

Summary
According to in vitro assays, T cells are thought to kill rapidly and efficiently, but the efficacy and dynamics of cytotoxic T lymphocyte (CTL)-mediated killing of virus-infected cells in vivo remains elusive. We used two-photon microscopy to quantify CTL-mediated killing in mice infected with herpesviruses or poxviruses. On average, one CTL killed 2–16 virus-infected cells per day as determined by real-time imaging and by mathematical modeling. In contrast, upon virus-induced MHC class I downmodulation, CTLs failed to destroy their targets. During killing, CTLs remained migratory and formed motile kinapses rather than static synapses with targets. Viruses encoding the calcium sensor GCaMP6s revealed strong heterogeneity in individual CTL functional capacity. Furthermore, the probability of death of infected cells increased for those contacted by more than two CTLs, indicative of CTL cooperation. Thus, direct visualization of CTLs during killing of virus-infected cells reveals crucial parameters of CD8+ T cell immunity.
Graphical Abstract
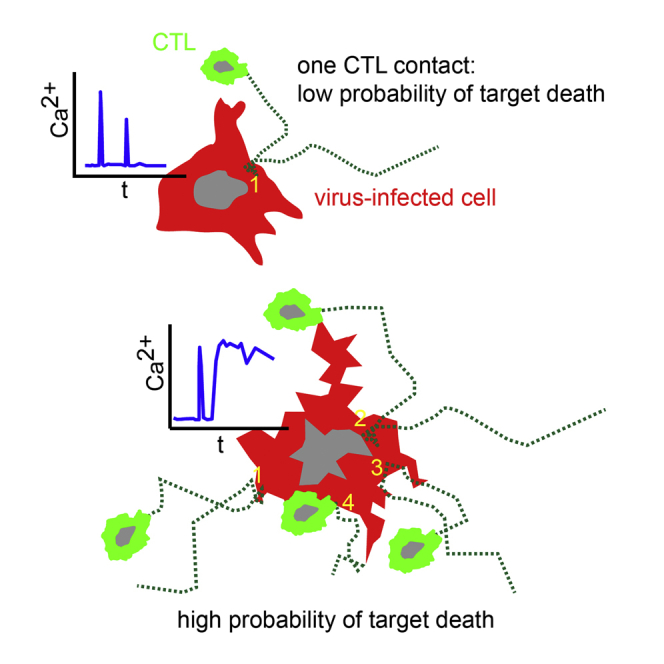
Highlights
-
•
Two-photon imaging indicates that CTLs kill 2–16 virus-infected cells per day
-
•
CTLs form kinapses rather than stable synapses when killing virus-infected cells
-
•
Some CTL contacts trigger long-lasting calcium fluxes in virus-infected cells
-
•
CTLs can cooperate during killing of virus-infected cells
According to in vitro assays, T cells are thought to kill rapidly and efficiently. Using two-photon microscopy, Forster and colleagues have found that killing capacities of single cytotoxic T lymphocytes (CTLs) in vivo are heterogeneous and limited. Quantification of target-cell-death probabilities identified efficient cooperative killing when multiple CTLs attacked a virus-infected cell.
Introduction
CD8+ cytotoxic T lymphocytes (CTLs) play an essential role in combating viral infections (Zhang and Bevan, 2011). During activation by antigen-presenting cells, CTLs integrate T cell receptor (TCR), co-stimulatory, and cytokine receptor signaling to fine-tune proliferation and differentiation and establish various effector cell subtypes characterized by the expression of different surface markers and cytokine production abilities (Marchingo et al., 2014). Together, these mechanisms allow the generation of CTL responses that can defend the host organism during primary infection and provide protective immunity against reinfection.
Primed CTLs are able to detect viral peptides restricted to major histocompatibility complex class I (MHC-I) and establish a TCR-triggered immunological synapse with their targets to secrete the content of their cytotoxic granules toward the infected cell (Dustin, 2008). The targeted secretion of several effector proteins, such as granzymes and perforin, triggers the cell-death machinery in the infected cell while leaving antigen-negative bystander cells intact (Lopez et al., 2012). Furthermore, CTLs secrete various cytokines that contribute to antiviral immunity. However, it remains unclear how important the contact-dependent killing of target cells is in relation to these indirect mechanisms of control.
