

Abstract
The epidermal growth factor receptor (EGFR) signaling plays a key role in the initiation, progression, growth and metastases of colorectal carcinoma (CRC). Monoclonal antibody against EGFR (aEGFR; Cetuximab) has been used in treating CRC but some CRCs appeared to be resistant to aEGFR therapy, with undetermined mechanisms. Here, we studied the effects of aEGFR on CRC cells in vitro. We found that aEGFR dose-dependently activated Beclin-1 in 2 CRC cell lines, HT29 and SW480. Inhibition of autophagy significantly increased the aEGFR-induced CRC cell death in an CCK-8 assay. Moreover, microRNA (miR)-216b levels were significantly downregulated in aEGFR-treated CRC cells. Bioinformatics study showed that miR-216b targeted the 3’-UTR of Beclin-1 mRNA to inhibit its translation, which was confirmed by luciferase reporter assay. Together, these data suggest that aEGFR may decrease miR-216b levels in CRC cells, which subsequently upregulates Beclin-1 to increase CRC cell autophagy to antagonize aEGFR-induced cell death. Strategies that increase miR-216b levels or inhibit cell autophagy may improve the outcome of aEGFR treatment in CRC therapy.
Keywords: Colorectal carcinoma (CRC), aEGFR, autophagy, Beclin-1, miR-216b
Introduction
Colorectal carcinoma (CRC) originate from the epithelial cells in the colon or rectum of the gastrointestinal tract, and are the third most prevalent cancer worldwide [1-3]. Although the primary CRC are highly curable, distal metastases of CRC to the liver, lungs or other sites may substantially increase the difficulties for treatments [4].
The epidermal growth factor receptor (EGFR) has been recognized as an important mediator in CRC initiation and progression. This membrane-bound receptor tyrosine kinase (RTK) has therefore become a key target of therapeutic strategies designed to treat metastatic CRC, in particular with monoclonal antibodies (mAbs) against the extracellular domain of the receptor [5-8]. KRAS is an effector molecule responsible for signal transduction from ligand-bound EGFR to the nucleus. Activating mutations in KRAS are recognized as a strong predictor of resistance to EGFR-targeted mAbs. Cetuximab is an EGFR chemeric human-murine monoclonal antibody (aEGFR). In 2009, the FDA approved cetuximab for treatment of colon cancer with wild-type KRAS, since it had little or no effect in colorectal tumors harboring a KRAS mutation [9]. In spite of all abovementioned approaches, the effects of aEGFR on CSCs in the chemotherapy of CRC remain unclear.
Autophagy is a catabolic biological event characterized by the degradation of the cellular compartments and their recycling in order to improve cell survival upon harsh living environment [10-13]. Among all autophagy-associated proteins, autophagy-associated protein 6 (ATG6, or Beclin-1) is a key regulator for autophagy [14]. Recent studies have demonstrated a critical role of autophagy in the tumor initiation, growth and metastases, and especially in the mechanisms underlying chemo-resistance of tumor cells during chemotherapy [15-19]. Nevertheless, autophagy has not been shown associated with resistance of CRC cells against aEGFR therapy.
MicroRNAs (miRNAs) are a class of non-coding small RNAs that regulate the protein translation of target mRNA, through its base-pairing with the 3’-untranslated region (3’-UTR) [20,21]. There are accumulating evidence to show that miRNAs play very important roles in tumor [22-24]. Among all miRNAs, the involvement of miR-216b as a tumor suppressor in the carcinogenesis of various cancers as just acknowledged recently [25-28]. However, the involvement of miR-216b in the chemo-resistance of CRC against aEGFR has not been studied.
Here, we found that aEGFR dose-dependently activated Beclin-1 in 2 CRC cell lines, HT29 and SW480. Inhibition of autophagy significantly increased the aEGFR-induced CRC cell death in an CCK-8 assay. Moreover, microRNA (miR)-216b levels were significantly downregulated in aEGFR-treated CRC cells. Bioinformatics study showed that miR-216b targeted the 3’-UTR of Beclin-1 mRNA to inhibit its translation, which was confirmed by luciferase reporter assay.
