

Abstract
Pharmacologic preconditioning is an intriguing and emerging approach adopted to prevent injury of ischemia/reperfusion. Neuroprotection is the cardinal effect of these pleiotropic actions of berberine. Here we investigated that whether berberine could acts as a preconditioning stimuli contributing to attenuate hypoxia-induced neurons death as well. Male Sprague-Dawley rats of middle cerebral artery occlusion (MCAO) and rat primary cortical neurons undergoing oxygen and glucose deprivation (OGD) were preconditioned with berberine (40 mg/kg, for 24 h in vivo, and 10-6 mol/L, for 2 h in vitro, respectively). The neurological deficits and cerebral water contents of MCAO rats were evaluated. The autophagy and apoptosis were further determined in primary neurons in vitro. Berberine preconditioning (BP) was then shown to ameliorate the neurological deficits, decrease cerebral water content and promote neurogenesis of MCAO rats. Decreased LDH release from OGD-treated neurons was observed via BP, which was blocked by LY294002 (20 µmol/L), GSK690693 (10 µmol/L), or YC-1 (25 µmol/L). Furthermore, BP stimulated autophagy and inhibited apoptosis via modulated the autophagy-associated proteins LC 3, Beclin-1 and p62, and apoptosis-modulating proteins caspase 3, caspase 8, caspase 9, PARP and BCL-2/Bax. In conclusion, berberine acts as a stimulus of preconditioning that exhibits neuroprotection via promoting autophagy and decreasing anoxia-induced apoptosis.
Keywords: Berberine, preconditioning, apoptosis, autophagy
Introduction
Pharmacological preconditioning is a promising approach for treating cerebral ischemia. Similar with ischemic preconditioning (IP), pharmacological preconditioning triggers intensively endogenous protective mechanisms attenuating the damage of the subsequent ischemia in both brain and heart. Beyond the hypoxia-induced ischemic tolerance, preconditioning with chemical agents might be more favorable and convenient for realizing in clinic. The transcription factor hypoxia inducible factor-1 (HIF-1) that senses hypoxia and transactivates those down-stream genes associated with energy metabolism and neurogenesis acts as pivotal modulator during IP [1]. It has been well documented that chemical agents stabilizing HIF-1 in brain stimulate ischemic tolerance. Stimuli such as lipopolysaccharide deferoxamine, and FTY-720 initiate the endogenous protective mechanisms in a HIF-1-dependent manner [2]. Other agents as the volatile anesthetics and KATP channel openers establish the IP phenotype through the transducers nitric oxide synthase, Akt, and KATP channels [3]. However, chemical agents that have intriguing safety and stability profile and show neuroprotective effects are the more promising candidates for pharmacologic preconditioning, especially those from plants.
Autophagy, a vital catabolic process within the lysosome, is an essential cytoprotective response and prosurvival pathway in both normal physiology and diseases such as cerebral ischemia and neurodegeneration. Although the role of autophagy is still controversial, autophagy has been reported to be upregulated after ischemia/reperfusion [4]. Furthermore, Sciarretta et al. indicate that autophagy is beneficial during ischemia but harmful during reperfusion and IP attenuate ischemic injury of neuron via inducing autophagy [5]. Interestingly, deferoxamine, the stabilizer of HIF-1 and stimulus of ischemia tolerance, induced autophagy in a HIF-1-mediated manner [6]. Then, the moderate autophagy during the initiation of ischemia or even ahead of hypoxia exhibits its fundamental role of maintaining cellular homeostasis and protecting neuron against anoxia insult.
Berberine is an isoquinoline alkaloid that shows beneficial effect in various neurodegeneration and neuropsychiatric disease. Neuroprotection of berberine against cerebral ischemia was reported to accomplish through multiple mechanisms that include antagonizing N-methyl-d-aspartate receptor, blocking voltage-dependent potassium currents and inhibiting inflammatory response [7]. Our previous investigation further indicated that berberine inhibited hypoxia-induced apoptosis by modulating HIF-1 levels [8]. Then, the present study was designed to explore whether berberine protect the neuron attacked by hypoxia via preconditioning. Berberine preconditioning was conducted 24 h before the anoxia injury on neuron. The damage of neuron, especially the apoptosis, was determined in vivo and in vitro, which was prevented through the berberine preconditioning. Meanwhile, autophagy was triggered by the stimulating of berberine.
