

Abstract
Deregulation of phenotypic modulation in VSMCs is the initial stage of atherosclerosis, especially in diabetes. Functional deficiency of IRAK4 inhibits the formation of vascular lesions in ApoE-/- mice. Therefore, in this study, we examined the functions of IRAK4 in the regulation of VSMCs differentiation and phenotypic modulation at the levels of transcription and translation in T2D rats. The T2D rat model was generated by feeding a high-fat diet and injecting a low dose of streptozotocin intraperitoneally. VSMCs were isolated from the thoracic aortas of the T2D rats. VSMCs proliferation and migration were measured using water soluble tetrazolium salt-1 assay, 5-ethynyl-29-deoxyuridine staining and migration assay. IRAK4 was knocked down by siRNA and inhibited by an IRAK1/4 inhibitor. The mRNAs and proteins of signal molecules and phenotypic markers were detected by qRT-PCR and western blotting. The results demonstrated that LPS significantly increased viability, cell migration rate and amount of DNA in VSMCs. The IRAK4 inhibitor also reduced LPS-mediated protein expression of myosin heavy chain and nuclear factor κB p65 subunit and increased smooth muscle 22α expression. Moreover, IRAK4 knock-down reduced the LPS-mediated expression of mRNAs for myosin heavy chain, nuclear factor κB p65 subunit, and monocyte chemoattractant protein-1 (MCP-1), but increased the mRNA of smooth muscle 22α in VSMCs. The activation of IRAK4 phenotypically modulated VSMCs from differentiation to dedifferentiation. Inactivation of IRAK4 exerts a protective effect on VSMCs differentiation and inhibits inflammation. IRAK4 could therefore be a target for interventions to prevent and treat the initial phase of atherosclerosis.
Keywords: Type 2 diabetes rat model, Vascular smooth muscle cells, IRAK4, phenotypic modulation, MCP-1
Introduction
Vascular smooth muscle cells (VSMCs) are highly differentiated and usually located in the arterial media. They control blood vessel tone and blood pressure through their contractility [1]. Unlike other differentiated cells, VSMCs can switch from a differentiated (also known as mature) to a dedifferentiated (also known as immature) state in response to vascular injury and disease [2]. Mature VSMCs are characterized by contractility, low rates of proliferation and migration, low extracellular matrix synthesis activity, and high levels of differentiation marker gene expression [3]. On the contrary, immature VSMCs are characterized by synthetic ability, higher rates of proliferation, migration and secretion of extracellular matrix, and higher expression of dedifferentiation marker genes [4].
Environmental stimuli can induce adverse phenotypic switching of VSMCs, which contribute to the development and progression of vascular diseases. Previous studies have demonstrated that phenotypic modulation of VSMCs is critical in major diseases, including atherosclerosis and hypertension [5-7]. Among these, atherosclerosis, which is responsible for major mortality worldwide, best illustrates the effects of phenotypic modulation of VSMCs. Atherosclerosis is the underlying cause of most myocardial infarction, which is associated with the rupture of atherosclerotic plaques [8-10]. Within the atherosclerotic plaques, dedifferentiated VSMCs induces the expression of various matrix metalloproteinases and inflammatory mediators, such as monocyte chemoattractant protein-1 (MCP-1). The expression of these mediators along with those produced by macrophages can promote plaque rupture and thrombosis [11-13].
Phenotypic modulation of VSMCs can be controlled by different environmental stimuli. Previous studies have demonstrated that VSMCs stimulated by PDGF, lysophosphatidic acid, inorganic phosphate, proatherogenic oxidized phospholipids, and inflammatory cytokines such as IL-1β, underwent rapid phenotypic modulation from differentiation to dedifferentiation [3,14]. Moreover, studies have confirmed that pathways involving ERK, P38 MAPKs and Akt are also involved in these processes [14]. Recently, it has been demonstrated that lysine-specific demethylase-1 and microRNA-125b tend to regulate the phenotypic modulation of VSMCs in diabetes [15,16]. However, the relationship between interleukin-1 (IL-1) receptor-associated kinase 4 (IRAK4) and the modulation has not yet been investigated, neither in diabetic nor in non-diabetic individuals.
In our previous study, we found greater vascular neointima formation in Goto-Kakizaki (GK) rats than in Wistar rats following the balloon injury [17]. Deregulation of the phenotypic modulation of VMSCs is the initial stage of atherosclerosis and neointima formation, especially in diabetes. To explore the molecular mechanism and signaling pathways regulating the modulation in diabetes patients could help target the vascular complications. Direct genetic evidence indicates that a functional deficiency of IRAK4 inhibits the formation of vascular lesions in ApoE-deficient mice [18]. IRAK4 has the potential to regulate multiple aspects of vascular disease. In the present study, we explored the anti-atherosclerosis effects of IRAK4 via phenotypic modulation of VSMCs in type 2 diabetes (T2D) rats.
Materials and methods
Generation of T2D rat model
Four-week-old male Spraque-Dawley rats were purchased from the Xu Zhou Medical College Laboratory Animal Institution and fed with a high-fat diet (HFD; 58% fat, 25% protein, and 17% carbohydrate, as a percentage of total kcal). The animals were provided by the Institute of Laboratory Animal of Xuzhou Medical College. The experimental protocols, especially the ethical animal care and use of animals, were approved by the Ethical Committee in Xuzhou Medical College. After two weeks of HFD feeding, the rats were injected intraperitoneally with a low dose of streptozotocin (STZ; 35 mg/kg; Sigma, USA) [19,20]. The control rats were also subjected to the same procedure without STZ. One week after the STZ injection, the rats with non-fasting plasma glucose ≥ 16.7 mmol/L were considered diabetic and were used for further studies.
VSMCs culture
VSMCs were harvested from the thoracic aortas of the T2D rats using media explant technique. For experiments, VSMCs were used with passage numbers ranging from 3 to 5. They were maintained in Dulbecco’s modified Eagle medium (DMEM, Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin. The VSMCs were serum-starved for 24 h before treatment or stimulation with reagents.