Materials and methods
Cell lines and reagents
HT29 and SW480 are two human CRC lines purchased from ATCC (ATCC, Rockville, MD, USA), and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 20% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) in a humidified chamber with 5% CO2 at 37°C. HT-29 is a colorectal adenocarcinoma from a 44 year-old female, and has been describe before [29]. SW480 is a colorectal adenocarcinoma from a 50 year-old male, and has been describe before [30]. aEGFR (Taxol, Sigma-Aldrich, St. Louis, MO, USA) was prepared in a stock of 100 mg/ml and applied to the cultured CRC cells at 0.2 mg/ml, 0.5 mg/ml and 1 mg/ml, respectively. 3-Methyladenine (3-MA, Sigma-Aldrich) was prepared and used fresh at a concentration of 5 mmol/l.
Cell transfection
MiRNAs mimics (miR-216b) and miRNAs antisense oligonucleotides (as-miR-216b) were obtained from Origene (Beijing, China). For a control of these plasmids for modifying miR-216b levels, a plasmid carrying a null sequence (null) was used. These constructs were generated and cloned into the TOPO plasmid (Invitrogen, Carlsbad, CA, USA). The plasmids were transfected into cells at a concentration of 50 nmol/l using Lipofectamine-2000 (Invitrogen), receiving a nearly 100% transfection efficiency. The cells were analyzed after 24 hours, according to the manufacturer’s instruction.
MicroRNA target prediction and 3’-UTR luciferase-reporter assay
MiRNAs targets were predicted with the algorithms TargetSan (https://www.targetscan.org) [31]. The Beclin-1 3’-UTR reporter plasmid (pRL-Beclin-1) was purchased from Creative Biogene (Shirley, NY, USA). CRC cells were co-transfected with pRL-Beclin-1 and miR-216b/as-miR-216b/null by Lipofectamine 2000 (5×104 cells per well). Cells were collected 48 hours after transfection for assay using the dual-luciferase reporter assay system gene assay kit (Promega, Beijing, China), according to the manufacturer’s instructions.
Cell counting kit-8 (CCK-8) assay
The CCK-8 detection kit (Sigma-Aldrich) was used to measure cell viability according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well microplate at a density of 5000/ml. After 24h, cells were treated with resveratrol. Subsequently, CCK-8 solution (20 ml/well) was added and the plate was incubated at 37°C for 2 h. The viable cells were counted by absorbance measurements with a monochromator microplate reader at a wavelength of 450 nm. The optical density value was reported as the percentage of cell viability in relation to the control group (set as 100%).
Western blot
Protein was extracted from the cultured cells with RIPA lysis buffer (Sigma-Aldrich) on ice. The supernatants were collected after centrifugation at 12000× g at 4°C for 20 min. Protein concentration was determined using a BCA protein assay kit (Bio-rad, China), and the proteins were separated on SDS-polyacrylamide gels, and then transferred to a PVDF membrane. The membrane blots were first probed with a primary antibody. After incubation with horseradish peroxidase-conjugated second antibody, autoradiograms were prepared using the enhanced chemiluminescent system to visualize the protein antigen. The signals were recorded using X-ray film. Primary antibodies were rabbit anti-Beclin-1 and anti- α-tubulin (Cell Signaling, San Jose, CA, USA). Secondary antibody is HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). α-tubulin was used as a protein loading control. The protein levels were first normalized to α-tubulin, and then normalized to the experimental controls.
Quantitative real-time PCR (RT-qPCR)
Total RNA was extracted from cultured cells using miRNeasy kit (Qiagen), for cDNA synthesis. Quantitative real-time PCR (RT-qPCR) was performed in duplicates with QuantiTect SYBR Green CRCR Kit (Qiagen). All primers were purchased from Qiagen. Data were collected and analyzed with 2-ΔΔCt method for quantification. Values of genes were first normalized against α-tubulin, and then compared to experimental controls.
Statistical analysis
All statistical analyses were carried out using the SPSS 18.0 statistical software package. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. All values are depicted as mean ± standard deviation and are considered significant if p < 0.05.
Results
aEGFR dose-dependently increases Beclin-1 levels in CRC cells
We examined the effects of aEGFR on CRC cell autophagy using 2 CRC cell lines, HT29 and SW480. We gave aEGFR at different doses (0.2 mg/ml, 0.5 mg/ml and 1 mg/ml) to these cells, with or without an autophagy inhibitor 3-MA (5 mmol/l). We found that aEGFR dose-dependently upregulated Beclin-1 in HT29 cells, which was significantly suppressed by 3-MA (Figure 1A). However, the Beclin-1 mRNA was not affected by aEGFR (Figure 1B). Similarly, we found that aEGFR dose-dependently upregulated Beclin-1 in SW480 cells, which was significantly suppressed by 3-MA (Figure 1C). However, the Beclin-1 mRNA was not affected by aEGFR (Figure 1D). These data suggest that aEGFR dose-dependently increases Beclin-1 levels and cell autophagy in CRC cells, which may contribute to its chemo-resistance against aEGFR. Moreover, the regulation of Beclin-1 by aEGFR may be at post-transcriptional level.