The efficiency of CTL-mediated contact-dependent killing of different cell types has been studied extensively in vitro. Such studies have suggested that CTLs can rapidly, serially, and even simultaneously kill multiple target cells within minutes (Wiedemann et al., 2006). However, assays of in vitro killing have a limited capacity to reflect the situation of how CTLs sense, reach, and interact with infected cells in a three-dimensional tissue in vivo. Whereas many assays of in vitro killing involve CTLs and targets co-cultured in suspension or as cell pellets, access to infected cells is likely to be limited in the intact tissue in which only selected cells are infected with viruses. Also, the extracellular matrix and bystander cells might exert multiple, often suppressive effects on CTL function (Zhang and Bevan, 2011). In addition, in co-culture killing assays, CTLs are brought passively together with target cells, whereas in vivo killing requires active CTL sensing and migration (Germain et al., 2012). Thus, it remains unclear how fast and how robustly virus-infected cells are killed by single CTLs in different virus-infected tissues (Elemans et al., 2014, Elemans et al., 2012, Hickman et al., 2015, Hogan et al., 2014).
In the current study, we quantified CTL killing kinetics by two-photon microscopy in mice infected with murine cytomegalovirus (MCMV) or modified vaccinia virus Ankara (MVA). To this end, we used ex vivo two-photon imaging of explanted lymph nodes and in vivo imaging of intact skin together with transgenic and natural CTLs and virus-expressed functional reporter systems. Importantly, we found that not every contact between CTLs and target cells led to a perforin-dependent Ca2+ flux and target-cell death. Using datasets on single-cell tracking, we estimated the average per capita killing rates (PCKRs: the number of targets killed per CTL per day) of transgenic and endogenous CTLs that kill different types of cells infected with several strains and species of viruses. In contrast to the conventional theory of “highly efficient” killing, our results consistently showed that PCKRs in vivo were overall limited to a value of about 2–16 infected cells killed per CTL per day. Furthermore, we observed that viral MHC-I immune evasion strongly reduced CTL-mediated antigen-specific contact-dependent killing in vivo. Finally, we showed that by increasing the probability of target-cell death after multiple encounters, CTLs could cooperate during killing of virus-infected cells.
Results
Single-Cell Visualization Allows for Quantification of Virus-Infected Cells
To determine killing kinetics of CTLs in vivo, we infected mice with MCMV reporter strains (Marquardt et al., 2011). MCMV-2D expresses the red fluorescent protein (FP) mCherry and a secretable Gaussia luciferase, whereas MCMV-3D additionally expresses the ovalbumin (OVA)-derived SIINFEKL peptide epitope. MCMV downmodulates surface MHC-I expression in infected cells, possibly leaving CTLs incapable of recognizing their targets (Gold et al., 2004, Hansen et al., 2010, Krmpotic et al., 1999). We therefore also used MCMV-3D-ΔvRAP, which lacks m06 and m152, two viral regulators of antigen presentation (vRAP) genes interfering with MHC-I recognition (Ziegler et al., 1997), to study the effect of MHC-I downregulation on CTL effector function.
To determine MCMV tropism after subcutaneous (s.c.) footpad infection of C57BL/6 (B6) mice with MCMV-3D, we used fluorescence microscopy to detect bright-mCherry-expressing cells in the draining popliteal lymph node at days 1 and 2 after infection. All MCMV-infected cells lacked expression of CD45 and CD169 and were located below the subcapsular sinus floor on the afferent side of the lymph node, approximately 20–30 μm beneath the capsule (Figures 1A–1E). Some MCMV-infected cells expressed the fibroblast markers podoplanin (gp38) and ER-TR7-antigen, but not the cell-adhesion molecule MAdCAM (data not shown). Thus, the MCMV strains used in this study initially infect fibroblast- or pericyte-like stromal cells, but not macrophages or dendritic cells. The number of mCherry+ virus-infected cells as determined by microscopy strongly correlated with the MCMV-encoded Gaussia luciferase activity (Figure 1F and data not shown). Therefore, we used the number of intact virus-infected cells to determine infected cell densities throughout this study. Taken together, different microscopy techniques allowed for a clear and unbiased quantification of the density of intact virus-infected cells in defined micro-anatomical regions.
Figure 1.

