

Abstract
Exact mechanism of cerebral ischemic stroke remains unclear. The calcium-sensing receptor (CaSR), a G-protein coupled receptor, has been reported to participate in the pathology of myocardial ischemia-reperfusion (I/R) injury and myocardial hypertrophy. Nevertheless, only a limited number of studies have been conducted to investigate the role of CaSR in cerebral ischemic stroke. This study was to investigate the effect of CaSR activation on cerebral ischemic stroke. Male adult Kunming mice were subjected to 2-h focal cerebral ischemia followed by 22-h reperfusion. Then, the brain was collected, and the expression of CaSR, JNK, p38, Bcl-2, and Bax was detected by Western blot assay. The morphology of neurons in the brain was evaluated by HE staining. Neurological function was scored, and the infarct volume was determined by TTC (triphenyltetrazolium chloride) staining. Results showed that ischemia/reperfusion (I/R) increased CaSR expression and induced neuronal apoptosis in the brain. Gadolinium trichloride (GdCl3), an agonist of CaSR, further deteriorated neurological dysfunction, increased infarct volume, enhanced CaSR expression, and promoted neuronal apoptosis. In addition, GdCl3 unregulated expression of Bax, p-JNK, and p-p38, and down-regulated Bcl-2 expression during I/R, which were attenuated by NPS2390, an inhibitor of CaSR. In conclusion, the CaSR activation promotes apoptosis in focal cerebral I/R in mice, which may be related to the activation of JNK/p38 MAPK signalling pathway. Targeting CaSR may be a novel strategy for the prevention and treatment of cerebral ischemic stroke.
Keywords: Calcium-sensing receptor (CaSR), focal cerebral ischemia-reperfusion, mice, MAPK, apoptosis
Introduction
Ischemic stroke is one of the major causes of human death and significantly increases the social and family burden throughout the world [1]. Restoration of the cerebral flow is fundamental for survival after ischemic stroke. However, the reperfusion after recanalization may further induce tissue damage through some mechanisms involving excitotoxicity, calcium overload, inflammation, apoptosis, and blood-brain barrier (BBB) injury [2,3], but the exact mechanism is still poorly understood. Although the survival and quality of life have been improved in ischemic stroke patients with the development of medical technologies, the mortality and disability remain at a high level in many countries.
In 1993, Brown et al. cloned the calcium-sensing receptors (CaSR), a seven transmembrane family of class C G protein-coupled receptors (GPCRs), from the parathyroid chief cells [4]. The human CaSR is a polypeptide composed of 1078 amino acids. It has an NH2-terminal extracellular domain (ECD), a 7-transmembrane region, and an intracellular COOH-terminal tail [5]. CaSR is extensively expressed in many tissues and organs, including the stomach, kidney, bone marrow, thyroid, and smooth muscle [6-8]. CaSR has been reported to play an essential role in modulating systemic calcium homeostasis, gene expression, apoptosis and differentiation [9].
CaSR is expressed in many regions of the brain at widely varying levels. Recent evidence suggests that it is able to maintain the Ca2+ concentration and ion permeability in the brain [10]. CaSR can be activated by both extracellular Ca2+ and many ligands, including divalent and trivalent cations, L-amino acids, and polyamines [11]. CaSR activation may induce the activation of some intracellular signaling pathways, including mitogen-activated protein kinases (MAPKs). In adult rat heart, Wang et al. reported for the first time that extracellular calcium increased the intracellular free calcium [Ca2+]i through the Gq-PI3-PLC pathway [12].
It has been revealed that the CaSR activation is involved in cardiac ischemia-reperfusion (I/R) injury and may promote ventricular cardiomyocyte apoptosis in neonatal rats through activating MAPK signalling pathway [13]. However, few studies have been conductced to investigate the role of CaSR in cerebral ischemic stroke. This study was to establish a mouse model of focal cerebral ischemia-reperfusion with previosuly reported method and investigate the expression of CaSR as well as the effect of CaSR activation on the potential intracellular signaling pathways in cerebral ischemic stroke.
Materials and methods
Materials
Gadolinium trichloride (GdCl3), 2,3,5-triphenyltetrazolium chloride (TTC) and NPS2390 were purchased from Sigma-Aldrich Co. LLC. MO, USA. The anti-CaSR antibody was from Santa Cruz Biotechnology®, Inc., TX, USA. Antibodies against JNK/p-JNK and p38/p-p38 were purchased from Bioworld Technology Inc, MN, USA. Antibodies against Bax, Bcl-2 and actin were obtained from Beijing Zhongshan Jinqiao Biotechnology Co., Ltd, Beijing, China.
Animals
56 healthy male adult Kunming mice weighing 23-25 g were purchased from the Experimental Animal Center of Anhui Medical University (License: SCXK [Wan] 2011-002). The animals were allowed to accommodate the environment for one week before experiments. Animals were housed with a 12 h light/dark cycle and given ad libitum access to food and water. The experimental procedures used in this study were reviewed and approved by the Animal Experiment Ethics Committee of Anhui Medical University.