VSMCs viability
The effect of the IRAK1/4 inhibitor or LPS on VSMCs viability was determined by tetrazolium salt reduction method using the water-soluble tetrazolium salt-1 (WST-1) assay according to the manufacturer’s instructions. Briefly, 1×104 cells were seeded in a 96-well plate in a final volume of 100 µL and cultured overnight. The cells were pretreated with IRAK1/4 inhibitor (1 µM) for 1 h and then were stimulated with 10 µg/mL LPS for 24 h. WST-1 (10 µL) was added and VSMCs were incubated for an additional 2 h. The absorbance at 450 nm was taken as the measure of the number of living cells. All the treatments were performed in triplicates, and the experiment was repeated three times.
Migration
Cell migration was assayed using a modification of the Boyden chamber method involving a polycarbonate filter (Corning, USA) with an 8.0 µm diameter pore. Migration activity was expressed as the mean number of migrated cells in three different fields.
5-ethynyl-29-deoxyuridine (Edu) staining
VSMCs were seeded in 6-well plates and stimulated with LPS (10 µg/mL) in the presence or absence of IRAK1/4 inhibitor (1 µM) for 24 h. Each well was incubated with serum-free DMEM, supplemented with 50 µM EDU (Rui Bo Guangzhou Biotechnology Co. Ltd., China) for 2 h. The cells were fixed with PBS, containing 4% paraformaldehyde, followed by washing with 2 mg/mL glycine in double distilled water for 10 min. They were permeabilized with 0.5% Triton X-100 in PBS for 10 min and washed with PBS for 5 min. Apollo reaction buffer, catalyst, fluorescent dyes, and buffer additives were dissolved in deionized water and shaken to generate the staining solution. The sections were incubated for 30 min in the dark and then washed twice with PBS for 10 min. For DNA staining, the sections were stained with 500 µl 1 X Hoechst33342 for 30 min in the dark. The wells were washed twice with PBS for 3 min and examined immediately under a fluorescence microscope (magnification 6400). All the procedures were carried out at room temperature. An Olympus BX51 microscope (Olympus, Japan) was used to detect EdU-positive cells.
siRNA-IRAK4 transfection
Transfection of siRNA was performed according to the manufacturer’s instructions. Lipofectamine 2000 (Invitrogen, USA) was used as the transfection reagent. The following siRNA oligos (Genepharma, China) were used: IRAK4: sense GUGACAACCUGUGCUUAGUTT, and antisense ACUAAGCACAGGUUGUCACTT. Cells grown on 6-well plates were transfected for experiments.
Enzyme-linked immunosorbent assay (ELISA)
To measure the generation of MCP-1, VSMCs were treated with LPS (10 µg/mL) or IRAK1/4 inhibitor for 24 h. The culture medium was collected and centrifuged (6000 g, 5 min at 4°C) and the supernatants were frozen at -80°C pending analysis. MCP-1 activity was determined using a commercial ELISA kit (Rui Bo Guangzhou Biotechnology Co. Ltd., China) according to the manufacturer’s standard protocol.
Isolation of mRNA and quantitative real-time reverse-transcription polymerase chain (qRT-PCR)
Following the treatments, mRNA was isolated from VSMCs cultures using a TRIzol reagent kit (Invitrogen, CA, USA). cDNAs were obtained using Script M-MLV (Tiangen Biochemical Technology, China). Real-time PCR was performed using 2 × SuperRealPreMix (SYBR Green, Tiangen Biochemical Technology, China) and 50 × ROX Reference Dye on an ABI7500 QPCR System. The PCR results were normalized to the expression levels of β-actin prior to further analysis. Relative gene expression was quantified by the comparative CT method (2-ΔΔCT). Rat β-actin was used as the internal reference gene. Primer sequences for IRAK4 were 5’-CCTGGCACATGAGATGCAAGATT-3’ (forward) and 5’-CGTGCAAGCCCAAAGTCAGATAT-3’ (reverse); for nuclear factor κB (NF-κB) p65 5’- GAAGCAACAGCTCACGGAGGA-3’ (forward) and 5’-TGTTCTGGAAGTTGAGGAAGGCC-3’ (reverse); for myosin heavy chain(MYH) 5’-GAAAGCGATGGTCAACAAAGAT-3’ (forward) and 5’-ACACAGAAGAGTCCCGAGTAAG-3’ (reverse); for smooth muscle 22α (SM22α) 5’-GAGCGG-CTAGTGGAGTGGATTG-3’ (forward) and 5’-AGGCACCTTCACTGGCTTGGAT-3’ (reverse); for MCP-1 5’-CTGTGCTGACCCCAATAAGGAAT-3’ (forward) and 5’-AGGTGGTTGTGGAAAAGAGAGTG-3’ (reverse); and for β-actin 5’-CCCATCTATGAGGGTTACGC-3’ (forward) and 5’-TTTAATGTCACGCACGATTTC-3’ (reverse).
Western blotting
Western blotting was used to determine the levels of IRAK4, NF-κB p65, MYH, SM22α, and activation of p-IRAK4 and p-NF-κB p65 in VSMCs upon LPS (10 µg/mL, Sigma, USA) stimulation. Cells were lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology, China), and the protein content was determined using BCA protein assay reagent (Beyotime Institute of Biotechnology, China). Proteins (25 µg) were resolved by electrophoresis on 6% or 10% SDS-polyacrylamide gels (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes wereblocked with 5% BSA andprobed with primary antibodies, including anti-IRAK4 (Cell Signaling, USA), anti-NF-κB p65 (Santa Cruz, USA), anti-MYH (Santa Cruz, USA), anti-SM22α (Santa Cruz, USA), anti-p-IRAK4 (Cell Signaling, USA), and anti-p-NF-κB p65 (Santa Cruz, USA). The membranes were incubated overnight at 4°C, washed in PBS-0.1% Tween 20, and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After successive washes, the membranes were developed using an ECL kit. Mouse anti-β-actin was used as a loading control. Protein expression levels were quantified as optical densities.