Figure 1.
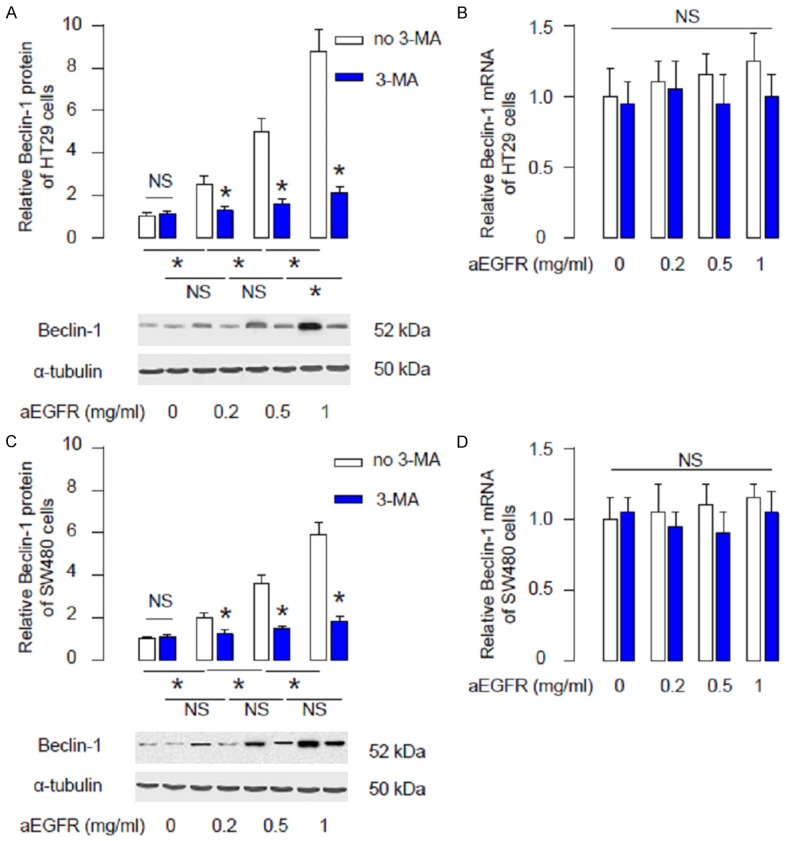
aEGFR dose-dependently increases Beclin-1 levels in CRC cells. (A, B) Beclin-1 levels in HT29 cells after aEGFR treatment (0.2 mg/ml, 0.5 mg/ml and 1 mg/ml) with/without 3-MA (5 mmol/l), shown by representative Western blots (A), and by RT-qPCR for mRNA (B). (C, D) Beclin-1 levels in SW480 cells after aEGFR treatment (0.2 mg/ml, 0.5 mg/ml and 1 mg/ml) with/without 3-MA (5 mmol/l), shown by representative Western blots (C), and by RT-qPCR for mRNA (D). *p<0.05. NS: non-significant. N=5.
Suppression of autophagy increases aEGFR-induced CRC cell death
Next, we examined the effects of aEGFR on CRC cell death when autophagy is inhibited. We found that aEGFR dose-dependently decreased HT29 cell survival in an CCK-8 assay, while these effects were further augmented when autophagy was suppressed by 3-MA (Figure 2A). Similarly, we found that aEGFR dose-dependently decreased SW480 cell survival, while these effects were further augmented when autophagy was suppressed by 3-MA (Figure 2B). These data suggest that while aEGFR dose-dependently increases CRC cell death, aEGFR also dose-dependently increases CRC cell autophagy, which attenuates aEGFR-induced cell death.
Figure 2.