Experimental protocol
The animals were randomly assigned to 4 groups (n = 14 per group): (1) Sham-operated group (2) I/R group: mice were subjected to a middle cerebral artery occlusion (MCAO) for 2h followed by 22-h reperfusion (3) I/R + GdCl3 (a specific activator of CaSR) group: mice underwent I/R injury and GdCl3 (16 mg/kg) were infused via the tail vein at 30 min and 120 min after ischemia; (4) I/R + NPS2390 (an inhibitor of CaSR) group: mice underwent I/R injury and were infused subcutaneously with NPS2390 (1.5 mg/kg) at 30 min and 120 min after ischemia. After reperfusion, the mice were subjected to following tests.
Establishment of MCAO model in mice
Focal cerebral I/R was induced with monofilaments according to described previously [14,15]. Male Kunming mice were weighed and intraperitoneally anesthetized with 7% chloral hydrate (0.05 ml/10 g). A midline incision was made in the neck, and the right common carotid artery (CCA), the external carotid artery (ECA), and the internal carotid artery (ICA) were exposed. The ECA and the CCA were ligated using 6-0 sutures, while the ICA was occluded with a clamp. Thereafter, a silicone-coated 6-0 nylon monofilament was advanced into the ICA though a small hole in the CCA until the tip blocked the origin of the middle cerebral artery (MCA; about 10 mm away from the carotid bifurcation). The surgical incisions were closed with 6-0 sutures. During the operation, the rectal temperature was maintained at 37°C. After 120 min, the monofilament was slowly withdrawn for reperfusion and the mice were put back into cages and housed at 26 ± 2°C for 22 h. Similar procedures without MCA occlusion were performed in mice of sham-operated group.
Neurofunction evaluation
Twenty-four hours following the surgery, the neurological function was examined blindly according to previously described [14]: 0 - no neural function deficit; 1 - left forepaw weakness; 2 - turns to the left; 3 - falling to the contralateral side; 4 - unable to walk spontaneously and even coma.
Measurement of cerebral infarct volume
The brain was carefully removed after 22-h reperfusion, and sectioned into 2-mm coronal sclices. The brain slices were then incubated in 1% TTC in dark (37°C, 20 min) and then fixed in 10% formalin. Photographs of the brain slices were taken with a digital camera (Nikon, Tokyo, Japan) and the infarct area of each coronal slice was analyzed using the Image J analysis software. The ratio of infarct volume was calculated using the following formula [16]. Infarct volume ratio = (∑Infarct area × thickness)/(∑whole brain area × thickness) × 100%.
Histopathological examination
At the end of each experiment, the brain was collected, fixed in 10% formalin, embedded in paraffin, cut into sections (4 µm in thickness), deparaffinized, and stained with haematoxylin and eosin (H&E). The histological examination was performed under a light microscope.
Western blot assay
The brain was collected and washed with normal saline (NS) at 4°C. The hippocampus and cortex of the injured hemisphere were then harvested and stored at -80°C until assay. The hippocampus and cortex were homogenized, independently, in cold lysis buffer containing PMSF. After centrifugation for 10 min at 12,000 rpm at 4°C, the supernatant was collected. The protein concentration was measured with the BCA protein assay kit (Beyotime Institute of Biotechnology, Jiangsu, China). The proteins were mixed with loading buffer, loaded onto 8%, 10%, 12%, or 15% gels and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes by wet electrophoresis (100 mA for 120 min), which were then blocked with 5% milk at room temperature for 1 h. The membranes were incubated overnight at 4°C with primary antibodies: anti-CaSR (1:100), anti-p-JNK/JNK (1:1,000), anti-p-p38/p38 (1:1,000), anti-Bax (1:800), anti-Bcl-2 (1:800), and anti-β-actin (1:1,000). After washing with TBST, the membranes were exposed to anti-mouse or anti-rabbit secondary antibodies (1:10,000) at 37°C for 2 h. The visualization was performed using an enhanced chemiluminescence system (ECL). β-actin was used as an internal control. There were 6 samples in each group.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) and then analysed with SPSS version 15.0 for Windows. Comparisons were done with one-way analysis of variance (ANOVA). A value of P<0.05 was considered statistically significant.
Results
Neurological function and cerebral infarct volume
All the mice except for those in the sham group presented neurological deficits (Figure 1B). The neurofunction score of I/R group was 1.83 ± 0.41, which increased significantly after administration of Gdcl3 (2.83 ± 0.41, P<0.01 vs I/R group). However, the neurolofunction score of NPS2390 group reduced dramatically (1.17 ± 0.40, P<0.05 vs I/R group).
Figure 1.