Statistical analysis
The results are expressed as mean ± standard error of the mean (SEM). Statistical significance was estimated using one-way analysis of variance (ANOVA) for multiple comparisons, while a two-tailed Student’s t-test was used to compare the data from only two groups. Differences were considered significant at P < 0.05.
Results
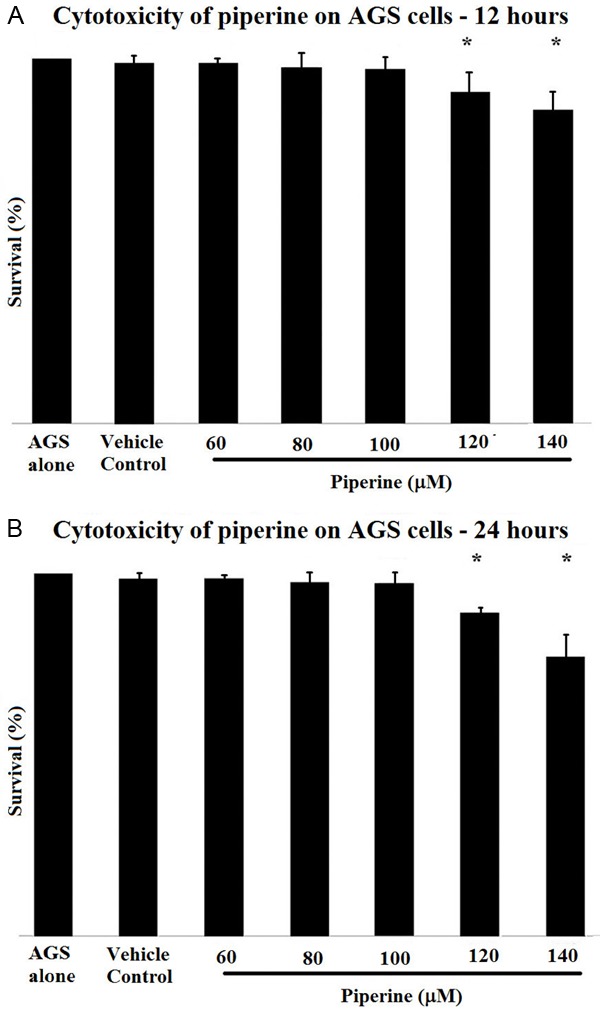
Effects of IRAK4 on LPS-induced proliferation and migration of VSMCs
Serum-starved VSMCs were incubated with 10 µg/mL LPS for 24 h or 6 h toassess the effect of IRAK4 on LPS-stimulated VSMCs viability or migration. Treatment with 10 µg/mL LPS resulted in increased proliferation and migration of VSMCs than unstimulated cells. However, inactive IRAK4 resulted in significant (P < 0.01) decrease in LPS-stimulatedcell viability and migration (Figures 1 and 2).
Figure 1.
Inhibitive effects of IRAK1/4 inhibitor on VSMCs viability induced by LPS. VSMCs were stimulated by 10 µg/mL LPS for 24 h after IRAK1/4 (1 µmol/L) inhibitor pretreated for 1 h. WST-1 assays were performed to measure cell viability. Dataisshown as the mean ± SEM of 3 independent experiments. **P < 0.01 compared with the control group; ##P < 0.01 compared with the LPS group.
Figure 2.
Inhibitory effects of IRAK1/4 inhibitor on LPS-induced VSMC migration. After being incubated with 1 µM IRAK1/4 inhibitor for 1 h, VSMCs were stimulated with 10 µg/mL LPS. The VSMCs migration rate was determined by Transwell chamber. Bright-field images of randomly selected squares per group (×100). The cell migration rate of the control group was taken as 1. ***P < 0.001 compared with the control group; ###P < 0.001 compared with the LPS group; &&P < 0.01 compared with the LPS + IRAK1/4 inhibitor group.
The levels of DNA synthesis were determined by EDU staining. Stimulation of VSMCs with 10 µg/mL LPS resulted in a significant increase in DNAlevel; however, 1 µmol/L IRAK1/4 inhibitor significantly inhibited this increase (Figure 3).
Figure 3.
Inhibitory effects of IRAK1/4 inhibitor on LPS-induced VSMCs proliferation. VSMCs were pretreated with IRAK1/4 inhibitor (1 µmol/L) for 1 h before adding LPS (10 µg/mL). Each well was treated with 500 µL EdU (50 mmol/mL) and incubated for 2 h. After incubation, whole-cell extracts were prepared, and detected by EdU assays described in Materials and Methods section. **P < 0.01 compared with the control group; ***P < 0.001 compared with the control group; ###P < 0.001 compared with the LPS group; &P < 0.05 compared with the LPS + IRAK1/4 inhibitor group.
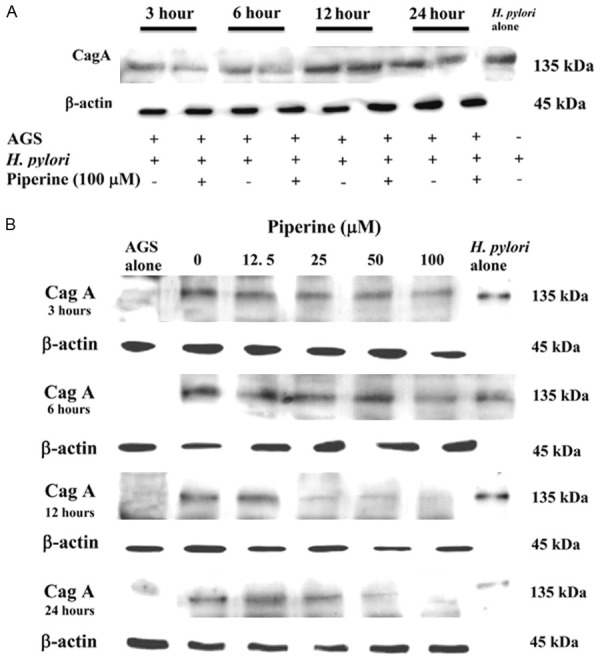
Effects of IRAK4 on LPS-stimulated VSMCs dedifferentiation
We assessed specific VSMCs markers using western blotting to determine whether IRAK4 influenced LPS-mediated phenotypic modulation of VSMCs. The results demonstrated that IRAK1/4 inhibitor reduced LPS-mediated MYH protein expression but increased the expression of SM22α (Figure 4A and 4B).
