

Abstract
Parkinson’s disease (PD) is the second most common age-related neurodegenerative disease. MicroRNA-7 (miR-7) displays neuroprotective properties against PD. However, the biological roles of miR-7 and its underlying molecular mechanisms in PD remain unclear. We demonstrated herein that 1-methyl-4-phenylpyridinium ion (MPP+) confers toxic effects on dopaminergic neuron in a dose-dependent manner in a cellular PD model, although this phenomenon is attenuated by miR-7 treatment. Introduction of miR-7 inhibits MPP+-induced neuronal apoptosis as reflected by the reduced terminal transferase-mediated dUTP nick end labeling-positive rate, mitochondrial permeability potential, caspase 3 activity, and nucleosomal enrichment factor. Bax and sirtuin 2 (Sirt2) are the direct targets of miR-7. Moreover, the effects of miR-7 were counteracted by Bax and Sirt2 overexpression, respectively. The altered molecular expressions downstream of Bax and Sirt2 are also involved in miR-7 regulation of the MPP+-triggered neuronal apoptosis. These findings have implications on the potential application of miR-7 in PD treatment.
Keywords: MicroRNA-7, Parkinson’s disease, apoptosis, Bax, Sirt2
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease and is primarily characterized by the loss of dopaminergic (DA) neurons [1]. To date, several lines of evidence from PD research have revealed that the pathophysiologic mechanisms underlying neurodegeneration are related to mitochondrial dysfunction and oxidative stress, which could cause apoptosis of DA neurons [2,3]. Thus, strategies aimed at reducing loss of DA neurons caused by apoptosis might provide an effective cure for PD patients.
MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is the most commonly used DA neurotoxin that disrupts the nigrostriatal DA pathway and causes permanent Parkinsonism, which are similar to those observed in PD patients [4]. MTTP also induces selective loss of DA neurons in substantia nigra of mice [5,6]. Methyl-4- phenylpyridinium (MPP+), the active metabolite of MPTP, is concentrated within the mitochondria of DA neurons where it potently inhibits the complex I of the electron transport system [7]. Consequently, MPP+ results in loss of mitochondrial membrane potential, information of reactive oxygen species (ROS), inactivation of caspase 9 and caspase 3 [8], and ultimately programmed cell death.
MicroRNAs (miRNAs) are a class of highly conserved small RNAs that regulate diverse cellular processes by base pairing with the 3’-untranslated regions (UTRs) of their target mRNAs [9]. Growing amount of evidence has suggested that miRNA dysfunction contributes to neurodegenerative disorders, including PD [10-12]. Junn E et al. [13] reported that miR-7 reduces neurotoxicity and apoptosis caused by α-synuclein (α-syn), a key protein in PD pathogenesis. MiR-7 inhibits MPP+-induced neuron death by targeting RelA [14]. In addition, miR-7 protects neurons from MPP+-triggered death by upregulating the mTOR pathway [15]. However, the biological function and the underlying molecular mechanism of the miR-7-elicited PD suppression remain unexplored.
This study investigated the potential function of miR-7 in MPP+-treated neurons. Dual-luciferase reporter assays showed that Bax and sirtuin 2 (Sirt2) are directly targeted by miR-7. MiR-7 restoration significantly inhibits MPP+-induced neuronal apoptosis, and this effect is abrogated by overexpression of Sirt2 or Bax. Analyses of the molecular mechanisms involved in these processes reveal that miR-7 reduces the MPP+-elicited apoptotic effects on neurons by inhibiting the expression of several pro-apoptotic molecules. These findings revealed that miR-7 exerts a neuroprotective effect against the MPP+-induced apoptosis by targeting Bax and Sirt2.
Materials and methods
Cell culture
The human DA neuroblastoma cell line SH-SY5Y was purchased from the American Type Culture Collection. The SH-SY5Y cell line was characterized by the provider using a gene profiling analysis and was used no later than 6 months after receipt. The cell line was maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen). The cultures were incubated at 37°C in 5% CO2 and 95% air, and the cells from passages 2-4 were used in our experiments.
Cell transfection
The miR-7 and miR-negative control (NC) mimics were synthesized by Qiagen (Hilden, Germany). The wild-type (WT) 3’-UTR of Bax, Sirt2, and two variants containing mutations in the putative miR-7 binding sites were synthesized by Qiagen and were inserted downstream of the firefly luciferase gene in the pGL3 vector (Promega, Madison, WI, USA). The plasmids pcDNA3.1-Bax and pcDNA3.1-Sirt2 (open reading frame) were purchased from Addgene (Cambridge, MA, USA). All constructs were confirmed by DNA sequencing. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. The cells were transfected using 100 nM miR-7 or miR-NC mimics in 6-well plates. For plasmid transfections, 3 μg of DNA was used.