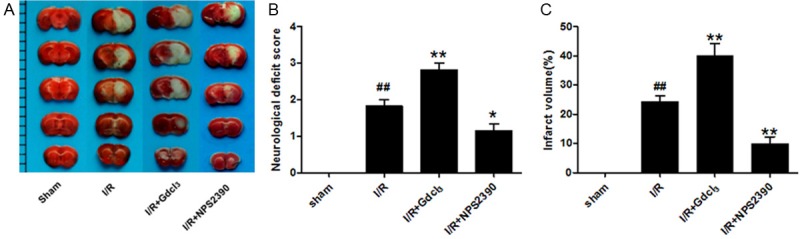
The cerebral infarct volume and neurological function in different groups. A. TTC-staining of brain slices (2 mm for each scale); B. Quantitative analysis of cerebral infarct volume; C. Quantitative analysis of neurological function. a: Sham group, b: I/R group, c: I/R + Gdcl3 group, d: I/R + NPS2390 group (n = 6 per group). ##P<0.01 vs sham group; *P<0.05, **P<0.01 vs I/R group.
After TTC staining, the normal tissues were stained deep red while the infarct area was pale grey (Figure 1A). No infarction was observed in sham group. The cerebral I/R caused a significant increase in the infarct volume. After administration of Gdcl3, the infarct volume increased significantly, while NPS2390 dramatically decreased the infarct volume (Figure 1C, P<0.01 vs I/R group). The cerebral infarct volume of I/R group, Gdcl3 group and NPS2390 group was 24.34 ± 5.06%, 40.07 ± 10.21%, and 9.89 ± 5.81%, respectively.
Histological changes in the brain
After HE staining, the hippocampal CA1 region, CA3 region, and cortex (Figure 2) were observed under a light microscope. The brains of sham group had no evident pathological changes (Figure 2e, 2i, 2m). Neurons in the hippocampal CA1 region of I/R group exhibited anachromasis and pyknosis of nuclei as well as shrunken cell body. There were deformation and swelling of the nerve cells, interstitial edema, and tissue damage in I/R group. The neurons were sparse and showed disordered structures. Moreover, a large number of neurons lost structural integrity with obvious karyopyknosis and disintegration of nucleoli. Gdcl3 significantly aggravated the abnormalities caused by I/R, especially in the hippocampal CA1 region (Figure 2g, 2k, 2o), while NPS2390 markedly inhibited cellular swelling and nuclear loss in the hippocampal CA1 region, CA3 region, and cortex (Figure 2h, 2l, 2p).
Figure 2.

Pathological changes of brain tissues after cerebral I/R. (A) Hematoxylin and eosin (HE) staining of the hippocampal CA1 region (a-h) and CA3 region (a-d, i-l) in sham (a, e, i), I/R (b, f, j), I/R + Gdcl3 (c, g, k) and I/R + NPS2390 (d, h, l) groups. (B) Hematoxylin and eosin (HE) staining of the cortex in sham (m), I/R (n), I/R + Gdcl3 (o) and I/R + NPS2390 (p) groups. Scale bar = 50 μm.
Expression of CaSR
The CaSR expression in the cortex and the hippocampus was measured by Western blot assay. The CaSR protein had the molecular weight of 160 kDa. The CaSR expression was at a low level in sham group, but significantly increased in I/R group in both cortex and hippocampus (P<0.05, P<0.01 vs sham group). The CaSR expression was markedly higher in I/R + Gdcl3 group than in I/R group (P<0.05), while NPS2390 decreased the CaSR expression (P<0.05, P<0.01 vs I/R group). (Figure 3A and 3B).
Figure 3.
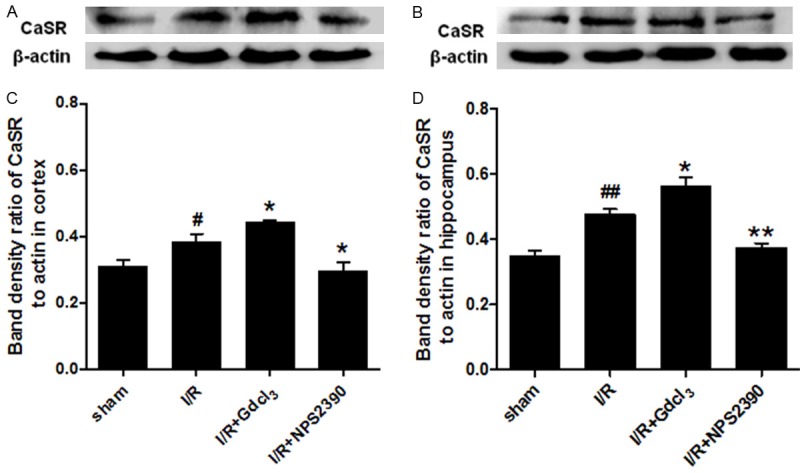
CaSR expression in the cortex (A, C) and hippocampus (B, D) after cerebral I/R (Western blot assay) (n = 6 per group), #P<0.05, ##P<0.01 vs sham group; *P<0.05, **P<0.01 vs I/R group.