Methods
Animal model of middle cerebral artery occlusion (MCAO)
Adult male Sprague-Dawley rats (280-300 g) from Shanghai Laboratory Animal Center of the Chinese Academy of Sciences (Shanghai, PRC) were kept at 20-21°C and 55 ± 5% relative humidity in a 12/12 h light/dark cycle. Standard rodent chow was provided ad libitum throughout the study. Transient focal cerebral ischemia models were prepared through MCAO according to the intraluminal suture method [9]. After 2 h of MCAO, blood flow was restored and reperfusion was allowed for 24 h, 72 h, and 7 d. Sham-operated control animals underwent the same procedure without the suture insertion. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Care and Use Committee of Nanjing University of Chinese Medicine.
Cerebral cortical neurons
The preparation and maintenance of dissociated neuronal cell cultures from embryonic day 18 rat (Sprague Dawley) embryos were performed as described previously [10]. Neuronal cells cultured for 8 d were identified by immunostaining with neurofilament antibody and Cy3-labeled secondary antibody, and were adopted for the following investigations. In the following preconditioning and oxygen-glucose deprivation (OGD) treatments, neuronal cells were starved in fetal bovine serum-free culture medium.
OGD treatment
Neuronal cell injury was induced by OGD to mimic cerebral ischemia 24 h after BP [11]. Cerebral cortical neurons were rinsed with Hanks’ balanced salt solution (HBSS) and incubated with glucose- and serum-free medium (Essential Medium, MEM) supplemented with 25% HBSS, 1 mM L-glutamine, 3.75 μg/mL amphotericin B, and 30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) saturated with 95% N2/5% CO2 in an airtight chamber flushed with 95% N2/5% CO2. OGD exposure lasted for 30 min. Control neurons were treated with glucose-containing and serum-free normoxic medium for the same period of time. At the end of the attack, the medium was replaced with glucose-containing and serum-free normoxic medium, and the cerebral cortical neurons were removed and placed in an incubator at 37°C and 100% humidity in 95% air/5% CO2 for recovery. Neuronal injury or viability was evaluated 24 h later.
Berberine preconditioning (BP)
Berberine was administrated intragastrically to MCAO rats as a single dose (40 mg/kg) 24 h before ischemia/reperfusion insult. BP for 2 h in vitro was performed in neurons 24 h before the 2-h OGD treatment. Saline (NaCl, 0.9%) or phosphate buffered saline (PBS) was used as the vehicle for the sham and MCAO groups in vivo, or the blank and OGD groups in vitro.
Neuroscore test of MCAO rats
A seven-point neuroscore test was used to assess the post-ischemic deficits of MCAO rats without or with BP at 24 h and 72 h [12]. The test was conducted by an investigator who was blinded to the treatment groups. The scale was as follows: Grade 6, normal extension of both forelimbs towards the floor when lifted; Grade 5, consistent flexion of the forelimb contralateral to the injured hemisphere; Grade 4, dysfunctional rats with consistently reduced resistance to a lateral push towards the paretic side; Grade 3, circling towards the paretic side if pulled and lifted by the tail; Grade 2, circling towards the paretic side if pulled by the tail; Grade 1, circling spontaneously towards the paretic side; and Grade 0, no spontaneous motion.
Brain water content measurement
Each brain section from the sacrificed rats was weighed before and after drying for 24 h at 100°C. The water content is expressed as the percentage change between the wet weight (WW) and dry weight (DW): [(WW - DW)/WW] × 100.
Lactate dehydrogenase (LDH) assay
The activity of LDH in the culture medium of neurons exposed to BP and OGD treatment was analyzed with a standardized colorimetric enzyme kit. The protective efficacy of BP on neurons was expressed as the percentage of the maximal LDH activity without intervention. Following berberine or vehicle preconditioning for 2 h, the neurons were cultured in serum-free medium for another 22 h. Before OGD exposure, the neurons were incubated with LY294002 (20 µmol/L), GSK690693 (10 µmol/L), or YC-1 (25 µmol/L) for 30 min, respectively. The LDH assay was performed 24 h after OGD.
ΔΨm analysis
The mitochondrial-specific cationic dye 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-benzimidazolyl-carbocyanine iodide (JC-1, Molecular Probes, Eugene, OR) was used to monitor the dissipation of the mitochondrial membrane potential, which accumulated in the mitochondria in a potential-dependent manner [13]. Cerebral cortical neurons with or without 2-h BP were incubated with JC-1 (10 μmol/L) for 30 min at 37°C 24 h post OGD treatment, which was performed 22 h after preconditioning. Fluorescence was then monitored via confocal laser scanning microscopy (Carl Zeiss, Jena, Germany).