Cell viability assay
Twenty-four hours after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, cell viability was measured using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). In accordance with the manufacturer’s instructions for CCK-8, the harvested cells were seeded into 96-well plates at a density of 1 × 103 cells/well with a final volume of 100 μL. The cells were cultured for 1, 2, 3, or 4 days after transfection. CCK-8 solution (10 μL) was added into each well, and the absorbance at 450 nm was measured after incubation for 2 h at 37°C to calculate the number of viable cells.
Lactate dehydrogenase release assay
Cytotoxicity was quantitatively evaluated by measuring the lactate dehydrogenase (LDH) release. Twenty-four hours after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, gentle centrifugation was performed and the supernatant was subsequently collected. The level of LDH in the culture media was assessed using commercially available kits (Jiancheng Bioengineering Research Institute, Nanjing, Jiansu, China) according to the manufacturer’s instructions.
Measurement of intracellular ROS
The formation of intracellular ROS was evaluated using the fluorescent probe 2’,7’-dichlorofluorescein diacetate (DCFH-DA), which readily permeates cells and is hydrolyzed into fluorescent dichlorofluorescein upon interacting with ROS. Briefly, at 4 h after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, the cells were incubated with 10 mM DCF-DA at 37°C for 30 min in the dark. Cellular fluorescence was measured at 485 nm excitation and 535 nm emission using a fluorescence microplate reader (Saire2; Tecan, Switzerland). The images were prepared using Adobe Photoshop (ver. 8.0).
Dual-luciferase reporter assay
The cells were co-transfected with reporter constructs, an internal control vector (pGL4.73), and a synthetic miR-7 or miR-NC mimic. Forty-eight hours after transfection, the luciferase activity was determined using the Dual-Luciferase Reporter Assay System (Promega) and a luminometer (Glomax 20/20; Promega) and was normalized to the activity of Renilla luciferase driven by a constitutively expressed promoter in the phRL vector. The basal promoter activity was measured as the fold-change relative to that observed for the basic pGL3 vector alone.
RNA extraction and quantitative real-time PCR
Twenty-four hours after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, the total RNA was extracted from 1 × 105 cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. The harvested RNA was diluted to a concentration of 1 μg/μL, packaged, and then preserved at -80°C. cDNA was generated using Reverse Transcription Kit (Promega, Madison, WI, USA). Quantitative real-time polymerase chain reaction (qPCR) was performed using a standard IQTM SYBR Green Supermix Kit (Bio-Rad, Berkeley, USA), and the PCR specific amplification was assessed by Mastercycler®ep Realplex (Eppendorf, Hamburg, Germany). β-actin was used as endogenous control. The relative levels of Bax and Sirt2 were calculated using the comparative 2-ΔΔCt method. The primers used for PCR amplification are listed as follows: for Bax, forward 5’-CTGTGTCTACCGAGGGTGC T-3’, reverse 5’-CGCTCCAACGGACTTTAACA-3’; for β-actin, 5’-TCCTAGCACCAT GAAGATCAAGATC-3’, reverse 5’-CTGCTTGCTGATCCACATCTG-3’.
Western blot analysis
Forty-eight hours after the SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmids, proteins were extracted using lysis buffer, separated by sodium dodecyl-sulfate polyacrylamidegel electrophoresis, transferred onto nitrocellulose membranes (Millipore, Bedford, MA, USA), and then subjected to immunoblot analyses. Blotting was performed using primary antibodies targeting Sirt2, Bim (all obtained from Abnova, Taiwan, China), cytochrome c (Cyt c), Bcl-2 (all obtained from Cell Signaling Technology, Danvers, MA, USA), Bax, cleaved (Cl)-caspase 3, and β-actin (all obtained from Sigma). The signals were detected using a horseradish peroxidase-conjugated secondary antibody (Sigma), and the bands were visualized using an enhanced chemiluminescence kit (Santa Cruz, Dallas, TX, USA). The protein bands were quantified using Quantity One software (Bio-Rad, USA).
Apoptosis detection by flow cytometry
Apoptosis was determined using Annexin V-fluorescein isothiocyanate (FITC) and propridium iodide (PI) staining. Forty-eight hours after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, the cells were harvested, centrifuged, and resuspended in binding buffer. Approximately 10 μL of ready-to-use Annexin V-FITC (BD Bioscience, MA, USA) was added into the mixture, incubated at 37°C for 15 min, and then counterstained with 5 μL PI in the dark for 30 min. Annexin V-FITC and PI fluorescence were assessed using BD FACSCalibur flow cytometer (BD Bioscience), and the results were analyzed using the CellQuest software (BD Bioscience).