Figure 4.
Effect of IRAK4 on the translation and transcription of LPS-mediated VSMCs specific markers. (1) VSMCs were pretreated with 1 µM IRAK1/4 inhibitor for 1 h followed by 24 h 10 µg/mL LPS stimulation. The cell lysates were analyzed by western blot analysis against anti-MYH and anti-SM22α (A and B). Data represented the mean ± SEM of triplicate samples from a single experiment, and the results were representative of three independent experiments. *P < 0.05 compared with the control group; **P < 0.01 compared with the control group; #P < 0.05 compared with the LPS group; ###P < 0.001 compared with the LPS group; &P < 0.05 compared with the LPS + IRAK1/4 inhibitor group; &&P < 0.01 compared with the LPS + IRAK1/4 inhibitor group. (2) VSMCs were untransfected or transfected with siR-IRAK4 for 48 h, and then stimulated by LPS (10 µg/mL) for 2 h. Total RNA was subjected to RT-PCR to measure the relative expression of MYH and SM22α (C and D). Data represents the mean ± SEM of triplicate samples from a single experiment, and the results are representative of three independent experiments. **P < 0.01 compared with the control group; ***P < 0.001 compared with the control group; ###P < 0.001 compared with the LPS group; &P < 0.05 compared with the LPS + siR-IRAK4 group; &&P < 0.01 compared with the LPS + siR-IRAK4 group.
We also assessed specific VSMCs markers using qRT-PCR. The cells, transfected with siRNA-IRAK4 for 48 h, were treated with 10 µg/mL LPS for 24 h and then the mRNA expression of MYH and SM22α was examined. Transfection of siRNA-IRAK4 significantly inhibited the LPS-mediated mRNA expression of the differentiation marker, MYH, and promoted the dedifferentiation marker, SM22α (Figure 4C and 4D).
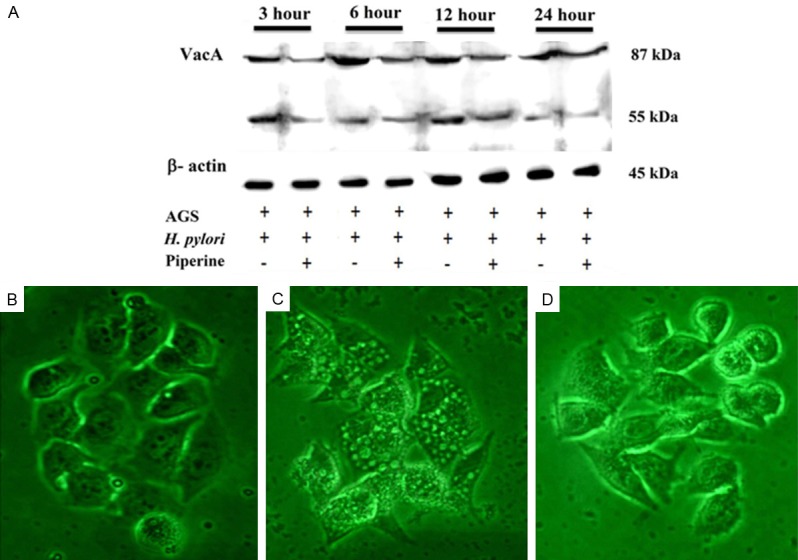
Effects of IRAK4 in NF-κB activation
To study the role of IRAK4 in LPS-induced NF-κB activation, we assessed NF-κB activity using western blotting. The results demonstrated thatthe IRAK1/4 inhibitor reduced LPS-mediated NF-κB protein expression (Figure 5A and 5B).
Figure 5.
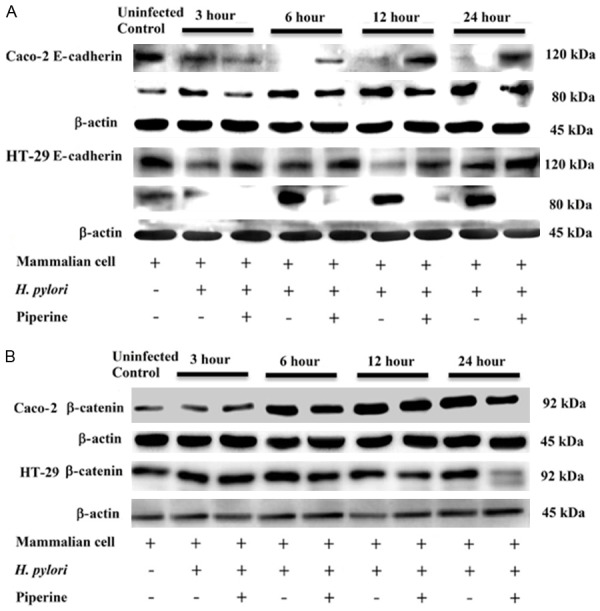
Effect of IRAK4 kinase activity on the activation of NF-κB. (1) VSMCs were pretreated with 1 µM IRAK1/4 inhibitor for 1 h, followed by 10 µg/mL LPS stimulation for 2 h. The cell lysates were analyzed by western blot analysis against anti-IRAK4, anti-p-IRAK4, anti-NF-κB p65, and anti-p-NF-κB p65 (A and B). Data represents the mean ± SEM of triplicate samples from a single experiment, and the results are representative of three independent experiments. ***P < 0.001 compared with the control group; #P < 0.05 compared with the LPS group; ##P < 0.01 compared with the LPS group; ###P < 0.001 compared with the LPS group. (2) VSMCs were untransfected or transfected with siR-IRAK4 for 48 h, and then stimulated by LPS (10 µg/mL) for 2 h. Total RNA was subjected to RT-PCR to measure the relative expression of IRAK4, NF-κB p65 (C and D). The expression of IRAK4 and NF-κB p65 normalized to β-actin expressionis demonstrated. Data represents the mean ± SEM of triplicate samples from a single experiment, and the results are representative of three independent experiments. *P < 0.05 compared with the control group; ***P < 0.001 compared with the control group; #P < 0.05 compared with the LPS group; ##P < 0.01 compared with the LPS group; ###P < 0.001 compared with the LPS group; &&&P < 0.001 compared with the LPS + siR-IRAK4 group.
