 2% to 5%, and treatment lasts for 5 or 7 days to induce acute colitis model. Treatment with 5% DSS in drinking water for 7 days is widely used to induce severe acute colitis in BALB/c or C57BL/6 mice, while treatment with 2%-2.5% DSS in drinking water for 5-10 days is employed to induce mild acute colitis [18,19]. During the anti-VEGFR-3 antibody-treatment, DAI was higher in mice with acute colitis induced by 7-day 5% DSS treatment than in mice induced by 10-day 2% DSS treatment in the first cycle of chronic colitis induction. However, different from the findings in chronic colitis, anti-VEGFR-3 antibody-treatment led to significantly enlarged LVs and lymphatic vessel dysfunction in acute colitis, but the growth of LVs remained unchanged.
This may be related several factors. First, the lymphatic changes vary in different severities of inflammation and different time points of acute versus chronic colitis (7 days acute colitismodel vs. 33 days chronic colitis model) [20]. In our study, mice with acute colitis induces by 7-day 5% DSS treatmenthad more severe intestinal inflammation than those with acute colitis induced by 10-day 2% DSS treatment according to the DAI scores. In addition, 7-day 5% DSS treatment induced more evident angiogenesis, and cause more severe submucosal edema and inflammatory cells infiltration. In a relatively short time, the functional changes were prevalent in the lymphatic endothelium, including dilation, increased permeability and activation of the endothelium. These changes will be subsequently accompanied by vessel remodeling [21]. Lymphangiogenic expansion may represent a protective/adaptive response rather than aggravating inflammation at least in acute phase of IBD [20]. This may explain the functional changes in lymphatic vessels with expansion in acute colitis. During early inflammation, lymphangiogenesis constitutes a positive response to injury by increasing lymph flow and drainage of inflammatory mediators and immune cells from inflammatory tissues into LNs. The proliferation and survival of LECs depend on signaling through VEGFR-2 and -3 mediated VEGF-C and -D signaling [22]. After blocking VEGFR-3 signaling, LECs function and LVs drainage are destroyed. In acute phase of inflammation, lymphatics show decreased function and distorted expansion. In chronic colitis, the number of LVs decreased accompanied by vessels remodeling. Reduction in LVD and LVs dysfunction overwhelm the intestinal inflammation, leading to aggravation of intestinal inflammation. These findings are consistent with those reported by Helgenberger et al. [10]. The acute skin inflammation is also characterized by the lymphatic enlargement but not the sprouting lymphangiogenesis even in VEGF-C or VEGF-D transgenic mice [10].
Second, the roles of different types of immune cells and cytokines are distinct in acute versus chronic colitis models. Significant increase in MPO+ neutrophils, macrophages and DCs in acute colitishas the potential to increase growth factors and cytokines, mediating the expansion of both vasculatures [20]. Under inflammatory conditions, large quantities of immune cells migrate through blood vessels and gather at the inflammatory colon tissues.Macrophages are one of the most important types of immune cells during IAL, and DCs have a major role in the adaptive immunity as antigen presenting cells (APCs). Their migration from colon tissues to regional LNs through lymphatic vessels is an important process for inflammation resolution. VEGFR-3 signaling mediates the migration of macrophages and DCs toward DLNs [23]. Unlike in human diseases, T and B cells are not required for the development of colitis in mice, just as in our study. When the growth and function of IAL are inhibited by anti-VEGFR-3 antibody in chronic colitis, the migration of these cells is suppressed and the adaptive immunity is affected. Reciprocal interaction between lymphatic vessels and immune cells in response to inflammation during IAL in colitis ultimately aggravates intestinal inflammation due to anti-lymphangiogenesis treatment [3,24].
Because the LVs function was destroyed by VEGFR-3 blocking, immune cells accumulated, and cytokines (such asTNF-α, IL-1β, IL-6, and IL-12) significantly increased in acute colitis. In chronic colitis, IL-4 and other cytokines also elevated. The pleiotropic activities of these cytokines are driven by a mixed of Th1/Th17 cells and Th2 cells. Therefore, their influence on DSS-induced pathology in the colon is rather complex [25]. Cytokines may also affect the expression of lymphatic VEGFRs with dysregulation of lymphatic proliferation, migration and survival [26]. However, no study has been undertaken to investigate the relationship of expressions of lymphangiogenic growth factors and their receptors with lymphatics function after blocking VEGFR-3 signaling. The roles of VEGFR-3 signaling in different severities of acute colitis induced by various concentrations of DSS are also needed to be further explored.
Conclusion
In summary, our study confirms that blocking VEGFR-3 signaling in acute and chronic colitis aggravates the intestinal inflammation by reducing the lymphatic drainage, inducing edema, increasing inflammatory cells infiltration and elevating pro-inflammatory cytokines. Moreover, lymphatic vessels show distinct changes in acute and chronic colitis after blocking VEGFR-3 signaling. Thus, it is possible to treat IBD by improving lymphatic function to limit intestinal inflammation via activating VEGFR-3 signaling, particularly in chronic colitis. However, it may not be feasible for severe acute colitis. Activation of VEGF-C/VEGFR-3 pathway in severe acute colitis may not improve the inflammation as in chronic colitis because new lymphatics may not sprout, and severe inflammation is overloaded to lymphatics drainage. Therefore, to modulate VEGF-C/VEGFR-3 signaling is more feasible to improve the intestinal inflammation in IAL of mild chronic inflammation.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No 81200260).