Nucleosomal enrichment factor assay
Forty-eight hours after the SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmids, cell apoptosis was quantified using a nucleosomal fragmentation kit (Cell Death Detection ELISAPLUS; Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s protocol. The absorbance values were normalized to those of control cells to derive the nucleosomal enrichment factor.
Quantitative caspase 3 activity assay
Caspase 3 activity assay was conducted using the Caspase 3/CPP32 Colorimetric Assay Kit (Biovision, Palo Alto, CA) following the standard protocols. Forty-eight hours after the SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmids, the cells were harvested, washed with cold PBS, and then incubated with 50 μL of chilled lysis buffer on ice for 10 min. After centrifugation at 10,000 × g, protein (150 μg) was added into 2 × 50 μL of reaction buffer containing 5 μL of N-acetyl-Asp-Glu-Val-Asp-pNA substrate (200 μM final concentration). After incubation for 1-2 h at room temperature, N-acetyl-Asp-Glu-Val-Asp-pNA cleavage was monitored using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA). The absorbance (405 nm) of each well was detected to monitor the enzyme-catalyzed pNA release.
Mitochondrial permeability potential
Loss of mitochondrial potential can be detected by 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolylcarbocyanine iodide staining, commonly known as JC-1. Forty-eight hours after the SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmids, the cells were stained with the cationic dye JC-1 (MitoPT, Immunohistochemistry Technologies, Bloomington, MN), which exhibits potential-dependent accumulation in mitochondria. At low membrane potentials, JC-1 continues to exist as a monomer and produces green fluorescence (emission at 527 nm). At high membrane potentials or concentrations, JC-1 forms J aggregates (emission at 590 nm) and produces red fluorescence.
Terminal transferase-mediated dUTP nick end labeling assay
A terminal transferase-mediated dUTP nick end labeling (TUNEL) assay was performed to detect DNA strand breaks in situ according to the manufacturer’s protocol (Roche, Mannheim, BW, Germany). Forty-eight hours after transfecting RNA oligonucleotides to the SH-SY5Y cells with or without MPP+ treatment, the cells seeded on glass slides were fixed with 80% glycerol at room temperature, rinsed with PBS (pH 7.4), and then permeabilized with 2% Triton X-100. The FITC-labeled terminal deoxynucleotidyl transferase (TdT) nucleotide mix (Promega, Madison, WI, USA) was added into each slide and incubated at 37°C for 60 min. The slides were rinsed twice in PBS and then counterstained with 10 mg/mL DAPI (Sigma). The FITC-labeled TdT was omitted in the nucleotide mix of the negative control. The TUNEL-positive cells were imaged and mounted by fluorescence microscopy (Carl Zeiss, Weimar, Germany) and were ultimately expressed as a percentage of the total cells (DAPI staining).
Statistical analysis
Statistical analyses were performed using SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA). Student’s t-tests were used to analyze the results expressed as the mean ± standard derivation (SD), and Fisher’s exact test was used for comparisons of qualitative variables. P < 0.05 was considered significant.
Results
MiR-7 provides protection against MPP+-induced neurotoxicity
MiR-7 represses MPTP-induced neurotoxicity in a PD mouse model [12]. We investigated whether miR-7 reduces the neurotoxicity caused by MPP+ insult in DA neurons. Figure 1A shows that MPP+ treatment significantly reduces neuronal viability in a dose-dependent manner, which is inhibited by miR-7. Oxidative stress and cytotoxicity play key roles in PD pathogenesis [16]. MPP+ treatment expectedly increases ROS levels (Figure 1B) and LDH release (Figure 1C) in SH-SY5Y cells, and this increase is reduced by miR-7. These findings demonstrated that miR-7 reduces MPP+-induced neurotoxicity.
Figure 1.
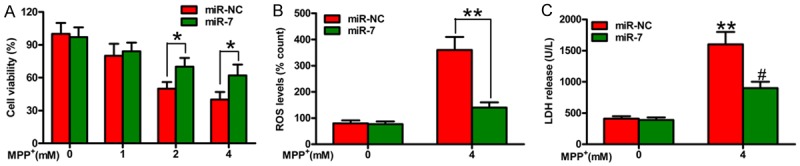
MiR-7 protects the neurons against MPP+-induced toxicity. The SH-SY5Y cells with or without MPP+ treatment were transfected with 100 nM miR-7 or miR-NC mimic. A. CCK-8 assay of the cells for the indicated number of days. B. ROS production in variously treated SH-SY5Y cells. C. LDH release in variously treated SH-SY5Y cells. Data are represented as the mean ± SD of three replicates. *P< 0.05, #P< 0.05, and **P< 0.01.