We also assessed NF-κB activation using qRT-PCR. VSMCs, transfected with siRNA-IRAK4 for 48 h, were treated with 10 µg/mL LPS for 2 h and then the expression of IRAK4 and NF-κB mRNAs was examined. Transfection of siRNA-IRAK4 partially inhibited the LPS-mediated expression of NF-κB Mrna (Figure 5C and 5D).
Effect of IRAK4 on LPS-mediated MCP-1 expression
We assessed the expression of MCP-1 mRNA using qRT-PCR. VSMCs, transfected with siRNA-IRAK4 for 48 h, were treated with 10 µg/mL LPS for 2 h and then the expression of MCP-1 mRNA was examined. Transfection of siRNA-IRAK4 partially inhibited the LPS-mediated expression of mRNA (Figure 6A).
Figure 6.
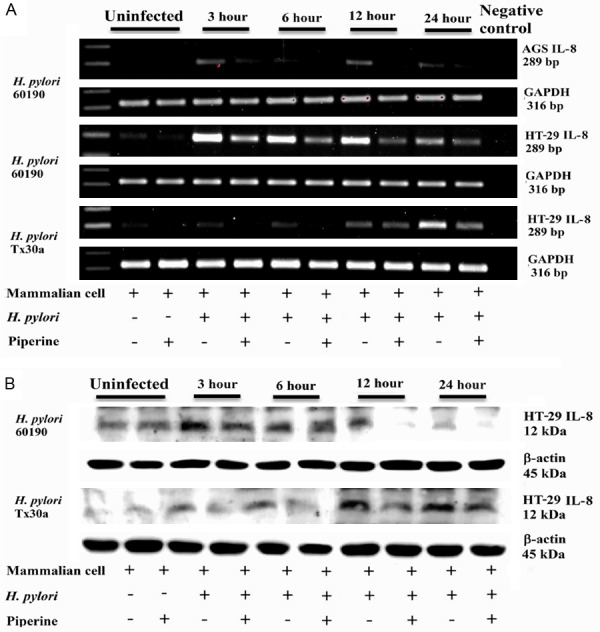
Effect of IRAK4 on LPS-mediated MCP-1 expression. (1) VSMCs were untransfected or transfected with siR-IRAK4 for 48 h, and then stimulated by LPS (10 µg/mL) for 2 h. Total RNA was subjected to RT-PCR to measure the relative expression of MCP-1 (A). The expression of MCP-1 normalized to β-actin expressionis demonstrated. (2) VSMCs were pretreated by IRAK1/4 inhibitor for 1 h, and then stimulated by LPS (10 µg/mL) for 24 h. Cell culture medium was collected, centrifuged (6000 g, 5 min at 4°C) and supernatants were analyzed to determine MCP-1 activity (B). Data represents the mean ± SEM of triplicate samples from a single experiment, and the results are representative of three independent experiments. *P < 0.05 compared with the control group; ***P < 0.001 compared with the control group; #P < 0.05 compared with the LPS group; ##P < 0.01 compared with the LPS group; ###P < 0.001 compared with the LPS group; &&&P < 0.01 compared with the LPS + IRAK1/4 inhibitor group.
Analysis of ELISA results demonstrated that IRAK1/4 inhibitor reduced LPS-mediated MCP-1 expression (Figure 6B).
Discussion
VSMCs, constituents of the vascular wall, are usually located in the arterial media and sustain a differentiated nonproliferative state. Stress or injury to arteries induces the VSMCs to migrate into the intimal layer of the arterial wall, where they switch from a differentiated to a dedifferentiated state with enhanced proliferation and synthesis of extracellular matrix proteins. In the dedifferentiated state, a concomitant downregulation of VSMCs-specific differentiation markersoccurs [7].
In T2D, vascular complications, such as cardiovascular disease are the main causes of high morbidity and mortality [21]. Cardiovascular diseasesare caused by atherosclerosis and are usuallyaccelerated by diabetes [22]. Atherosclerosis is the underlying cause for most of themyocardial infarctions, which are associated with the rupture of atherosclerotic plaques [8,9]. Previousstudies have confirmed that the development of atherosclerotic plaques is associated with an inflammatory reaction at the site of injury, migration, and proliferation of VSMCs, extracellular matrix formation, insulin resistance and hyperglycemia [9,23-26]. These processesare related to the phenotypic switching of VSMCs from differentiation to dedifferentiation, which is characterized by markedly increased proliferation, migration, and synthesis of extracellular matrix components [3]. In other words, the phenotypic modulation of VSMCs plays a vital role in the development of atherosclerosis.
In our previous study, we also reported increased synthesis of DNA in the vascular neointima in Goto-Kakizaki (GK) rats than in Wistar rats followingballoon injury [17]. Further, we demonstratedthat IRAK1/4 inhibitor inhibited neotintimal formation in Wistar rats following balloon injury [27]. Previousstudies have shownthat the mechanism of neointimal proliferation is associated with migration, proliferation of VSMCs, and extracellular matrix formation [28]. We have shown that IRAK1/4 inhibitor decreased LPS-stimulatedcell proliferation and migration, there by demonstrating the potent reductive effect of IRAK4 on LPS-stimulated VSMCs proliferation and migration.
Phenotypic modulation of VSMCs is characterized by the downregulation of differentiation marker genes and upregulation of genes involvedin inflammation, proliferation, and extracellular matrix [29]. MYH is a phenotypic marker of dedifferentiated proliferative VSMCs, whereas SM22α is a marker of differentiated contractile VSMCs [30]. IRAK1/4 inhibitor increased the level of MYH protein, but decreased the protein level of SM22α under LPS stimulation. Further, we knocked down IRAK4 with siRNA, which was accompanied by changes in the VSMCs-specific markers MYH and SM22α. Our study results, which were consistent with the results of western blotting, demonstrated that downregulation of IRAK4 inhibited LPS-mediated VSMCs dedifferentiation.