Immunofluorescence assay of LC3 in neurons
The formalin-fixed neurons were incubated with rabbit anti-LC3 antibody (diluted 1:100; CST, Boston, MA) overnight at 4°C. Goat anti-rabbit secondary antibody (tetramethyl rhodamin isothiocyanate conjugated; diluted 1:200; CST) was added for 30 min at room temperature. The nuclei and cytoskeleton were stained by 4’6-diamidino-2-phenylindole and rhodamine phalloidin (Sigma-Aldrich), respectively, before obtaining the images. Fluorescence was observed under a confocal laser scanning microscope (Carl Zeiss). The neurons were treated without or with berberine for 2 h.
Autophagy of neurons analyzed by electron microscopy
Neurons treated without or with berberine for 1, 2, or 4 h were rinsed with ice-cold PBS twice, and then collected by centrifugation at 600 × g for 10 min. The pellet was immersed in fixative (2.5% glutaraldehyde with 2% paraformaldehyde in 0.1 mol/L cacodylate buffer, pH 7.4) at 4°C overnight. After removing the fixative, the pellet was washed in 0.05 mol/L cacodylate buffer and post-fixed in 1% OsO4 in 0.05 mol/L cacodylate buffer for 90 min. Following ethyl alcohol dehydration and resin infiltration, ultrathin sections of the embedded pellet were obtained with a Leica Ultramicrotome UCT (Leica Microsystems, Wetzlar, Germany), stained with uranyl acetate and lead citrate, and visualized with a JEM1010 transmission electron microscope (JEOL, Tokyo, Japan).
Western blotting analysis
The proteins (30 μg/per lane) from neurons with or without BP were separated by 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). The blotted membranes were incubated with the corresponding primary antibodies (Clv-caspase 3, caspase 3, caspase 8, caspase 9, Bcl-2, Bax, Clv-poly (ADP-ribose) polymerase [PARP], PARP, LC3, p62, Beclin-1; Abcam, Cambridge, UK) and the appropriate secondary antibodies. Then, the blotted proteins were detected by the enhanced chemiluminescence system (Applygen, Beijing, China). Immunoreactive bands were visualized by autoradiography, and the density of these bands was densitometrically quantified via a Gel Pro analyzer (Media Cybernetics, USA). Clv-PARP, PARP, clv-caspase 3, caspase 3, caspase 8, caspase 9, Bcl-2, and Bax were analyzed in blank, OGD-treated, and berberine-preconditioned neurons 24 h after OGD. LC3, p62, and Beclin-1 were determined in neurons at 0, 1, 2, and 4 h from the beginning of BP.
Data analysis and statistical procedures
Results are expressed as the mean ± the standard error of the mean (SEM). Data were analyzed using one-way analyses of variance (ANOVA). Newman-Keuls post-hoc analyses were conducted for pairwise multiple comparisons when significance was reached by ANOVA. The difference was considered statistically significant if P < 0.05.
Results
BP attenuated brain injury in MCAO rats
Berberine’s neuroprotective effects via preconditioning were first evaluated in the MCAO rats. Preconditioning was performed 24 h before MCAO and the assessments were conducted after reperfusion for 24 h, 72 h, and 7 d (Figure 1A). Compared to MCAO rats without BP, BP significantly improved neurological scores at 24 and 72 h following MCAO (Figure 1B). The cerebral water contents resulted from ischemia/reperfusion were also pronouncedly decreased by BP (Figure 1C). Moreover, compared with MCAO model rats without berberine, an increased number of BrdU-labeled neurons in berberine-preconditioned MCAO rats indicated neuronal proliferation after 7 d of MCAO (Figure 1D). These results suggest that BP can ameliorate the cerebral injury induced by ischemia/reperfusion and provoke neurogenesis.
Figure 1.