MiR-7 reduces MPP+-enhanced neuronal apoptosis
We further investigated whether miR-7 regulates neuron survival by orchestrating apoptosis. Flow cytometry assay showed that MPP+ treatment promotes apoptosis of the SH-SY5Y cells, which is attenuated by miR-7 administration (Figure 2A and 2B). To further confirm these observations, we performed TUNEL assay and found that the percentage of the TUNEL-positive neurons in the MPP+ group is higher than that in the MPP+ + miR-7 group (Figure 2C). In addition, miR-7 inhibits the MPP+-induced SH-SY5Y cell apoptosis by reducing caspase 3 activity (Figure 2D), MMP (Figure 2E), and nucleosomal enrichment fragmentation (Figure 2F). Overall, miR-7 reduces significantly the MPP+-induced neuronal apoptosis.
Figure 2.

MiR-7 inhibits the apoptosis of MPP+-challenged neurons. The SH-SY5Y cells with or without MPP+ treatment were transfected with 100 nM miR-7 or miR-NC mimic. (A) FACS assay was performed to detect the apoptotic cells. (B) The apoptotic rate was calculated from the FACS data. (C-F) TUNEL (C), caspase 3 activity (D), MMP (E), and nucleosomal enrichment fragmentation (F) assays of the cells transfected with miR-7 or miR-NC show the effects on apoptosis of the SH-SY5Y cells with or without MPP+ treatment. Representative images are shown. Data are represented as the mean ± SD of three replicates. *P< 0.05, #P< 0.05, and **P< 0.01.
Bax and Sirt2 are direct targets of miR-7
We subsequently searched for the candidate target genes of miR-7 using the publicly available databases, namely, TargetScan, miRanda, and PicTar (Figure 3A). The complementary miR-7 sequences were identified in the 3’-UTR of the Bax and Sirt2 mRNA (Figure 3B). Therefore, these genes were selected for further analysis. Dual reporter assays revealed that the introduction of miR-7 in the SH-SY5Y cells suppresses the activity of the luciferase reporter fused to the WT 3’-UTR of Bax or Sirt2 but does not suppress that of a reporter fused to a mutant (MUT) version of the 3’-UTR (Figure 3C and 3D). We then detected the mRNA and protein levels of Bax and Sirt2 in the SH-SY5Y cells with or without MPP+ treatment that were transfected with or without miR-7; the findings showed that miR-7 significantly reduces the mRNA and protein expression levels of Bax and Sirt2 (Figure 3E-J) in MPP+-challenged SH-SY5Y cells. These results suggested that Bax and Sirt2 are the direct targets of miR-7 in a cellular PD model.
Figure 3.

Bax and Sirt2 are the direct targets of miR-7. (A) The potential targets of miR-7 were predicted by integrating the results of three algorithms (TargetScan, PicTar, and miRanda). (B) The predicted binding sites of miR-7 in the 3’-UTR of Bax (upper) and Sirt2 (bottom). (C, D) The results of luciferase reporter assays performed at 24 h after co-transfection of SH-SY5Y cells with a pGL3 construct containing the WT or MUT Bax 3’-UTR region (C) or the WT or MUT Sirt2 3’-UTR region and miR-7 mimics (D). Data were normalized to those from cells co-transfected with pGL3 and miR-NC. (E, F) qPCR (E) and Western blot (F) analyses of Bax expression in mock-treated or MPP+-treated SH-SY5Y cells transfected with miR-7 or miR-NC mimics. (G) Relative protein band densities of Bax. (H, I) qPCR (H) and Western blot (I) analyses of Sirt2 expression in mock-treated or MPP+-treated SH-SY5Y cells transfected with miR-7 or miR-NC mimics. (J) Relative protein band densities of Sirt2. (E-J) The Bax and Sirt2 mRNA levels were normalized to that of GAPDH, and the Bax and Sirt2 protein levels were normalized to that of β-actin. Representative Western blots are shown. Data are represented as the mean ± SD of three replicates. *P< 0.05, #P< 0.05, and **P< 0.01.
MiR-7 attenuates MPP+-induced neuronal apoptosis by repressing Sirt2 expression
To confirm that Sirt2 is a functional target of miR-7, we transfected the MPP+-treated SH-SY5Y cells with miR-NC, miR-7 mimic, or a plasmid expressing Sirt2. The protein expressions of Sirt2 and its downstream molecule Bim in different groups were detected by Western blot analysis (Figure 4A-C). The data revealed that miR-7 inhibits Sirt2 and Bim expression (Figure 4A-C). Sirt2 activates Bim, resulting in the increase of caspase 3 activity in PD. Figure 4D shows that Sirt2 overexpression rescues the miR-7-mediated inhibition of MPP+-enhanced caspase 3 activity in SH-SY5Y cells. These results prove that Sirt2 is a downstream functional target of miR-7 in MPP+-induced neuronal apoptosis.