Bcl-2 and Bax expressions
In order to investigate the pathway through which GdCl3 promotes apoptosis, the expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 was detected in the cortex and hippocampus by Western blot assay. The Bax expression significantly increased in the cortex and hippocampus of I/R group as compared to the sham group (P<0.05). Gdcl3 treatment further increased Bax expression (P<0.05 vs I/R group), while Bax expression markedly reduced in NPS2390 group (P<0.05 vs I/R group). The Bcl-2 expression was decreased after cerebral I/R in the cortex and the hippocampus (P<0.05 and P<0.01 vs sham group, respectively), and GdCl3 further down-regulated Bcl-2 expression (P<0.05 vs I/R group), while NPS-2390 markedly increased Bcl-2 expression (P<0.05 vs I/R). (Figures 4 and 5).
Figure 4.
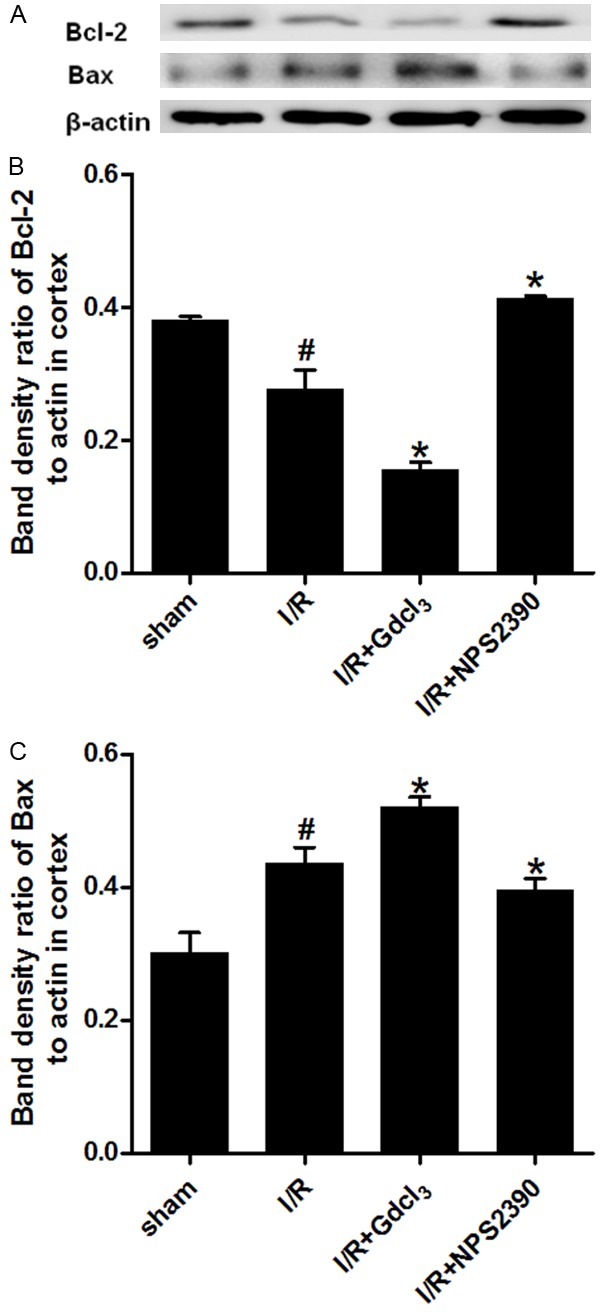
Bax and Bcl-2 expression in the cortex after cerebral I/R (Western blot assay). Western blot assay for Bax (A, C) and Bcl-2 expression (A, B) in the cortex (n = 6 per group). #P<0.05 vs sham group; *P<0.05 vs I/R group.
Figure 5.