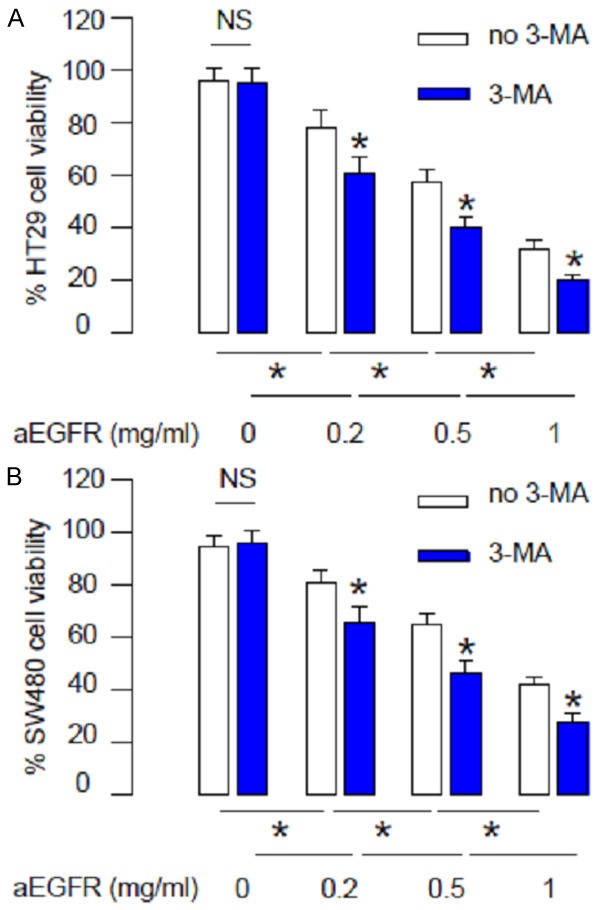
Suppression of autophagy increases aEGFR-induced CRC cell death. (A, B) aEGFR was given to HT29 (A) or SW480 cells (B) at different doses (0.2 mg/ml, 0.5 mg/ml and 1 mg/ml) with/without 3-MA (5 mmol/l). Cell viability was examined in an CCK-8 assay. *p<0.05. NS: non-significant. N=5.
aEGFR modulates miR-216b, which targets Beclin-1 to regulate CRC cell autophagy
Since miRNAs are critical regulators for protein translation, we performed bioinformatics studies to screen miRNAs that may target 3’-UTR of Beclin-1 mRNA, and were regulated by aEGFR treatment. Among all predicted binding miRNAs, we found that miR-216b targeted the 579-585 base pairs of 3’-UTR of Beclin-1 mRNA (Figure 3A). Moreover, miR-216b levels were found to be dose-dependently inhibited by aEGFR in HT29 cells (Figure 3B), and in SW480 cells (Figure 3C). Together, these data suggest that miR-216b may be a Beclin-1-targetting miRNA that is suppressed by aEGFR treatment in CRC cells.
Figure 3.
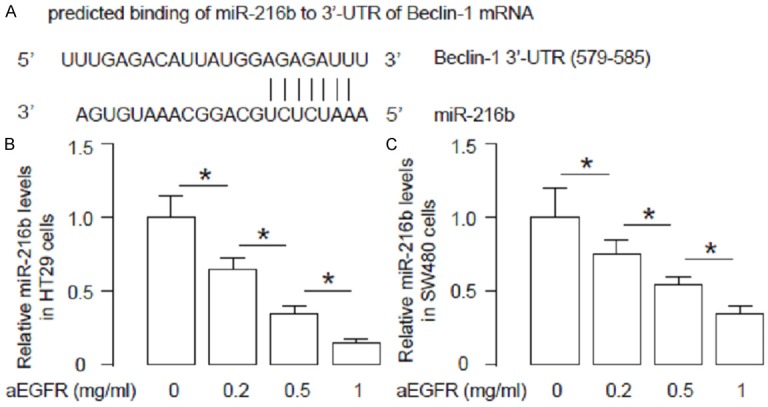
aEGFR modulates miR-216b, which targets Beclin-1 to regulate CRC cell autophagy. (A) Bioinformatics study for miRNAs that target 3’-UTR of Beclin-1 mRNA, showing that miR-216b targets the 579-585 base pair of 3’-UTR of Beclin-1 mRNA. (B, C) MiR-216b levels were found to be dose-dependently inhibited by aEGFR in HT29 cells (B), and in SW480 cells (C). *p<0.05. N=5.