Figure 4.
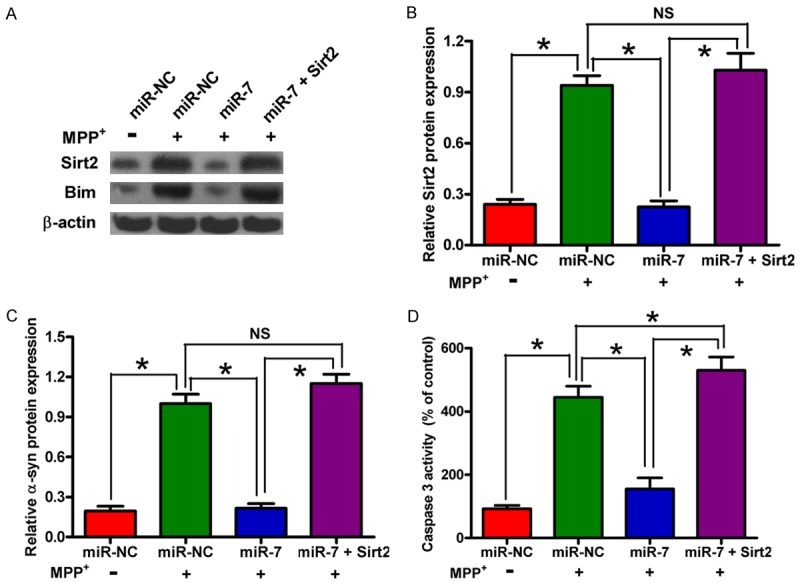
MiR-7 mitigates the MPP+-induced neuronal apoptosis by targeting Sirt2. The SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmid pcDNA3.1-Sirt2. (A) Western blot analyses of Sirt2 and Bim expression. β-actin was used as a control. (B, C) Relative protein band densities of Sirt2 (B) and Bim (C). (D) Caspase 3 activity assay for detection of neuronal apoptosis. Representative Western blots are shown. Data are represented as the mean ± SD of three replicates. *P< 0.05.
MiR-7 hampers MPP+-induced neuronal apoptosis by downregulating Bax
We investigated whether Bax is another functional target of miR-7. The SH-SY5Y cells were transfected with miR-NC, miR-7 mimic, or with a plasmid expressing Bax following MPP+ treatment. Overexpression of Bax counteracted the miR-7-elicited inhibition of the MPP+-induced SH-SY5Y cell apoptosis; this finding was evidenced by the enhanced nucleosomal enrichment fragmentation and caspase 3 activity and by the loss of MMP (Figure 5A-C). To explore the molecular mechanisms by which miR-7 exerts its protective effect against neuronal apoptosis caused by MPP+ insult, we examined the levels of Bcl-2, Bax, Cl-caspase 3, and Cyt c. Figure 5D shows that the miR-7 restoration in neurons reduces the MPP+-induced expression of the anti-apoptotic molecule Bcl-2 and enhances the MPP+-induced expression of pro-apoptotic molecules, such as Cl-caspase 3, Bax, and Cyt c; these effects are reversed by Bax overexpression. These results suggested that Bax is a downstream functional target of miR-7 in MPP+-induced neuronal apoptosis.
Figure 5.
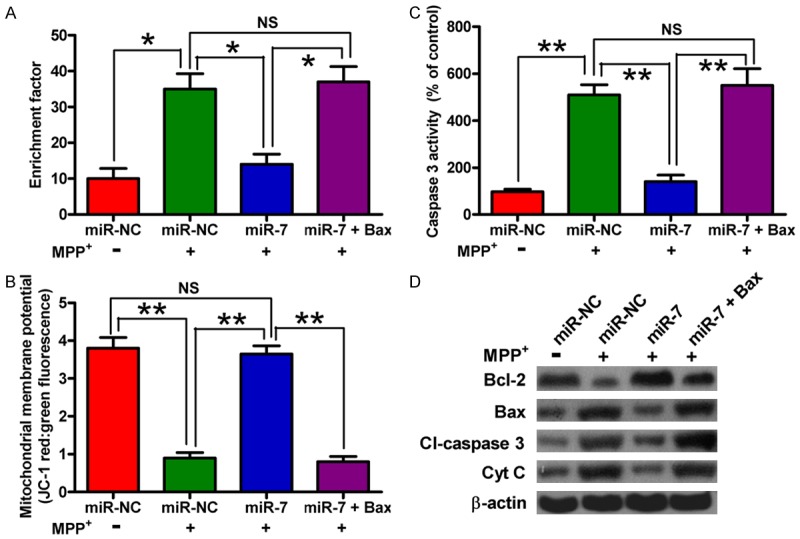
Restoration of Bax counteracts the inhibitory effects of miR-7 on MPP+-induced neuronal apoptosis. The SH-SY5Y cells with or without MPP+ treatment were transfected with RNA oligonucleotides or with or without plasmid pcDNA3.1-Bax. (A-C) The nucleosomal fragmentation (A), MMP (B), and caspase 3 activity (C) assays of the cells transfected with miR-7 or miR-NC showing the effects on apoptosis of mock-treated or MPP+-treated SH-SY5Y cells with or without introduction of Bax. (D) Western blot analyses of Bcl-2, Bax, Cl-caspase 3, and Cyt c expression. β-actin was used as control. Representative Western blots are shown. Data are represented as the mean ± SD of three replicates. *P< 0.05, and **P< 0.01.