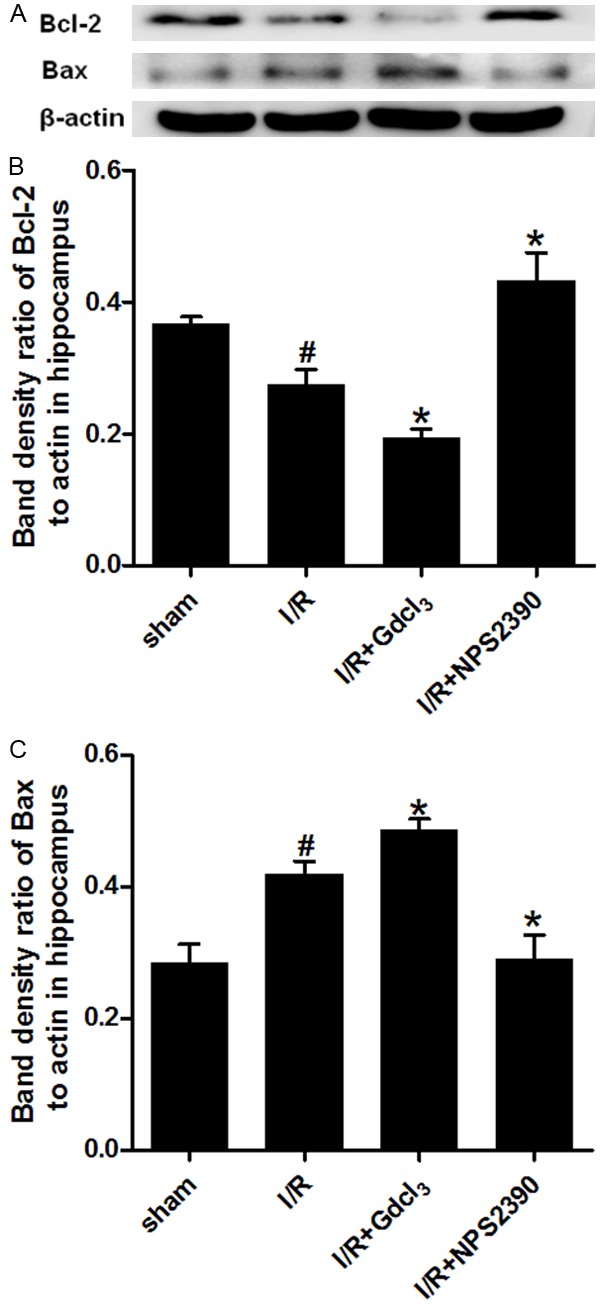
Bax and Bcl-2 expression in the hippocampus after cerebral I/R (Western blot assay). Western blot assay for Bax (A, C) and Bcl-2 expression (A, B) in the cortex (n = 6 per group). #P<0.05 vs sham group; *P<0.05 vs I/R group.
Expression of phosphorylated JNK and p38
The activity of MAPK signalling pathway was evaluated after CaSR activation in the cerebral I/R. The expressions of phosphorylated JNK (p-JNK) and phosphorylated p38 (p-p38) in the cortex and hippocampus were detected by Western blot assay. Our results showed that the expression of total JNK and p38 in the brain was comparable among groups. A low p-JNK expression was detected in the cortex and hippocampus of sham group, while p-JNK expression was up-regulated significantly after I/R (P<0.01 vs sham group). GdCl3 could further enhance the p-JNK expression induced by I/R (P<0.01 vs I/R group), but p-JNK expression was dramatically inhibited by NPS2390 (P<0.01, P<0.05 vs I/R group). Only a weak p-p38 expression was observed in sham group, but p-p38 expression markedly increased in the cortex and hippocampus after cerebral I/R (P<0.05, P<0.01 vs sham group). GdCl3 further increased the p-p38 expression in the cortex and hippocampus (P<0.05 vs I/R group), while NPS2390 significantly inhibited the p-p38 expression (P<0.05 vs I/R group) (Figures 6 and 7).
Figure 6.
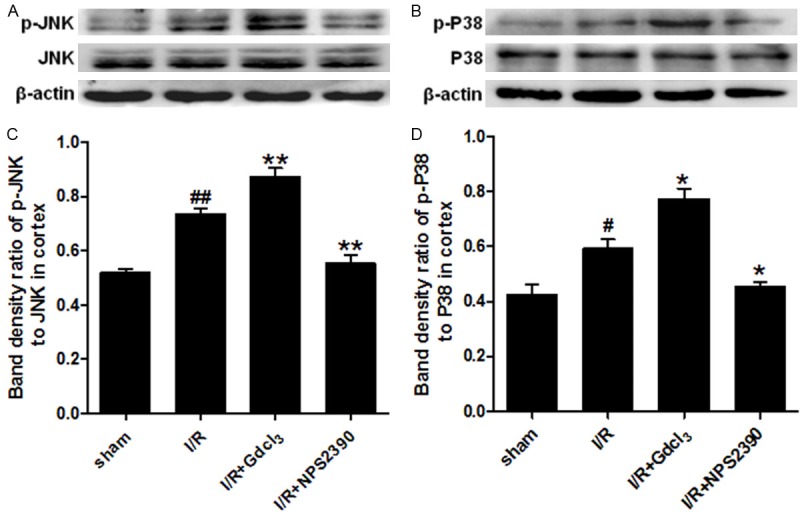
p-JNK and p-p38 expression in the cortex after cerebral I/R (Western blot assay). Western blot assay for p-JNK (A, C) and p-p38 (B, D) expression in the cortex (n = 6 per group). #P<0.05, ##P<0.01 vs sham group; *P<0.05, **P<0.01 vs I/R group.
Figure 7.