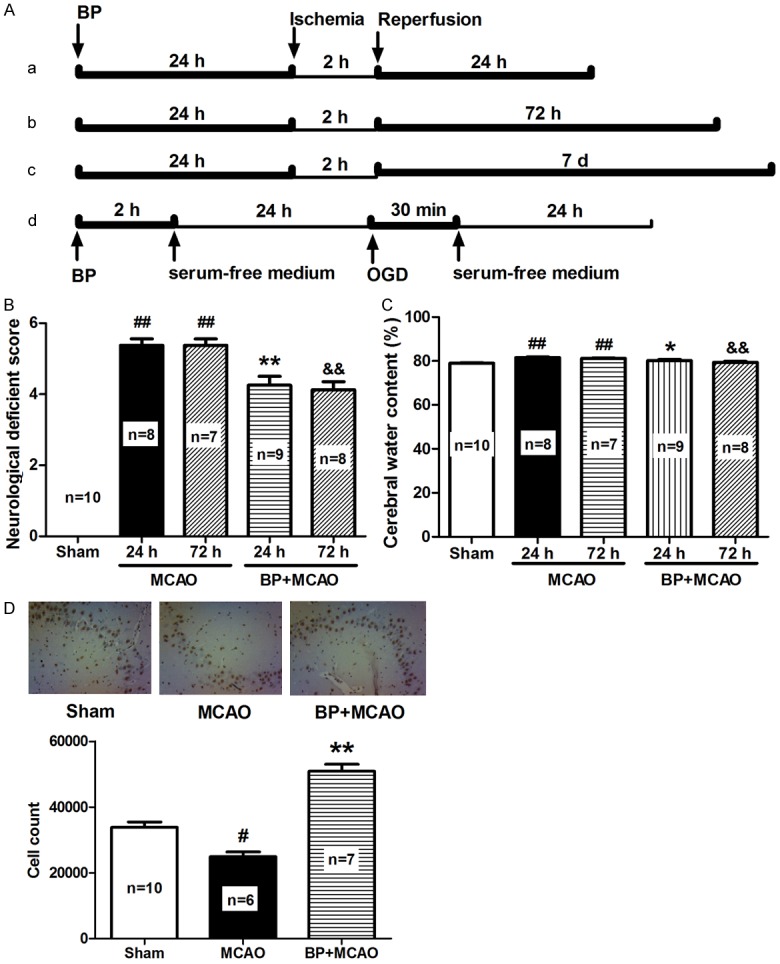
BP attenuates neuronal injury in MCAO rats. (A) Scheme used for BP. Rats were preconditioned with berberine 24 h before ischemia, subjected to ischemia for 2 h, and then reperfusion for 24, 72 h, or 7 d, respectively (a, b, c); the neurons was treated according to the procedure (d). (B) Neurological deficient score of MCAO rats subjected to BP was assayed 24 h after ischemia. (C) Cerebral water content of MCAO rats subjected to BP was determined 24 h after ischemia. (D) BrdU-labeled neurons in MCAO rats after reperfusion for 7 days with or without BP visualized by immunohistochemical assays (n = 3). All data are presented as the mean ± SEM. #P < 0.05, ##P < 0.01 vs. Sham; *P < 0.05, **P < 0.01 vs. MCAO 24 h; &&P < 0.01 vs. MCAO 72 h.
BP protected neurons against OGD-induced damage
The effects of BP on neurons under hypoxic conditions were further characterized via analyzing LDH release from OGD-treated primary cerebral cortical neurons. BP remarkably decreased LDH release from OGD-treated neurons at the concentrations from 10-5 to 10-9 ug/mL (Figure 2A), which was blocked by inhibiting of PI3K/Akt signaling and HIF-1 pathway (Figure 2B). Subsequently, OGD-provoked apoptosis was determined in the absence or presence of BP. With BP, the attenuated aggregation of JC-1 associated with the OGD-induced mitochondrial outer membrane permeabilization (Figure 2C). It’s postulated that PI3K/Akt-regulated HIF-1 pathway might be involved in the BP process.
Figure 2.