MiR-216b inhibits Beclin-1 protein translation in CRC cells
In order to verify that the binding of miR-216b to the 3’-UTR of Beclin-1 mRNA is functional, the intact 3’-UTR of Beclin-1 was cloned into a luciferase reporter plasmid. Then, miR-216b levels in HT29 and SW480 cells were modified by transfection of either miR-216b mimic, or antisense, or null as a control. The modification of miR-216b levels in HT29 and SW480 cells was confirmed by RT-qPCR (Figure 4A). We found that miR-216b overexpression significantly reduced the luciferase activity of 3’-UTR reporter for Beclin-1, while miR-216b depletion significantly increased the luciferase activity of 3’-UTR reporter for Beclin-1, in either HT29 cells or SW480 cells (Figure 4B). Thus, our data suggest that the binding of miR-216b to the 3’-UTR of Beclin-1 mRNA in CRC cells is functional. Together, the findings in the current study suggest that aEGFR may decrease miR-216b levels in CRC cells, which subsequently upregulates Beclin-1 to increase CRC cell autophagy to antagonize aEGFR-induced cell death. Strategies that increase miR-216b levels or inhibit cell autophagy may improve the outcome of aEGFR treatment in CRC therapy (Figure 5).
Figure 4.
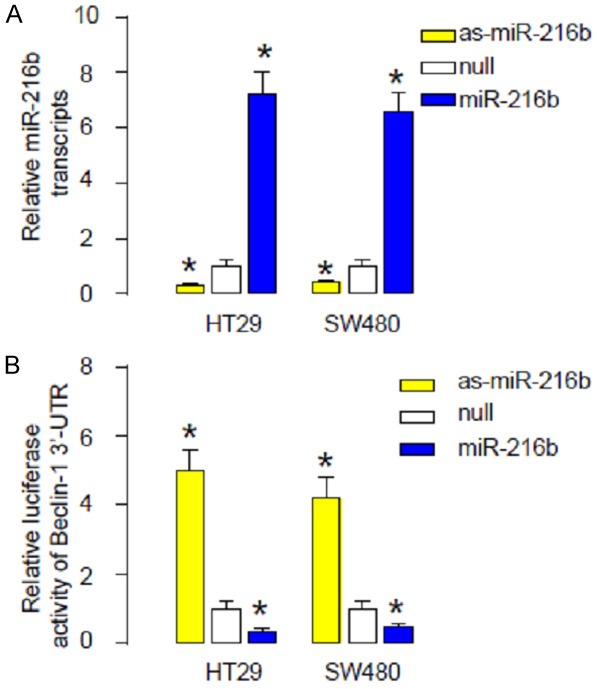
MiR-216b inhibits Beclin-1 protein translation in CRC cells. A. The miR-216b levels in HT29 and SW480 cells were modified and verified by RT-qPCR. B. In order to verify that the binding of miR-216b to the 3’-UTR of Beclin-1 mRNA is functional, the intact 3’-UTR of Beclin-1 was cloned into a luciferase reporter plasmid. MiR-216b overexpression significantly reduced the luciferase activity of 3’-UTR reporter for Beclin-1, while miR-216b depletion significantly increased the luciferase activity of 3’-UTR reporter for Beclin-1. *p<0.05. N=5.
Figure 5.
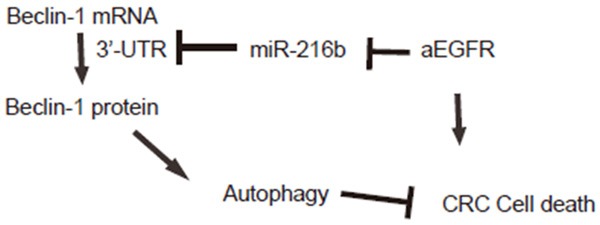
Schematic of the model. aEGFR may decrease miR-216b levels in CRC cells, which subsequently upregulates Beclin-1 to increase CRC cell autophagy to antagonize aEGFR-induced cell death.
Discussion
aEGFR induces mitotic arrest at concentrations typically used in human tumor cell (including CRC cells) culture, it was believed that aEGFR may have a therapeutic effect on human cancer. Nevertheless, some cancer cells appear to be resistant to aEGFR treatment. On the other hand, the compromised effects of aEGFR on cancer mitotic arrest and cell death may be resulting from the molecular responses of cancer cells to aEGFR, in which enhanced survival is reached by activation of cell autophagy. Numerous screens to identify cellular and molecular markers of resistance or sensitivity to paclitaxel have been performed by different researchers. However, so far a validated biomarker to be used in clinic for predicting the outcome of aEGFR therapy has not been achieved.