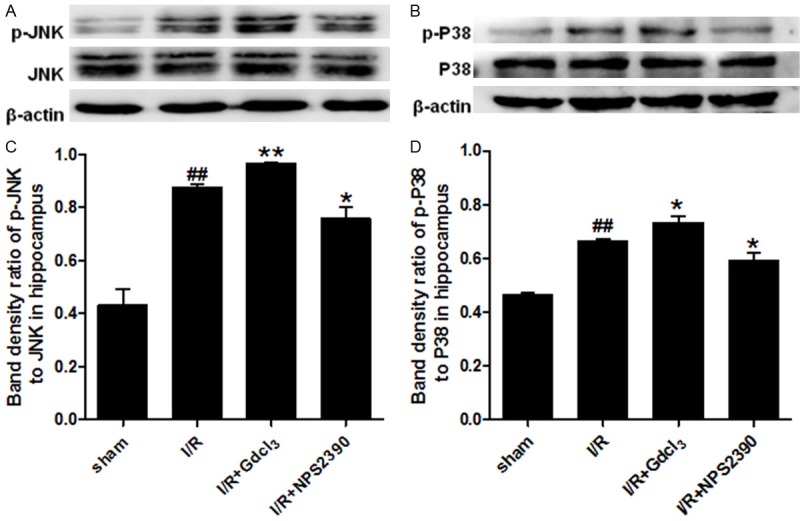
p-JNK and phosphor-p38 expression in the hippocampus after cerebral I/R (Western blot assay). Western blot assay for p-JNK (A, C) and p-P38 (B, D) expression in the hippocampus (n = 6 per group). #P<0.05, ##P<0.01 vs sham group; *P<0.05, **P<0.01 vs I/R group.
Discussion
Ischemic stroke significantly affects human health, but few medicines are available and the mortality of ischemic stroke is still at a high level. Recanalization seems to be fundamental for survival. However, it may cause reperfusion injury, even deteriorating the general ischemia. It is well known that calcium overload and apoptosis are involved in the pathogenesis of cerebral ischemic stroke. CaSR, a member of the superfamily of class C GPCRs, has been reported to participate in the pathology of myocardial I/R and myocardial hypertrophy [17]. However, little is known about the role of CaSR in the cerebral ischemic stroke. In the present study, a mouse model of focal cerebral I/R injury was established, and GdCl3 (a specific CaSR activator) as well as NPS2390 (a specific CaSR inhibitor) were employed, aiming to investigate the potential role and mechanisms of CaSR in the pathophysiological processes of cerebral I/R injury.
In this study, neurological deficit scores were evaluated and the infarct volume was determined by TTC (triphenyltetrazolium chloride) staining. Neuron apoptosis was examined by HE staining and CaSR expression was evaluated by western blotting. Our results showed cerebral I/R could significantly up-regulate the CaSR expression, cause neurological dysfunction and infarction, and induce neuronal apoptosis. These changes were further deteriorated by GdCl3, but improved by NPS2390. These findings suggest that CaSR may contribute to the development of cerebral ischemic stroke and the increased CaSR expression is closely related to the neuronal apoptosis during cerebral I/R.
There are two major forms of cell death after brain ischemia injury: necrosis and apoptosis [18]. A plethora of evidence has demonstrated that apoptosis is several times more common than necrosis. Apoptotic mechanisms are highly intricate and sophisticated, involving an energy-dependent cascade of molecular events [19]. To date, two central pathways for apoptosis induction have been described: the intrinsic pathway (or mitochondria-mediated pathway) and the extrinsic pathway (or death receptor-mediated pathway) [20]. The intrinsic pathway appears to be more complex and its molecular mechanism remains unclear. However, it is known that mitochondria play a pivotal role in this pathway [21]. Oxidative stress and calcium overload may damage the mitochondria, causing the release of cytochrome C and then activating the caspase cascade [22]. The Bcl-2 protein family is a significant participant in the mitochondrial pathway of apoptosis. Bcl-2 protein family includes two classes of regulatory proteins, pro-apoptotic proteins Bax and Bak, and anti-apoptosis proteins Bcl-2 and Bcl-x [23,24]. Bcl-2 is an important anti-apoptotic protein that protects cells against apoptosis [25]. Evidence has demonstrated that the Bcl-2 over-expression can inhibit the neuronal apoptosis [26,27]. In contrast, Bax that is regulated by the Bcl-2 family has been reported to play an essential role in regulating apoptotic cell death. The anti-apoptotic effect of Bcl-2 impedes the activity of pro-apoptotic Bax.
To investigate the neuronal apoptosis after cerebral I/R with respect to the activation of CaSR, the expression of Bcl-2 and Bax was detected by Western blot assay. Our results showed that I/R considerably increased the Bax expression but down-regulated the Bcl-2 expression. The CaSR agonist (GdCl3) or antagonist (NPS2390) further augmented or weakened the effects of I/R on the expression of apoptosis related proteins. These suggest that the pro-apoptotic role of CaSR in cerebral I/R is related to the down-regulation of Bcl-2 expression. Subsequently, the mechanism by which CaSR regulates neuronal apoptosis in focal cerebral I/R was further explored.