Discussion
MiRNAs are involved in various neurodegenerative disorders, including PD [17]. Although previous studies have reported that miR-7 represses the development and progression of PD by targeting a-syn and RelA [13,14], the underlying mechanisms by which miR-7 inhibits PD remain unclear. This study elucidates a novel neuroprotective mechanism of miR-7. The key findings are as follows (Figure 6): First, we found that miR-7 inhibits the MPP+-induced loss of viability and enhancement of ROS levels and LDH release in neurons. Second, miR-7 reduces the MPP+-induced neuronal apoptosis as evidenced by the decrease in the percentage of TUNEL-positive cells, caspase 3 activity, MMP, and nucleosomal enrichment factor. Third, we demonstrated that Bax and Sirt2 are directly targeted by miR-7. Fourth, the inhibitory effects of miR-7 on the MPP+-induced neuronal apoptosis are abrogated by overexpression of Sirt2 and Bax. Overall, these findings revealed that miR-7 exerts a neuroprotective effect against MPP+-elicited apoptosis by targeting Bax and Sirt2.
Figure 6.
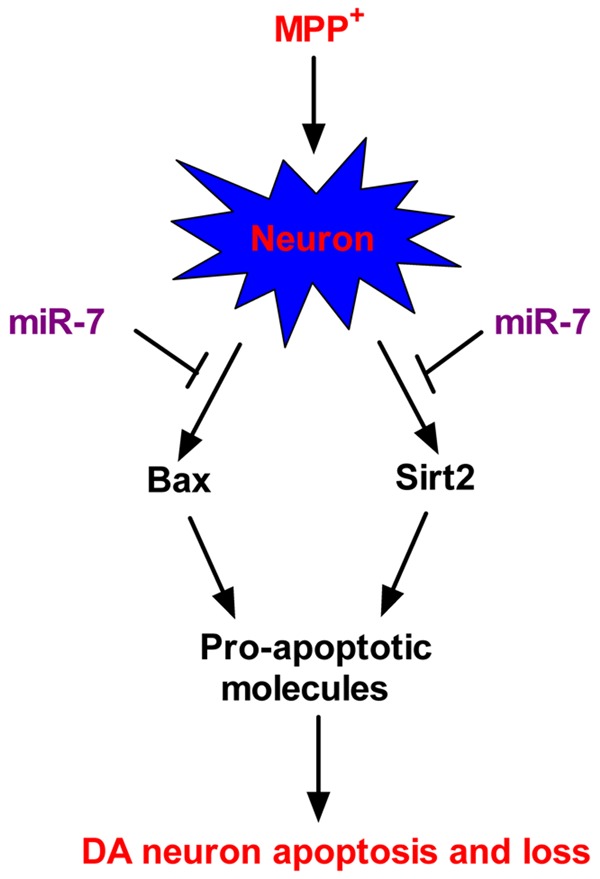
Proposed mechanism of the miR-7-mediated protection against MPP+-induced neuronal apoptosis. The cellular model of PD shows the MPP+-induced enhancement of Bax and Sirt2 expression in neurons. In the same conditions, upregulation of Bax further increases the expression of other pro-apoptotic molecules, and enhancement of Sirt2 also results in Bim elevation. MiR-7 provides protection against MPP+-induced neuronal apoptosis by targeting Bax and Sirt2, thereby suppressing the expression of their downstream pro-apoptotic factors. These anti-apoptotic benefits of miR-7 contribute to the maintenance of neuronal viability in a cellular PD model.