In the current study, we analyzed the effects of aEGFR on the cell viability and autophagy of CRC cells. We found that aEGFR dose-dependently decreases CRC cell viability. However, this aEGFR-induced CRC cell death appeared to be attenuated by enhanced autophagy-associated cell survival. In line with this view, it seemed that aEGFR induce both apoptotic cell death and autophagic cell survival in CRC cells, and autophagy appears to be a negative feedback of CRC cells to antagonize the toxicity of aEGFR. aEGFR generated a harsh environment for CRC cells. In order to survive, lung cancer cells decreased miR-216b levels to induce upregulation of Beclin-1 to augment autophagic cell survival. Here, we used 2 different CRC lines (HT29 and SW480) and achieved same result, which potentially excluded a possibility of cell-line dependence of the current findings.
Together, our data shed light on a previously non-appreciated signaling regulatory pathway that controls the effects of aEGFR against CRC cells. Future experiments may be applied to further dissect the details of the pathway, e.g. other involved key proteins, other involved miRNAs, and the clinic relevance. Our study also suggests that strategies that increase miR-216b levels or inhibit cell autophagy may improve the outcome of aEGFR treatment in CRC therapy.
Acknowledgements
This study was supported by Jilin province Natural Science Foundation of China 20160101115JC, and the provincal funds for health service of Jilin.
Disclosure of conflict of interest
None.
References
- 1.East JE, Dekker E. Colorectal cancer diagnosis in 2012: A new focus for CRC prevention--more serration, less inflammation. Nat Rev Gastroenterol Hepatol. 2013;10:69–70. doi: 10.1038/nrgastro.2012.245. [DOI] [PubMed] [Google Scholar]
- 2.Van Schaeybroeck S, Allen WL, Turkington RC, Johnston PG. Implementing prognostic and predictive biomarkers in CRC clinical trials. Nat Rev Clin Oncol. 2011;8:222–232. doi: 10.1038/nrclinonc.2011.15. [DOI] [PubMed] [Google Scholar]
- 3.Labianca R, Beretta GD, Mosconi S, Pessi MA, Milesi L. The development of clinical research in CRC. Ann Oncol. 2005;16(Suppl 4):iv37–43. doi: 10.1093/annonc/mdi906. [DOI] [PubMed] [Google Scholar]
- 4.Garza-Trevino EN, Said-Fernandez SL, Martinez-Rodriguez HG. Understanding the colon cancer stem cells and perspectives on treatment. Cancer Cell Int. 2015;15:2. doi: 10.1186/s12935-015-0163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: Anti-epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154:37–49. doi: 10.7326/0003-4819-154-1-201101040-00006. [DOI] [PubMed] [Google Scholar]
- 6.Dasari A, Messersmith WA. New strategies in colorectal cancer: biomarkers of response to epidermal growth factor receptor monoclonal antibodies and potential therapeutic targets in phosphoinositide 3-kinase and mitogen-activated protein kinase pathways. Clin Cancer Res. 2010;16:3811–3818. doi: 10.1158/1078-0432.CCR-09-2283. [DOI] [PubMed] [Google Scholar]
- 7.Patel DK. Clinical use of anti-epidermal growth factor receptor monoclonal antibodies in metastatic colorectal cancer. Pharmacotherapy. 2008;28:31S–41S. doi: 10.1592/phco.28.11-supp.31S. [DOI] [PubMed] [Google Scholar]
- 8.Jean GW, Shah SR. Epidermal growth factor receptor monoclonal antibodies for the treatment of metastatic colorectal cancer. Pharmacotherapy. 2008;28:742–754. doi: 10.1592/phco.28.6.742. [DOI] [PubMed] [Google Scholar]
- 9.Jones C, Taylor MA, McWilliams B. The role of cetuximab as first-line treatment of colorectal liver metastases. HPB (Oxford) 2013;15:11–17. doi: 10.1111/j.1477-2574.2012.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Liu H, Guan Y, Wang Q, Zhou F, Jie L, Ju J, Pu L, Du H, Wang X. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am J Transl Res. 2015;7:1574–1587. [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Wang Q, Yin FQ, Zhang W, Yan LH, Li L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am J Transl Res. 2015;7:1510–1527. [PMC free article] [PubMed] [Google Scholar]