MAPKs play a prominent role in the adaptation, proliferation, and differentiation of neurons. Moreover, the MAPK signalling pathway is essential for the regulation of inflammation and apoptosis during cerebral I/R. The MAPK signalling pathway is generally organised into three major subgroups, consisting of ERK1/2, C-Jun N-terminal kinases (JNKs) and p38 kinases [28-30]. Several studies have shown that the ERK1/2 activation usually protects neurons from apoptosis, whereas prolonged activation of p38 and/or JNK often causes neuronal cell death [31-35]. Tournier et al. found that the phosphorylation of JNK could regulate downstream signalling pathways associated with apoptosis in ischemic injury [36]. p38 MAPK has been shown to be activated following focal cerebral ischemia [37]. However, the role of p38/MAPK in stroke is still controversial. Although the inhibition of p38 MAPK is able to protect neurons from apoptosis in permanent and transient MCAO [38,39], in one study, p38 was proven to deteriorate brain injury [40]. Moreover, p38 has been reported to mediate the Bax translocation in nitric oxide-induced apoptosis in neurons [41].
In order to investigate the relationship between MAPK pathway and CaSR activation following cerebral I/R, the expression of JNK and p38 was detected. Our results showed that cerebral I/R and GdCl3 had no influence on the expression of total JNK and p38 but significantly increased the phosphorylation of JNK and p38, which was accompanied by the elevated apoptosis and brain injury. However, NPS2390 markedly attenuated the phosphorylation of JNK and p38, which was accompanied by the attenuation of apoptosis and brain injury. These findings suggest that CaSR is involved in cerebral ischemic stroke, and may exert pro-apoptotic effect in the mouse model of focal cerebral I/R.
Conclusion
In summary, our results demonstrate that CaSR is activated following focal cerebral I/R in mice and may promote neuronal apoptosis via up-regulating Bax expression and down-regulating Bcl-2 expression, which may be regulated by the JNK/p38 MAPK signalling pathway. Thus, strategies targeting CaSR may be helpful for the prevention and treatment of cerebral ischemic stroke.
Acknowledgements
This study performed in our laboratory was supported by Natural Science Foundation of Anhui Province (1408085MH174), National Natural Science Foundation of China (81470432) and Project of Educational Commission of Anhui Province, China (KJ2014A260).
Disclosure of conflict of interest
None.
References
- 1.Pang X, Li T, Feng L, Zhao J, Zhang X, Liu J. Ellagic acid-induced thrombotic focal cerebral ischemic model in rats. J Pharmacol Toxicol Methods. 2014;69:217–222. doi: 10.1016/j.vascn.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 2010;17:197–218. doi: 10.1016/j.pathophys.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Auriel E, Bornstein NM. Neuroprotection in acute ischemic stroke--current status. J Cell Mol Med. 2010;14:2200–2202. doi: 10.1111/j.1582-4934.2010.01135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 5.Bai M. Structure and function of the extracellular calcium-sensing receptor (Review) Int J Mol Med. 1999;4:115–125. doi: 10.3892/ijmm.4.2.115. [DOI] [PubMed] [Google Scholar]
- 6.Bai M, Trivedi S, Lane CR, Yang Y, Quinn SJ, Brown EM. Protein kinase C phosphorylation of threonine at position 888 in Ca2+o-sensing receptor (CaR) inhibits coupling to Ca2+ store release. J Biol Chem. 1998;273:21267–21275. doi: 10.1074/jbc.273.33.21267. [DOI] [PubMed] [Google Scholar]
- 7.Jung SY, Kwak JO, Kim HW, Kim DS, Ryu SD, Ko CB, Cha SH. Calcium sensing receptor forms complex with and is up-regulated by caveolin-1 in cultured human osteosarcoma (Saos-2) cells. Exp Mol Med. 2005;37:91–100. doi: 10.1038/emm.2005.13. [DOI] [PubMed] [Google Scholar]
- 8.Bevilacqua M, Dominguez LJ, Righini V, Valdes V, Toscano R, Sangaletti O, Vago T, Baldi G, Barrella M, Bianchi-Porro G. Increased gastrin and calcitonin secretion after oral calcium or peptones administration in patients with hypercalciuria: a clue to an alteration in calciumsensing receptor activity. J Clin Endocrinol Metab. 2005;90:1489–1494. doi: 10.1210/jc.2004-0045. [DOI] [PubMed] [Google Scholar]
- 9.Smajilovic S, Tfelt-Hansen J. Calcium acts as a first messenger through the calcium-sensing receptor in the cardiovascular system. Cardiovasc Res. 2007;75:457–467. doi: 10.1016/j.cardiores.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 11.Fellner SK, Parker L. Ionic strength and the polyvalent cation receptor of shark rectal gland and artery. J Exp Zool A Comp Exp Biol. 2004;301:235–239. doi: 10.1002/jez.a.20029. [DOI] [PubMed] [Google Scholar]