Atherosclerosis is an immunoinflammatory disease [31,32]. Innate immunity is a first-line defense against a causative organism. Stimulation of the innate immune system by TLRs activates the acquired immune system by inducing theproduction of proinflammatory cytokines [33]. The IL-1R-TLR super family plays key roles in immune responses [34]. IRAK4, a member of the IRAK family, is critical in Toll-like receptor mediated innate immune responses, which ischaracterized by the up regulation of inflammatory genes expresionin multiple target cells [33]. Previous studies have clearly demonstrated a link between innate immunity and atherosclerosis [31,32]. Direct genetic evidence indicated that functional deficiency of IRAK4 inhibited the formation of vascular lesions in ApoE-deficient mice [18]. Furthermore, inactivation of IRAK4 kinase did not completely abolish NF-κB activation, but reduced the stability of LPS-mediated mRNAs, therebydecreasing the production of cytokine and chemokine [13]. We have shownthat IRAK1/4 inhibitor decreased LPS-mediated NF-κB activation. Moreover, we knocked down IRAK4 with siRNA, which was accompanied by the downregulation of NF-κB activity. Taken together, these findings indicated that IL-1R-TLR is involved in the phenotypic modulation of VSMCs in T2D rats.
The pathophysiological mechanism of atherosclerosis includes an inflammatory process and VSMCs proliferation [35]. While VSMCs proliferation is crucial for the formation of atherosclerotic plaques, plaque disruption is associated with the inflammatory process. Previous studies have demonstrated that rupture of atheromatous plaque occurs inhighly inflamed regions, where macrophages areaccumulated [36,37]. MCP-1, also known as chemokine (C-C motif) ligand 2 (CCL2), has been found to be critical in insulin resistance and recruitment of macrophages and monocytes into atherosclerotic tissue and other chronic inflammatory lesions [38,39]. It is abundantly produced in various inflammatory diseases, such as atherosclerosis and diabetes and plays an essential role in atherosclerosis and neointimal hyperplasia [40]. Recent studies have demonstratedthat MCP-1 promotes VSMCs migration and proliferation [41]. NF-κB is a crucial regulator of MCP-1 gene expression [42]. In T2D rat model, IRAK1/4 inhibitor decreased the secretion of MCP-1. Further, we knocked down IRAK4 with siRNA, which was accompanied by the decreasein MCP-1 expression. Downregulation of MCP-1 is associatedwith the inhibition of macrophage recruitment into early lesions and phenotypic modulation of VSMCs.
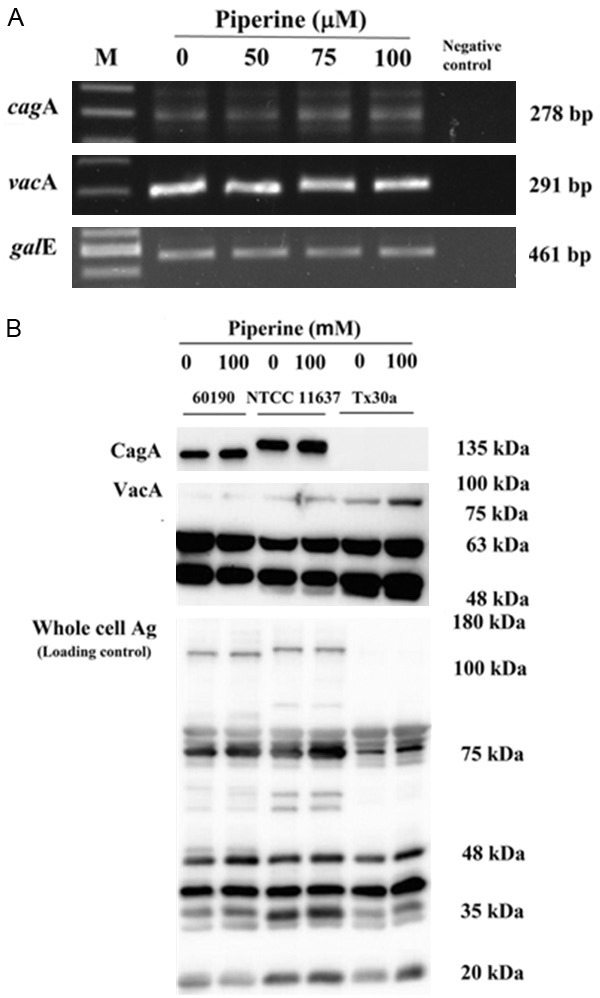
Conclusion
Our results have shown that IRAK4 is a potent regulator of VSMCs phenotype and can potentially regulate multiple aspects of VSMCs biology, including proliferation and migration, inflammation, and decreases morbidity and mortality associated with cardiovascular diseases. Our findings provide insights into a new class of intracellular receptor responsible for VSMCs differentiation and suggested that enhancement of IRAK4 expression could be a potential therapeutic approach foraltering the course of atherosclerosis. Further, our results also demonstratedthat inactivation of IRAK4 kinase decreases the expression of MCP-1, an anti-inflammatory molecule associated withplaque rupture (Figure 7). Therefore, IRAK4 could be a potential preventionand treatment of atherosclerosis.
Figure 7.