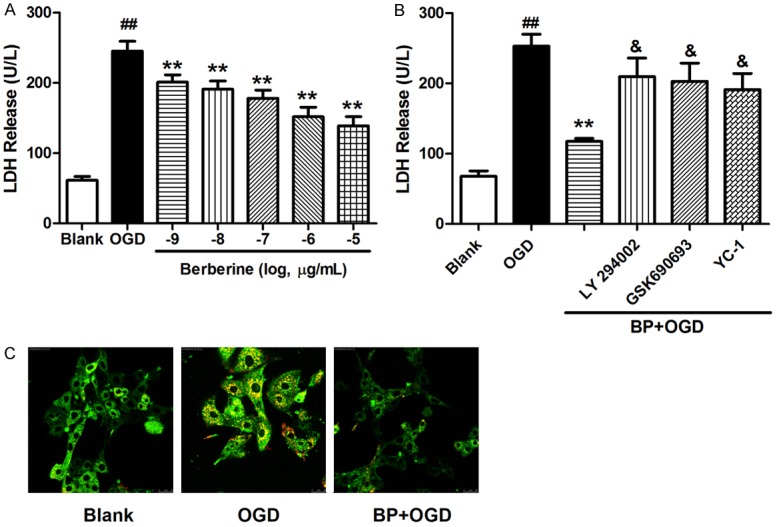
BP protected neurons against OGD. A. LDH release from OGD attacked neurons in 24 h with BP at concentrations from 10-9 to 10-5 μg/mL. B. Neurons firstly incubated with LY294002 (20 µmol/L), GSK690693 (10 µmol/L), or YC-1 (25 µmol/L) for 30 min were preconditioned with berberine for 2 h, and then subjected to OGD for 24 h. An LDH assay was performed after OGD. C. The mitochondrial membrane potential was indicated with JC-1 in control neurons (blank) or OGD-treated neurons with or without BP. All data are presented as the mean ± SEM n = 8. ##P < 0.01 vs. Blank; **P < 0.01 vs. OGD; &P < 0.05 vs. BP+OGD.
BP modulated hypoxia-induced apoptosis-associated proteins
Based on the indication of Figure 2C, the apoptosis-associated proteins were subsequently determined. Inhibition of caspase 3’s activation occurred in berberine-preconditioned neurons, as demonstrated by reducing caspase 3 and PARP cleavage (Figure 3A, 3B). Alternatively, the increased accumulation of caspase 8 and caspase 9, and the enhanced ratio of BCL-2/Bax were reversed by BP in neurons with OGD (Figure 3C, 3D). These results indicate that BP exerted neuroprotective effects against hypoxia/ischemia by inhibiting apoptosis.
Figure 3.
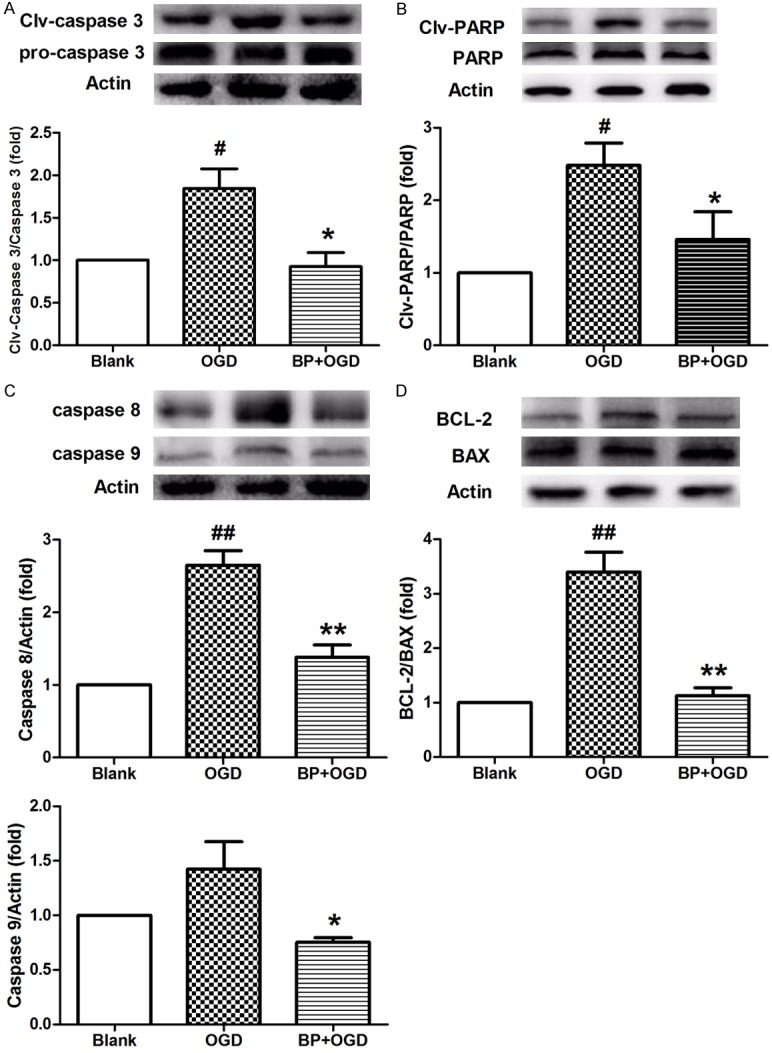
BP inhibits hypoxia-induced neuronal apoptosis. A and B. The activation of caspase 3, and the levels of PARP, Clv-PARP in OGD-treated neurons following BP were analyzed through Western blotting. C. The levels of caspase 8 and caspase 9 were determined by Western blotting in OGD-treated neurons following BP. D. The levels of BCL-2 and Bax were determined by Western blotting, and the ratio of BCL-2/Bax was calculated in OGD-treated neurons following BP. All data are presented as the mean ± SEM, n = 3. #P < 0.05, ##P < 0.01 vs. Blank; *P < 0.05, **P < 0.01 vs. OGD.