Sirt2 is a strong NAD-dependent protein deacetylase and is highly expressed in the brain. Sirt2 is also an abundant neuronal protein and accumulates in the central nervous system of aged mice [18]. Moreover, Sirt2 co-localizes with microtubules and functions as a-tubulin deacetylase, leading to PD aggravation. Outerio et al. [19] demonstrated that Sirt2 inhibitors reduce DA neuron death both in vitro and in a Drosophila model of PD. Sirt2 expression also increases in cells subjected to oxidative stress; in addition, Sirt2 enhancement promotes DA neuronal apoptosis when cells are under MPP+-induced severe stress by activating the pro-apoptotic factor Bim, subsequently increasing caspase 3 activity [20]. Therefore, Sirt2 inhibition possibly offers protection against PD. We found herein that miR-7 inhibits ROS production in MPP+-treated DA neuron, which possibly contributes to the reduced Sirt2 expression; however, this speculation requires clarification. We subsequently confirmed whether Sirt2 is a functional target of miR-7 in MPP+-stimulated neurons. MiR-7 reduces the levels of Sirt2 mRNA and protein and subsequently inhibits the activation of the pro-apoptotic molecules Bim and caspase 3, resulting in reduction of neuronal apoptosis in a cellular PD model; this finding is highly consistent with a previous result [21]. Caspase 3 is an active cell-death protease involved in the execution phase of apoptosis, where cells undergo morphological changes, such as DNA fragmentation and apoptotic body formation [22]. This study found that miR-7 reduces the percentage of TUNEL-positive cells, as well as the nucleosomal enrichment factor of the MPP+-challenged DA neurons, paralleling the reduction in caspase 3 activity.
The molecular pathogenesis of PD is speculated to be associated with mitochondrial dysfunction and activation of apoptotic cascade [2]. MPP+-induced neuronal death is mediated by the loss of mitochondrial membrane potential [23,24]. Activation of apoptotic cascade may play a role in MPP+-induced cell death by altering mitochondrial membrane permeability and by controlling the release of Cyt c from the mitochondria [25,26]. Caspase 3 activation by the released Cyt c is involved in MPP+-induced apoptosis [27-29]. Once activated, caspase 3 will induce nuclear DNA condensation and fragmentation and ultimately apoptosis [22]. Studies have shown that Bax is strongly upregulated in nigrostriatal DA neurons after exposure to MPTP [30-32]. In a chronic MPTP mouse model, the protein levels of Bax are increased and mitochondrial translocation of Bax is observed [30,33]. Bax-/- mice are resistant to MPTP-induced neurodegeneration in the substantia nigra [21]; in addition, inhibiting the increase in Bax levels or its mitochondrial translocation can reduce DA cell loss in MPTP-challenged mice [34]. Bax knockdown also reduces caspase 3 activity in PD. Bcl-2 overexpression protects DA neurons against MPTP-induced neurodegeneration [35,36]. The ratio of pro-apoptotic Bax to anti-apoptotic Bcl-2 increases in response to MPP+ treatment of DA neurons [37]. The present study demonstrated that miR-7 inhibits MPP+-induced neuronal apoptosis by targeting Bax. MiR-7 also reduces the expression of the pro-apoptotic Bax, Cyt c, and Cl-caspase 3 and increases the expression anti-apoptotic Bcl-2 in the MPP+-treated DA neurons. Moreover, miR-7 attenuates MPP+-enhanced neuronal apoptosis, as reflected in the reduction of MMP, caspase 3 activity, and nucleosomal enrichment factor. The anti-apoptotic effects of miR-7 are abrogated by overexpression of Bax, suggesting that Bax is a key player in the MPP+-induced neuronal apoptosis as it regulates the expression of other apoptosis-related factors. Perier et al. [34] showed that Bim, a downstream effector of Sirt2, mediates the translocation of Bax into the mitochondria, thereby releasing Cyt c, resulting in neuronal apoptosis in MPTP-treated mice. We validated herein that Bax and Sirt2 are functional targets of miR-7 in MPP+-treated DA neurons; however, we cannot exclude the possibility that downregulation of Bax by miR-7 is dependent on Sirt2 reduction.
In summary, the results presented herein collectively showed that miR-7 provides protection against MPP+-induced DA neuronal apoptosis by repressing Bax and Sirt2 expression, consequently reducing the expression of a series of pro-apoptotic molecules. These findings suggested that augmenting miR-7 levels is an effective disease-modifying strategy in PD.
Disclosure of conflict of interest
None.