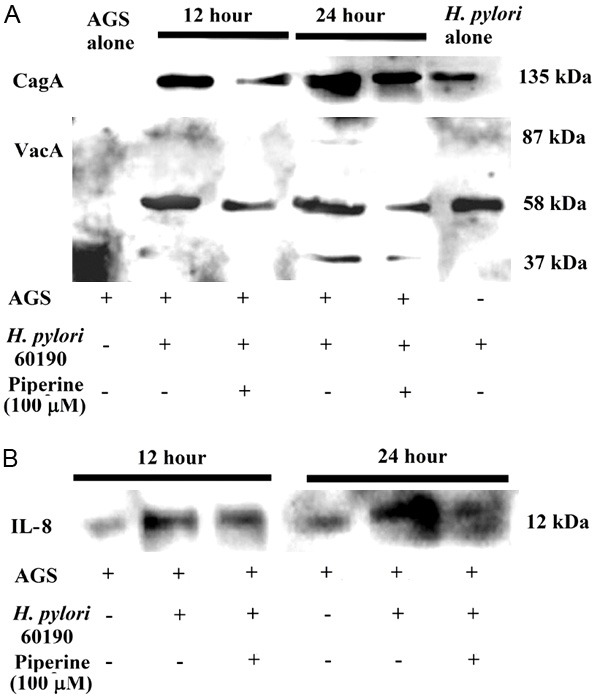
Proposed model for phenotypic modulation of VSMCs. Under LPS stimulation, the activation of IRAK4 promotes VSMCs phenotypic modulation from contractile type to synthetic type, which is characterized by markedly increased proliferation, migration, and secretion of inflammatory cytokine, MCP-1. MCP-1 can promote VSMCs migration and proliferation. NF-κB is a crucial regulatory factor of MCP-1 gene expression. IRAK4 is knockdown by siRNA and inhibited by IRAK1/4 inhibitor, which isaccompanied by the downregulation of NF-κB activity, phenotypic modulation from synthetic type to contractile type, and decrease in MCP-1 expression. These findings indicated that IL-1R-TLR participate inthe VSMCs phenotype modulation and downregulation of IRAK4, which inhibits LPS-mediated VSMCs dedifferentiation.
Collectively, our findings suggested that inactivation of IRAK4 promotes the conversion of LPS-stimulated VSMCs phenotype from dedifferentiation to differentiation. We confirm the findings in two levels of transcription and translation. Our study provided a new rationale for understanding the mechanisms of VSMCs phenotypic modulation involved in the progression of atherosclerosis in T2D.
References
- 1.Martin-Garrido A, Williams HC, Lee M, Seidel-Rogol B, Ci X, Dong JT, Lassegue B, Martin AS, Griendling KK. Transforming growth factor beta inhibits platelet derived growth factor-induced vascular smooth muscle cell proliferation via Akt-independent, Smad-mediated cyclin D1 downregulation. PLoS One. 2013;8:e79657. doi: 10.1371/journal.pone.0079657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol. 2011;31:2370–2377. doi: 10.1161/ATVBAHA.111.226670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee MH, Kwon BJ, Seo HJ, Yoo KE, Kim MS, Koo MA, Park JC. Resveratrol inhibits phenotype modulation by platelet derived growth factor-bb in rat aortic smooth muscle cells. Oxid Med Cell Longev. 2014;2014:572430. doi: 10.1155/2014/572430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang S, Song S, Lee J, Yoon J, Park J, Choi S, Park JK, Choi K, Choi C. Phenotypic modulation of primary vascular smooth muscle cells by short-term culture on micropatterned substrate. PLoS One. 2014;9:e88089. doi: 10.1371/journal.pone.0088089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C. MicroRNA and vascular smooth muscle cell phenotype: new therapy for atherosclerosis? Genome Med. 2009;1:85. doi: 10.1186/gm85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meloche J, Paulin R, Courboulin A, Lambert C, Barrier M, Bonnet P, Bisserier M, Roy M, Sussman MA, Agharazii M, Bonnet S. RAGE-dependent activation of the oncoprotein Pim1 plays a critical role in systemic vascular remodeling processes. Arterioscler Thromb Vasc Biol. 2011;31:2114–2124. doi: 10.1161/ATVBAHA.111.230573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudryavtseva O, Aalkjaer C, Matchkov VV. Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J. 2013;280:5488–5499. doi: 10.1111/febs.12414. [DOI] [PubMed] [Google Scholar]
- 8.Davies MK, Hollman A. Atherosclerosis and myocardial infarction. Heart. 1998;79:218. doi: 10.1136/hrt.79.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadeghi MM, Glover DK, Lanza GM, Fayad ZA, Johnson LL. Imaging atherosclerosis and vulnerable plaque. J Nucl Med. 2010;51(Suppl 1):51S–65S. doi: 10.2967/jnumed.109.068163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu LH, Huang L, Zhang X, Zhang P, Zhang SM, Guan H, Zhang Y, Zhu XY, Tian S, Deng K, Li H. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin Sci (Lond) 2015;129:129–145. doi: 10.1042/CS20140679. [DOI] [PubMed] [Google Scholar]
- 11.Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, Steer BM, Ingram AJ, Gupta M, Al-Omran M, Teoh H, Marsden PA, Verma S. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation. 2012;126:S81–90. doi: 10.1161/CIRCULATIONAHA.111.084186. [DOI] [PubMed] [Google Scholar]
- 12.von der Thusen JH, Borensztajn KS, Moimas S, van Heiningen S, Teeling P, van Berkel TJ, Biessen EA. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol. 2011;178:924–934. doi: 10.1016/j.ajpath.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 14.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 15.Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natarajan R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes. 2010;59:2904–2915. doi: 10.2337/db10-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao ZX, Cao LL, Luo Q, Yu L, Wang T, Wang XS, Kang LM, Liu HM. [Role of spleen tyrosine kinase in phenotypic modulation of vascular smooth muscle cell induced by platelet-derived growth factor-BB] . Zhonghua Er Ke Za Zhi. 2010;48:460–464. [PubMed] [Google Scholar]
- 17.Guo J, Li D, Bai S, Xu T, Zhou Z, Zhang Y. Detecting DNA synthesis of neointimal formation after catheter balloon injury in GK and in Wistar rats: using 5-ethynyl-2’-deoxyuridine. Cardiovasc Diabetol. 2012;11:150. doi: 10.1186/1475-2840-11-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rekhter M, Staschke K, Estridge T, Rutherford P, Jackson N, Gifford-Moore D, Foxworthy P, Reidy C, Huang XD, Kalbfleisch M, Hui K, Kuo MS, Gilmour R, Vlahos CJ. Genetic ablation of IRAK4 kinase activity inhibits vascular lesion formation. Biochem Biophys Res Commun. 2008;367:642–648. doi: 10.1016/j.bbrc.2007.12.186. [DOI] [PubMed] [Google Scholar]
- 19.Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–320. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Reed MJ, Meszaros K, Entes LJ, Claypool MD, Pinkett JG, Gadbois TM, Reaven GM. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism. 2000;49:1390–1394. doi: 10.1053/meta.2000.17721. [DOI] [PubMed] [Google Scholar]