BP initiated autophagy in neurons
Because of the close relationship between autophagy and apoptosis and the pivotal roles of autophagy in the pathophysiology process of central nervous system, we next investigated the phenomenon of autophagy in the berberine-treated neurons. Autophagosomes with double membranes were firstly observed in berberine-treated neurons at 2 h with transmission electron microscopy (Figure 4A). Aggregation of LC3-II converted from LC3-I via lipidation was then detected in neurons following berberine incubation at 2 h with a confocal microscope, which indicated the formation of autophagic vesicles (Figure 4B). The turnover of LC3 was further confirmed by western blotting, where berberine promoted the conversion from LC3-I to LC3-II at 1, 2, and 4 h (Figure 4C). Consistent with the activation of autophagy, significantly enhanced beclin-1, an autophagy-concerned protein, was detected in berberine-treated neurons at 1, 2, and 4 h (Figure 4D). The levels of p62, another hallmark of autophagy that interacts with LC3-II, were also increased in berberine-incubated neurons at 2 and 4 h. However, decreased p62 was observed at 1 h (Figure 4E). These data suggest the involvement of autophagy-inducing mechanisms against hypoxia/ischemia injury with BP.
Figure 4.

Berberine induces neuronal autophagy. (A) Autophagy activation was observed by a transmission electron microscope without or with berberine for 1, 2, and 4 h. (B and C) Accumulation of LC3-positive puncta (green) in neurons was visualized by immunofluorescence without or with berberine for 2 h (B), and the protein levels of LC3-I and LC3-II in neurons following berberine incubation at 1, 2, and 4 h were determined via western blotting (C). (D and E) Immunoblot analysis of beclin-1 (D) and p62 (E) protein levels in neurons incubated without or with berberine for 1, 2, and 4 h. All data are presented as the mean ± SEM, n = 3. **P < 0.01 vs. 0 h.
Discussion
Autophagy is characterized by the bulk degradation of cytoplasmic macromolecules and organelles in cells via the lysosomal system responded to stresses involving oxidative stress, infection, nutrient deprivation and ischemia, which is important to maintain the cellular metabolic turnover and homeostasis. In neurons, autophagy plays multiple roles based on the different pathophysiological stage. Upregulated autophagy was promoted by increased protein synthesis or transcription of lysosomal components, which was contributed to clear amyloid precursor protein products such as A-beta and C-terminal fragment against intraneuronal neurofibrillary tangles and the extracellular senile plaques [14]. With the development of neurodegeneration, failure of autophagy characterized by hyperactivated mTORC1 and decreased Beclin 1 protein levels has dysfunction of degradation of proteins with aberrant structure. Under condition of ischemia/reperfusion, however, enhanced autophagy is commonly demonstrated to protect tissues involving heart, renal against injury. Cardiac-specific loss of autophagy causes cardiomyopathy, and impaired autophagy has been found in the ischemia/reperfusion injury [15]. Inhibition of autophagy by 3-methyladenine (3-MA), an autophagy inhibitor, deteriorated ischemia/reperfusion injury of renal [16]. Thus, autophagy may provide a protective mechanism for cell survival under ischemia/reperfusion conditions. However, Sciarretta et al. indicate that autophagy is beneficial during ischemia but harmful during reperfusion [5]. This phenomenon was further explained by the biphasic role of ALDH2 in the ischemia/reperfusion models, which triggers autophagy through AMPK activation during ischemia, but it decreases autophagy through Akt-mTOR activation in reperfusion (Ma et al., 2011). In neurons with hypoxia or ischemia/reperfusion, inhibition of autophagy shows neuroprotection. 3-MA rescued PC12 cell from serum deprivation [17]. In the autophagy associated gene 7 (Atg7)-conditional knockout mice, pyramidal neuron death was prevented by Atg7 deficiency that attenuated hypoxia-induced autophagy in neonatal hippocampus [18]. It’s well accepted presently that basal and moderate autophagy have positive effect on brain attacked by anoxia, which might be accomplished through removing the damaged mitochondria, scavenging the abnormally aggregated proteins, and compensating energy deprivation. Therefore, autophagy is one of pivotal characteristics of tolerance against nutrient deprivation such as ischemia. On the other hand, accelerated or severe autophagy that degrades normal components or those protection neurons during hypoxia/reperfusion may be detrimental. Three pathways as HIF-1α dependent manner, AMPK-dependent mechanism and HIF-1α and AMPK-independent hypophosphorylation contribute to the inactivation of mTOR inducing the autophagy [19]. Both HIF-1α and AMPK are involved in the ischemic preconditioning performed in neural cells and rats of stroke. Chemical preconditioning stimulus as isoflurane induced autophagy in both in vitro and in vivo [20-21]. Metformin, a well-known hypoglycaemic agent, conferred neuroprotection via AMPK-dependent autophagy [22].