Disclosure of conflict of interest
None.
References
- 1.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. e42. doi: 10.1053/j.gastro.2011.10.001. quiz e30. [DOI] [PubMed] [Google Scholar]
- 2.Danese S. Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: ‘brothers in arms’. Gut. 2011;60:998–1008. doi: 10.1136/gut.2010.207480. [DOI] [PubMed] [Google Scholar]
- 3.Kim H, Kataru RP, Koh GY. Inflammation-associated lymphangiogenesis: a doubleedged sword? J Clin Invest. 2014;124:936–942. doi: 10.1172/JCI71607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halin C, Detmar M. Chapter 1. Inflammation, angiogenesis, and lymphangiogenesis. Methods Enzymol. 2008;445:1–25. doi: 10.1016/S0076-6879(08)03001-2. [DOI] [PubMed] [Google Scholar]
- 5.Guo R, Zhou Q, Proulx ST, Wood R, Ji RC, Ritchlin CT, Pytowski B, Zhu Z, Wang YJ, Schwarz EM, Xing L. Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. 2009;60:2666–2676. doi: 10.1002/art.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurisic G, Sundberg JP, Detmar M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm Bowel Dis. 2013;19:1983–1989. doi: 10.1097/MIB.0b013e31829292f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med. 2010;207:2255–2269. doi: 10.1084/jem.20100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, Genua M, Arena V, Spinelli A, Peyrin-Biroulet L, Fiocchi C, Danese S. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. 2014;124:3863–3878. doi: 10.1172/JCI72189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, Werner S, Alitalo K, Detmar M. An important role of lymphatic vessel activation in limiting acute inflammation. Blood. 2011;117:4667–4678. doi: 10.1182/blood-2010-10-316356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ongstad EL, Bouta EM, Roberts JE, Uzarski JS, Gibbs SE, Sabel MS, Cimmino VM, Roberts MA, Goldman J. Lymphangiogenesis-independent resolution of experimental edema. Am J Physiol Heart Circ Physiol. 2010;299:H46–54. doi: 10.1152/ajpheart.00008.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis. 2012;18:1900–1909. doi: 10.1002/ibd.22900. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu K, Kubo H, Yamaguchi K, Kawashima K, Ueda Y, Matsuo K, Awane M, Shimahara Y, Takabayashi A, Yamaoka Y, Satoh S. Suppression of VEGFR-3 signaling inhibits lymph node metastasis in gastric cancer. Cancer Sci. 2004;95:328–333. doi: 10.1111/j.1349-7006.2004.tb03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 16.Keuschnigg J, Henttinen T, Auvinen K, Karikoski M, Salmi M, Jalkanen S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood. 2009;114:478–484. doi: 10.1182/blood-2008-11-188763. [DOI] [PubMed] [Google Scholar]
- 17.Tammela T, Saaristo A, Holopainen T, Lyytikka J, Kotronen A, Pitkonen M, Abo-Ramadan U, Yla-Herttuala S, Petrova TV, Alitalo K. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat Med. 2007;13:1458–1466. doi: 10.1038/nm1689. [DOI] [PubMed] [Google Scholar]
- 18.Scaldaferri F, Vetrano S, Sans M, Arena V, Straface G, Stigliano E, Repici A, Sturm A, Malesci A, Panes J, Yla-Herttuala S, Fiocchi C, Danese S. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136:585–595. e585. doi: 10.1053/j.gastro.2008.09.064. [DOI] [PubMed] [Google Scholar]
- 19.Bento AF, Leite DF, Marcon R, Claudino RF, Dutra RC, Cola M, Martini AC, Calixto JB. Evaluation of chemical mediators and cellular response during acute and chronic gut inflammatory response induced by dextran sodium sulfate in mice. Biochem Pharmacol. 2012;84:1459–1469. doi: 10.1016/j.bcp.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Alexander JS, Chaitanya GV, Grisham MB, Boktor M. Emerging roles of lymphatics in inflammatory bowel disease. Ann N Y Acad Sci. 2010;1207(Suppl 1):E75–85. doi: 10.1111/j.1749-6632.2010.05757.x. [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 22.Chaitanya GV, Franks SE, Cromer W, Wells SR, Bienkowska M, Jennings MH, Ruddell A, Ando T, Wang Y, Gu Y, Sapp M, Mathis JM, Jordan PA, Minagar A, Alexander JS. Differential cytokine responses in human and mouse lymphatic endothelial cells to cytokines in vitro. Lymphat Res Biol. 2010;8:155–164. doi: 10.1089/lrb.2010.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nykanen AI, Sandelin H, Krebs R, Keranen MA, Tuuminen R, Karpanen T, Wu Y, Pytowski B, Koskinen PK, Yla-Herttuala S, Alitalo K, Lemstrom KB. Targeting lymphatic vessel activation and CCL21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation. 2010;121:1413–1422. doi: 10.1161/CIRCULATIONAHA.109.910703. [DOI] [PubMed] [Google Scholar]
- 24.Ji RC. Macrophages are important mediators of either tumor- or inflammation-induced lymphangiogenesis. Cell Mol Life Sci. 2012;69:897–914. doi: 10.1007/s00018-011-0848-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104:Unit 15.25. doi: 10.1002/0471142735.im1525s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RC. Lymphatic endothelial cells, inflammatory lymphangiogenesis, and prospective players. Curr Med Chem. 2007;14:2359–2368. doi: 10.2174/092986707781745541. [DOI] [PubMed] [Google Scholar]