- 13.Nandi SS, Duryee MJ, Shahshahan HR, Thiele GM, Anderson DR, Mishra PK. Induction of autophagy markers is associated with attenuation of miR-133a in diabetic heart failure patients undergoing mechanical unloading. Am J Transl Res. 2015;7:683–696. [PMC free article] [PubMed] [Google Scholar]
- 14.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang CY, Huang SP, Lin VC, Yu CC, Chang TY, Lu TL, Chiang HC, Bao BY. Genetic variants of the autophagy pathway as prognostic indicators for prostate cancer. Sci Rep. 2015;5:14045. doi: 10.1038/srep14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrow JM, Yang JC, Evans CP. Autophagy as a modulator and target in prostate cancer. Nat Rev Urol. 2014;11:508–516. doi: 10.1038/nrurol.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Han JJ, Tennakoon JB, Mehta FF, Merchant FA, Burns AR, Howe MK, McDonnell DP, Frigo DE. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27:280–295. doi: 10.1210/me.2012-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Leva G, Croce CM. miRNA profiling of cancer. Curr Opin Genet Dev. 2013;23:3–11. doi: 10.1016/j.gde.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereira DM, Rodrigues PM, Borralho PM, Rodrigues CM. Delivering the promise of miRNA cancer therapeutics. Drug Discov Today. 2013;18:282–289. doi: 10.1016/j.drudis.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Mei Q, Li F, Quan H, Liu Y, Xu H. Busulfan inhibits growth of human osteosarcoma through miR-200 family microRNAs in vitro and in vivo. Cancer Sci. 2014;105:755–762. doi: 10.1111/cas.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Xiao W, Sun J, Han D, Zhu Y. MiRNA-181c inhibits EGFR-signaling-dependent MMP9 activation via suppressing Akt phosphorylation in glioblastoma. Tumour Biol. 2014;35:8653–8658. doi: 10.1007/s13277-014-2131-6. [DOI] [PubMed] [Google Scholar]
- 24.Liu G, Jiang C, Li D, Wang R, Wang W. MiRNA-34a inhibits EGFR-signaling-dependent MMP7 activation in gastric cancer. Tumour Biol. 2014;35:9801–9806. doi: 10.1007/s13277-014-2273-6. [DOI] [PubMed] [Google Scholar]
- 25.Kim SY, Lee YH, Bae YS. MiR-186, miR-216b, miR-337-3p, and miR-760 cooperatively induce cellular senescence by targeting alpha subunit of protein kinase CKII in human colorectal cancer cells. Biochem Biophys Res Commun. 2012;429:173–179. doi: 10.1016/j.bbrc.2012.10.117. [DOI] [PubMed] [Google Scholar]
- 26.Liu FY, Zhou SJ, Deng YL, Zhang ZY, Zhang EL, Wu ZB, Huang ZY, Chen XP. MiR-216b is involved in pathogenesis and progression of hepatocellular carcinoma through HBx-miR-216b-IGF2BP2 signaling pathway. Cell Death Dis. 2015;6:e1670. doi: 10.1038/cddis.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng M, Tang H, Zhou Y, Zhou M, Xiong W, Zheng Y, Ye Q, Zeng X, Liao Q, Guo X, Li X, Ma J, Li G. miR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J Cell Sci. 2011;124:2997–3005. doi: 10.1242/jcs.085050. [DOI] [PubMed] [Google Scholar]
- 28.Wang F, Ying HQ, He BS, Pan YQ, Deng QW, Sun HL, Chen J, Liu X, Wang SK. Upregulated lncRNA-UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b and activation of FGFR1/ERK signaling pathway. Oncotarget. 2015;6:7899–7917. doi: 10.18632/oncotarget.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Kleist S, Chany E, Burtin P, King M, Fogh J. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J Natl Cancer Inst. 1975;55:555–560. doi: 10.1093/jnci/55.3.555. [DOI] [PubMed] [Google Scholar]
- 30.Leibovitz A, Stinson JC, McCombs WB 3rd, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976;36:4562–4569. [PubMed] [Google Scholar]
- 31.Coronnello C, Benos PV. ComiR: Combinatorial microRNA target prediction tool. Nucleic Acids Res. 2013;41:W159–164. doi: 10.1093/nar/gkt379. [DOI] [PMC free article] [PubMed] [Google Scholar]