Ischemic tolerance is an important phenomenon for protecting neuronal cells to against hypoxic injury, where autophagy and apoptosis procedures are initiated in response to anoxia stress or other stimulants. Autophagic activity commonly peaks at the beginning of stress process in stimulated neuronal cells and is considered a protective process, as it isolates potentially damaging reactions in the cell. In the present investigation, BP influenced the neuronal morphology by decreasing hypoxia-induced apoptosis and promoting LC3-dependent autophagy. HIF-1-mediated autophagy conventionally results from activation of BNIP3, a BH3-only protein, which is essential for decreasing the mitochondrial mass during hypoxia in a beclin-1-dependent manner [23]. During physiological hypoxia, HIF-1-triggered autophagy helps promote survival. With severe hypoxia or drastic glucose and amino-acid restriction, HIF-1-independent autophagy leads to cell death via the adenosine monophosphate-activated protein kinase-mTOR pathway or unfolded protein response pathway [24]. The autophagy results in survival or death depends on whether HIF-1 is the activated key target or whether it is accompanied by other stressors. Berberine stabilized HIF-1α without disturbing the nutrient supplements and metabolism in neurons. In contrast, berberine commonly promotes substance uptake (e.g., glucose) in muscle and adipose and provokes energy metabolism in tissue, which is beneficial for diabetes mellitus and obesity [25].
Although modulation of autophagy and apoptosis was demonstrated by BP in the present investigation, the relationship between autophagy and apoptosis was not exactly investigated in neurons following BP, which requires further study. Autophagy and apoptosis, however, shared several common modulators or signaling pathways. Apoptosis and autophagy are physiologically necessary pathways that are vital for cell homeostasis. Apoptosis facilitates type I programmed cell death, while the autophagic survival mechanism counteracts apoptosis. The bifunctional role of p53 is illustrated to trigger and inhibit both autophagy and apoptosis based on its subcellular localization, where the BH3-only proteins, Bad and (t)Bid, are involved. p53 directly modulates the expression of Bad and (t)Bid [26]. On the other hand, p53 plays important role in autophagy via binding to BCL-2 and Bax and interacting with mTOR [27]. BCL-2 acts as a switching factor for inhibiting autophagy via the directional action with Beclin-1 [28]. In this study, we demonstrated that BP decreased BCL-2 and Beclin-1, which might be responsible for the process of both apoptosis and autophagy.
In summary, the present study demonstrated that the activation of autophagy, as an endogenous neuroprotective mechanism, is responsible for the actions of BP on neurons in which PI3K/Akt signaling and HIF-1 pathway are involved. Our investigation provides novel indication that BP might be a preferable and rational therapeutic approach to ameliorate neuronal injury from hypoxia/ischemia.
Acknowledgements
This research was financially supported by the Administration of Traditional Chinese Medicine of Jiangsu Province [Project No. LZ13007]; National Natural Science Foundation of China [Project No. 81573635; 30873450]; A Project Funded by Natural Science Foundation of Jiangsu Province [Project No. BK 2012855; BY 2012036]; A Project Funded by Innovation Research Team of Nanjing University of Chinese Medicine; A Project Funded by the Six Talent Project in Jiangsu Province; and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure of conflict of interest
None.