References
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 3.Brown JM, Yamamoto BK. Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther. 2003;99:45–53. doi: 10.1016/s0163-7258(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 4.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 5.Blandini F, Armentero MT. Animal models of Parkinson’s disease. FEBS J. 2012;279:1156–1166. doi: 10.1111/j.1742-4658.2012.08491.x. [DOI] [PubMed] [Google Scholar]
- 6.Bezard E, Yue Z, Kirik D, Spillantini MG. Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov Disord. 2013;28:61–70. doi: 10.1002/mds.25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsay RR, Singer TP. Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J Biol Chem. 1986;261:7885–7. [PubMed] [Google Scholar]
- 8.Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 10.Yelamanchili SV, Fox HS. Defining larger roles for “tiny” RNA molecules: role of miRNAs in neurodegeneration research. J Neuroimmune Pharmacol. 2010;5:63–69. doi: 10.1007/s11481-009-9172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harraz MM, Dawson TM, Dawson VL. MicroRNAs in Parkinson’s disease. J Chem Neuroanat. 2011;42:127–130. doi: 10.1016/j.jchemneu.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi DC, Chae YJ, Kabaria S, Chaudhuri AD, Jain MR, Li H, Mouradian MM, Junn E. Mi croRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J Neurosci. 2014;34:12725–12737. doi: 10.1523/JNEUROSCI.0985-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fragkouli A, Doxakis E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front Cell Neurosci. 2014;8:182. doi: 10.3389/fncel.2014.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson’s disease. J Neurosci Res. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- 17.Ha TY. MicroRNAs in human diseases: from autoimmune diseases to skin, psychiatric and neurodegenerative diseases. Immune Netw. 2011;11:227–244. doi: 10.4110/in.2011.11.5.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maxwell MM, Tomkinson EM, Nobles J, Wizeman JW, Amore AM, Quinti L, Chopra V, Hersch SM, Kazantsev AG. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet. 2011;20:3986–3996. doi: 10.1093/hmg/ddr326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Outerio TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synucleinmediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, Arun A, Ellis L, Peritore C, Donmez G. SIRT2 enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigostriatal damage via apoptotic pathway. Front Aging Neurosci. 2014;6:184. doi: 10.3389/fnagi.2014.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun DS, Chang HH. Differential regulation of JNK in caspase-3-mediated apoptosis of MPP(+)-treated primary cortical neurons. Cell Bio Int. 2003;27:769–777. doi: 10.1016/s1065-6995(03)00165-3. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki K, Mizuno Y, Yoshida M. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-like compounds on mitochondrial respiration. Adv Neurol. 1990;53:215–218. [PubMed] [Google Scholar]
- 24.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998;809:12–17. doi: 10.1016/s0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 26.Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 27.Turmel H, Hartmann A, Parain K, Douhou A, Srinivasan A, Agid Y, Hirsch EC. Caspase-3 activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)- treated mice. Mov Disord. 2001;16:185–189. doi: 10.1002/mds.1037. [DOI] [PubMed] [Google Scholar]
- 28.Xu Q, Kanthasamy AG, Reddy MB. Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson’s disease. Toxicology. 2008;245:101–108. doi: 10.1016/j.tox.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 29.Zeng G, Tang T, Wu HJ, You WH, Luo JK, Lin Y, Liang QH, Li XQ, Huang X, Yang QD. Salvianolic acid B protects SH-SY5Y neuroblastoma cells from 1-methyl-4-phenylpyridinium-induced apoptosis. Biol Pharm Bull. 2010;33:1337–1342. doi: 10.1248/bpb.33.1337. [DOI] [PubMed] [Google Scholar]
- 30.Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee do Y, Lee KS, Lee HJ, Noh YH, Kim do H, Lee JY, Cho SH, Yoon OJ, Lee WB, Kim KY, Chung YH, Kim SS. Kynurenic acid attenuates MPP(+)-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur J Cell Biol. 2008;87:389–397. doi: 10.1016/j.ejcb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Slone SR, Lesort M, Yacoubian TA. 14-3-3theta protects against neurotoxicity in a cellular Parkinson’s disease model through inhibition of the apoptotic factor Bax. PLoS One. 2011;6:e21720. doi: 10.1371/journal.pone.0021720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perier C, Tieu K, Guégan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax- dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A. 2005;102:19126–31. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perier C, Bové J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, Przedborski S, Vila M. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:8161–8166. doi: 10.1073/pnas.0609874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Offen D, Beart PM, Cheung NS, Pascoe CJ, Hochman A, Gorodin S, Melamed E, Bernard R, Bernard O. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:5789–5794. doi: 10.1073/pnas.95.10.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou JC, Penney JB Jr, Hyman BT, Beal MF. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci. 1998;18:8145–8152. doi: 10.1523/JNEUROSCI.18-20-08145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Malley KL, Liu J, Lotharius J, Holtz W. Targeted expression of BCL-2 attenuates MPP+ but not 6-OHDA induced cell death in dopaminergic neurons. Neurobiol Dis. 2003;14:43–51. doi: 10.1016/s0969-9961(03)00013-5. [DOI] [PubMed] [Google Scholar]