- 21.Snell-Bergeon JK, Wadwa RP. Hypoglycemia, diabetes, and cardiovascular disease. Diabetes Technol Ther. 2012;14(Suppl 1):S51–58. doi: 10.1089/dia.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117. doi: 10.1186/1741-7015-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karrowni W, Li Y, Jones PG, Cresci S, Abdallah MS, Lanfear DE, Maddox TM, McGuire DK, Spertus JA, Horwitz PA. Insulin resistance is associated with significant clinical atherosclerosis in nondiabetic patients with acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2013;33:2245–2251. doi: 10.1161/ATVBAHA.113.301585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chait A, Bornfeldt KE. Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res. 2009;50(Suppl):S335–339. doi: 10.1194/jlr.R800059-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silvestre-Roig C, de Winther MP, Weber C, Daemen MJ, Lutgens E, Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114:214–226. doi: 10.1161/CIRCRESAHA.114.302355. [DOI] [PubMed] [Google Scholar]
- 26.Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J, Littlewood T, Bennett M. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res. 2015;116:816–826. doi: 10.1161/CIRCRESAHA.116.304921. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Wang Y, Hu W, Li D, Zhou Z, Pan D, Wu W, Xu T. Interleukin-1/toll-like receptor-induced nuclear factor kappa B signaling participates in intima hyperplasia after carotid artery balloon injury in goto-kakizaki rats: a potential target therapy pathway. PLoS One. 2014;9:e103794. doi: 10.1371/journal.pone.0103794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI. Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc Interv. 2011;4:1057–1066. doi: 10.1016/j.jcin.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang H, Hata A. MicroRNA regulation of smooth muscle gene expression and phenotype. Curr Opin Hematol. 2012;19:224–231. doi: 10.1097/MOH.0b013e3283523e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arakawa E, Hasegawa K, Irie J, Ide S, Ushiki J, Yamaguchi K, Oda S, Matsuda Y. L-ascorbic acid stimulates expression of smooth musclespecific markers in smooth muscle cells both in vitro and in vivo. J Cardiovasc Pharmacol. 2003;42:745–751. doi: 10.1097/00005344-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Loppnow H, Buerke M, Werdan K, Rose-John S. Contribution of vascular cell-derived cytokines to innate and inflammatory pathways in atherogenesis. J Cell Mol Med. 2011;15:484–500. doi: 10.1111/j.1582-4934.2010.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paramel GV, Folkersen L, Strawbridge RJ, Elmabsout AA, Sarndahl E, Lundman P, Jansson JH, Hansson GK, Sirsjo A, Fransen K. CARD8 gene encoding a protein of innate immunity is expressed in human atherosclerosis and associated with markers of inflammation. Clin Sci (Lond) 2013;125:401–407. doi: 10.1042/CS20120572. [DOI] [PubMed] [Google Scholar]
- 33.Valaperti A, Nishii M, Liu Y, Naito K, Chan M, Zhang L, Skurk C, Schultheiss HP, Wells GA, Eriksson U, Liu PP. Innate immune interleukin-1 receptor-associated kinase 4 exacerbates viral myocarditis by reducing CCR5(+) CD11b(+) monocyte migration and impairing interferon production. Circulation. 2013;128:1542–1554. doi: 10.1161/CIRCULATIONAHA.113.002275. [DOI] [PubMed] [Google Scholar]
- 34.Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, Latz E, Gilmour R, Staschke K, Chisolm G, Fox PL, DiCorleto PE, Smith JD, Li X. The critical role of IL-1 receptor-associated kinase 4-mediated NF-kappaB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol. 2011;186:2871–2880. doi: 10.4049/jimmunol.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maiuri MC, Grassia G, Platt AM, Carnuccio R, Ialenti A, Maffia P. Macrophage autophagy in atherosclerosis. Mediators Inflamm. 2013;2013:584715. doi: 10.1155/2013/584715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata N, Glass CK. Macrophages, oxysterols and atherosclerosis. Circ J. 2010;74:2045–2051. doi: 10.1253/circj.cj-10-0860. [DOI] [PubMed] [Google Scholar]
- 38.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I, van den Berg S, Romijn J, Rensen PC, Joosten LA, Netea MG, Kanneganti TD. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Tai HH. Thromboxane A receptor-mediated release of matrix metalloproteinase-1 (MMP-1) induces expression of monocyte chemoattractant protein-1 (MCP-1) by activation of protease-activated receptor 2 (PAR2) in A549 human lung adenocarcinoma cells. Mol Carcinog. 2013 doi: 10.1002/mc.22020. [DOI] [PubMed] [Google Scholar]
- 40.Fu C, Yu P, Tao M, Gupta T, Moldawer LL, Berceli SA, Jiang Z. Monocyte chemoattractant protein-1/CCR2 axis promotes vein graft neointimal hyperplasia through its signaling in graft-extrinsic cell populations. Arterioscler Thromb Vasc Biol. 2012;32:2418–2426. doi: 10.1161/ATVBAHA.112.255786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang YJ, Wu LS, Shu B, Qian MZ. [MCPIP1 mediates MCP-1-induced vascular smooth muscle cell proliferation] . Sheng Li Xue Bao. 2013;65:616–622. [PubMed] [Google Scholar]
- 42.Hildebrand DG, Alexander E, Horber S, Lehle S, Obermayer K, Munck NA, Rothfuss O, Frick JS, Morimatsu M, Schmitz I, Roth J, Ehrchen JM, Essmann F, Schulze-Osthoff K. IkappaBzeta is a transcriptional key regulator of CCL2/MCP-1. J Immunol. 2013;190:4812–4820. doi: 10.4049/jimmunol.1300089. [DOI] [PubMed] [Google Scholar]