References
- 1.Grimm C, Hermann DM, Bogdanova A, Hotop S, Kilic U, Wenzel A, Kilic E, Gassmann M. Neuroprotection by hypoxic preconditioning: HIF-1 and erythropoietin protect from retinal degeneration. Semin Cell Dev Biol. 2005;16:531–538. doi: 10.1016/j.semcdb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Nouri F, Salehinejad P, Nematollahi-Mahani SN, Kamarul T, Zarrindast MR, Sharifi AM. Deferoxamine Preconditioning of Neural-Like Cells Derived from Human Wharton’s Jelly Mesenchymal Stem Cells as a Strategy to Promote Their Tolerance and Therapeutic Potential: An In Vitro Study. Cell Mol Neurobiol. 2015 doi: 10.1007/s10571-015-0249-8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codaccioni JL, Velly LJ, Moubarik C, Bruder NJ, Pisano PS, Guillet BA. Sevoflurane preconditioning against focal cerebral ischemia: inhibition of apoptosis in the face of transient improvement of neurological outcome. Anesthesiology. 2009;110:1271–1278. doi: 10.1097/ALN.0b013e3181a1fe68. [DOI] [PubMed] [Google Scholar]
- 4.Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia--a potential target for neuroprotective strategies? Pharmacol Rep. 2012;64:1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- 5.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol. 2011;32:275–281. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T. Neuroprotection of deferoxamine on rotenoneinduced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem Int. 2010;57:198–205. doi: 10.1016/j.neuint.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Kulkarni SK, Dhir A. Berberine: a plant alkaloid with therapeutic potential for central nervous system disorders. Phytother Res. 2010;24:317–324. doi: 10.1002/ptr.2968. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Qian Z, Pan L, Li H, Zhu H. Hypoxiainducible factor 1 mediates the anti-apoptosis of berberine in neurons during hypoxia/ischemia. Acta Physiol Hung. 2012;99:311–323. doi: 10.1556/APhysiol.99.2012.3.8. [DOI] [PubMed] [Google Scholar]
- 9.Leonardo CC, Hall AA, Collier LA, Green SM, Willing AE, Pennypacker KR. Administration of a Sigma Receptor Agonist Delays MCAOInduced Neurodegeneration and White Matter Injury. Transl Stroke Res. 2010;1:135–145. doi: 10.1007/s12975-009-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zausinger S, Hungerhuber E, Baethmann A, Reulen H, Schmid-Elsaesser R. Neurological impairment in rats after transient middle cerebral artery occlusion: a comparative study under various treatment paradigms. Brain Res. 2000;863:94–105. doi: 10.1016/s0006-8993(00)02100-4. [DOI] [PubMed] [Google Scholar]
- 13.Ward MW. Quantitative analysis of membrane potentials. Methods Mol Biol. 2010;591:335–351. doi: 10.1007/978-1-60761-404-3_20. [DOI] [PubMed] [Google Scholar]
- 14.Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011;25:219–231. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51:584–593. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–1192. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol. 2001;64:233–246. doi: 10.1679/aohc.64.233. [DOI] [PubMed] [Google Scholar]
- 18.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 20.Li ZQ, Li LX, Mo N, Cao YY, Kuerban B, Liang YX, Fan DS, Chui DH, Guo XY. Duration-dependent regulation of autophagy by isoflurane exposure in aged rats. Neurosci Bull. 2015;31:505–513. doi: 10.1007/s12264-015-1549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng J, Drobish JK, Liang G, Wu Z, Liu C, Joseph DJ, Abdou H, Eckenhoff MF, Wei H. Anesthetic preconditioning inhibits isoflurane-mediated apoptosis in the developing rat brain. Anesth Analg. 2014;119:939–946. doi: 10.1213/ANE.0000000000000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, Zhang QQ, Gao L, Shi JQ, Zhang YD, Tan L. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171:3146–3157. doi: 10.1111/bph.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22:177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med. 2009;9:417–424. doi: 10.2174/156652409788167131. [DOI] [PubMed] [Google Scholar]
- 25.Xu M, Xiao Y, Yin J, Hou W, Yu X, Shen L, Liu F, Wei L, Jia W. Berberine promotes glucose consumption independently of AMP-activated protein kinase activation. PLoS One. 2014;9:e103702. doi: 10.1371/journal.pone.0103702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy. 2011;7:477–489. doi: 10.4161/auto.7.5.14811. [DOI] [PMC free article] [PubMed] [Google Scholar]