- 12.Wang R, Xu C, Zhao W, Zhang J, Cao K, Yang B, Wu L. Calcium and polyamine regulated calcium-sensing receptors in cardiac tissues. Eur J Biochem. 2003;270:2680–2688. doi: 10.1046/j.1432-1033.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- 13.Sun YH, Liu MN, Li H, Shi S, Zhao YJ, Wang R, Xu CQ. Calcium-sensing receptor induces rat neonatal ventricular cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;350:942–948. doi: 10.1016/j.bbrc.2006.09.142. [DOI] [PubMed] [Google Scholar]
- 14.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 15.Choi BI, Park D, Lee SH, Bae DK, Yang G, Yang YH, Kim TK, Choi EK, Lee HJ, Choi KC, Nahm SS, Kim YB. Neurobehavioural deficits correlate with the cerebral infarction volume of stroke animals: a comparative study on ischaemia-reperfusion and photothrombosis models. Environ Toxicol Pharmacol. 2012;33:60–69. doi: 10.1016/j.etap.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Jin YL, Dong LY, Wu CQ, Qin J, Li S, Wang CY, Shao X, Huang DK. Buyang Huanwu Decoction fraction protects against cerebral ischemia/reperfusion injury by attenuating the inflammatory response and cellular apoptosis. Neural Regen Res. 2013;8:197–207. doi: 10.3969/j.issn.1673-5374.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang LN, Wang C, Lin Y, Xi YH, Zhang WH, Zhao YJ, Li HZ, Tian Y, Lv YJ, Yang BF, Xu CQ. Involvement of calcium-sensing receptor in cardiac hypertrophy-induced by angiotensinII through calcineurin pathway in cultured neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2008;369:584–589. doi: 10.1016/j.bbrc.2008.02.053. [DOI] [PubMed] [Google Scholar]
- 18.Abbate A, Biondi-Zoccai GG, Baldi A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol. 2002;193:145–153. doi: 10.1002/jcp.10174. [DOI] [PubMed] [Google Scholar]
- 19.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol. 2000;32:1123–1136. doi: 10.1016/s1357-2725(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 21.Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Jin HW, Ichikawa H, Nomura K, Mukae K, Terayama R, Yamaai T, Sugimoto T. Activation of the caspase cascade underlies the rat trigeminal primary neuronal apoptosis induced by neonatal capsaicin administration. Arch Histol Cytol. 2005;68:301–310. doi: 10.1679/aohc.68.301. [DOI] [PubMed] [Google Scholar]
- 23.Kotipatruni RR, Dasari VR, Veeravalli KK, Dinh DH, Fassett D, Rao JS. p53- and Baxmediated apoptosis in injured rat spinal cord. Neurochem Res. 2011;36:2063–2074. doi: 10.1007/s11064-011-0530-2. [DOI] [PubMed] [Google Scholar]
- 24.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 25.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 26.Liu C, Shi Z, Fan L, Zhang C, Wang K, Wang B. Resveratrol improves neuron protection and functional recovery in rat model of spinal cord injury. Brain Res. 2011;1374:100–109. doi: 10.1016/j.brainres.2010.11.061. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Sun ZY, Zhang KM, Xu GQ, Li G. Bcl-2 in suppressing neuronal apoptosis after spinal cord injury. World J Emerg Med. 2011;2:38–44. [PMC free article] [PubMed] [Google Scholar]
- 28.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 29.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 30.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J Signal Transduct. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Q, Rau TF, Harris V, Johnson M, Poulsen DJ, Black SM. Increased p38 mitogen-activated protein kinase signaling is involved in the oxidative stress associated with oxygen and glucose deprivation in neonatal hippocampal slice cultures. Eur J Neurosci. 2011;34:1093–1101. doi: 10.1111/j.1460-9568.2011.07786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang YX, Xu XY, Su WL, Wang Q, Zhu WX, Chen F, Jin G, Liu YJ, Li YD, Sun YP, Gao WC, Ruan CP. Activation and clinical significance of p38 MAPK signaling pathway in patients with severe trauma. J Surg Res. 2010;161:119–125. doi: 10.1016/j.jss.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Chen X, Zhou L, Wu D, Che X, Yang G. Effects of extracellular signal-regulated kinase (ERK) on focal cerebral ischemia. Chin Med J (Engl) 2003;116:1497–1503. [PubMed] [Google Scholar]
- 34.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 35.Hetman M, Xia Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol Exp (Wars) 2000;60:531–545. doi: 10.55782/ane-2000-1374. [DOI] [PubMed] [Google Scholar]
- 36.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stressinduced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 37.Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- 38.Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- 39.Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28:1686–1696. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lennmyr F, Ericsson A, Gerwins P, Ahlstrom H, Terent A. Increased brain injury and vascular leakage after pretreatment with p38-inhibitor SB203580 in transient ischemia. Acta Neurol Scand. 2003;108:339–345. doi: 10.1034/j.1600-0404.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 41.Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]