Single-Cell Visualization Allows for Quantification of Virus-Infected Cell Numbers
(A) Detection of mCherry+ MCMV-infected cells (white) by epi-fluorescence microscopy in cortical region of the popliteal lymph node 48 hr after MCMV-3D footpad injection (106 plaque-forming units [PFU]) into B6 mice.
(B) One day after infection, MCMV-infected cells expressing mCherry (red) and second harmonic generation signals (SHG, blue) were observed by two-photon microscopy. Scale bar represents 20 μm.
(C) Non-infected lymph node imaged at an identical region. Scale bar represents 20 μm.
(D) MCMV-3D-infected cells (red) and counterstaining with anti-CD45 (upper panel; green) and anti-CD169 (lower panel; cyan). Pictures shown in (A)–(D) are representative of >15 experiments. Scale bar represents 20 μm.
(E) Distance of MCMV-infected cells to the lymph node capsule. Dots represent cells, and bars represent medians. Data were pooled from six mice from two independent experiments. ns, not significant.
(F) After infection with different doses of MCMV-3D, the number of MCMV-infected cells observed by microscopy was plotted against MCMV-expressed luciferase signals. The red line represents linear regression, and the 99% prediction interval is shown in gray.
CTLs Fail to Recognize Virus-Infected Cells Protected by Viral Immune Evasion
We tested the ability of CTLs to directly detect and kill cells infected by different variants of MCMV after adoptive transfer of TCR-transgenic, FP-expressing, SIINFEKL-Kb-specific OT-I cells into B6 recipients. These mice were immunized with SIINFEKL peptide and poly(I:C) or with OVA protein together with MVA. Both procedures induced expansion and maturation of OT-I CTLs within 4–6 days (Figures 2A and 2B). The percentage of expanded CD44hi OT-I CTLs present in blood reliably predicted the number of OT-I CTLs in lymph nodes 2 days later (Figure 2C), allowing the analysis of MCMV infection in mice harboring defined numbers of virus-specific CTLs.
Figure 2.

Cytotoxic T lymphocytes Fail to Kill Infected Cells Protected by Viral MHC-I Immune Evasion
Protocol for generation of CTLs: 105 naive FP-expressing OT-I cells were transferred into C57BL/6 mice and activated at day 1 by different protocols. After expansion, different MCMV strains were injected into the footpad, and two-photon microscopy was used for studying CTL behavior and the number of infected cells in the draining popliteal lymph node.
(A) On day 5 after priming, all GFP+ FP-OT-I CTLs expressed CD44 after expansion. Flow cytometry was gated on all blood CD8+ T cells.
(B) The percentage of FP-OT-I CTLs (of total leukocytes) in blood after priming by different protocols (data were pooled from at least six independent experiments).
(C) The percentage of OT-I CTLs in blood is plotted against the percentage of OT-I CTLs found in brachial lymph nodes. Dots represent lymph nodes, and the red line represents linear regression. Data were pooled from three independent experiments.
(D) One day after infection, OT-I CTLs in the popliteal lymph node were stained for surface CD69 expression. MCMV-2D (2D) was used as a SIINFEKL-negative control, and Kruskal-Wallis and Dunn’s tests were used for comparing multiple groups. Dots represent lymph nodes, and red lines represent medians. Data were pooled from at least three experiments. ∗∗p < 0.01; ns, not significant.
(E–G) One day after infection with the viruses indicated, OT-I CTLs were observed in regions harboring infected cells (upper panel), and numbers of infected cells were counted (lower panel). In the upper panels, 10 min tracks are shown in gray, and scale bars represent 20 μm. In the lower panels, dots represent lymph nodes, and lines represent linear regression (E and F) or one-phase exponential decay (G). Data were pooled from at least six experiments.
CTL priming was performed with SIINFEKL and poly(I:C) in (C)–(G).
One day after MCMV-2D infection, CTLs expectedly did not express the activation marker CD69, and ex vivo two-photon microscopy showed that CTLs failed to attach to or kill infected cells (Figures 2D and 2E; Movie S1). Upon infection with MCMV-3D, OT-I CTLs strongly upregulated the activation marker CD69 but remained highly motile and established only a few contacts with infected cells. Consequently, most MCMV-3D-infected cells remained intact, even in the presence of high numbers of activated CTLs (Figures 2D and 2F; Movie S1).
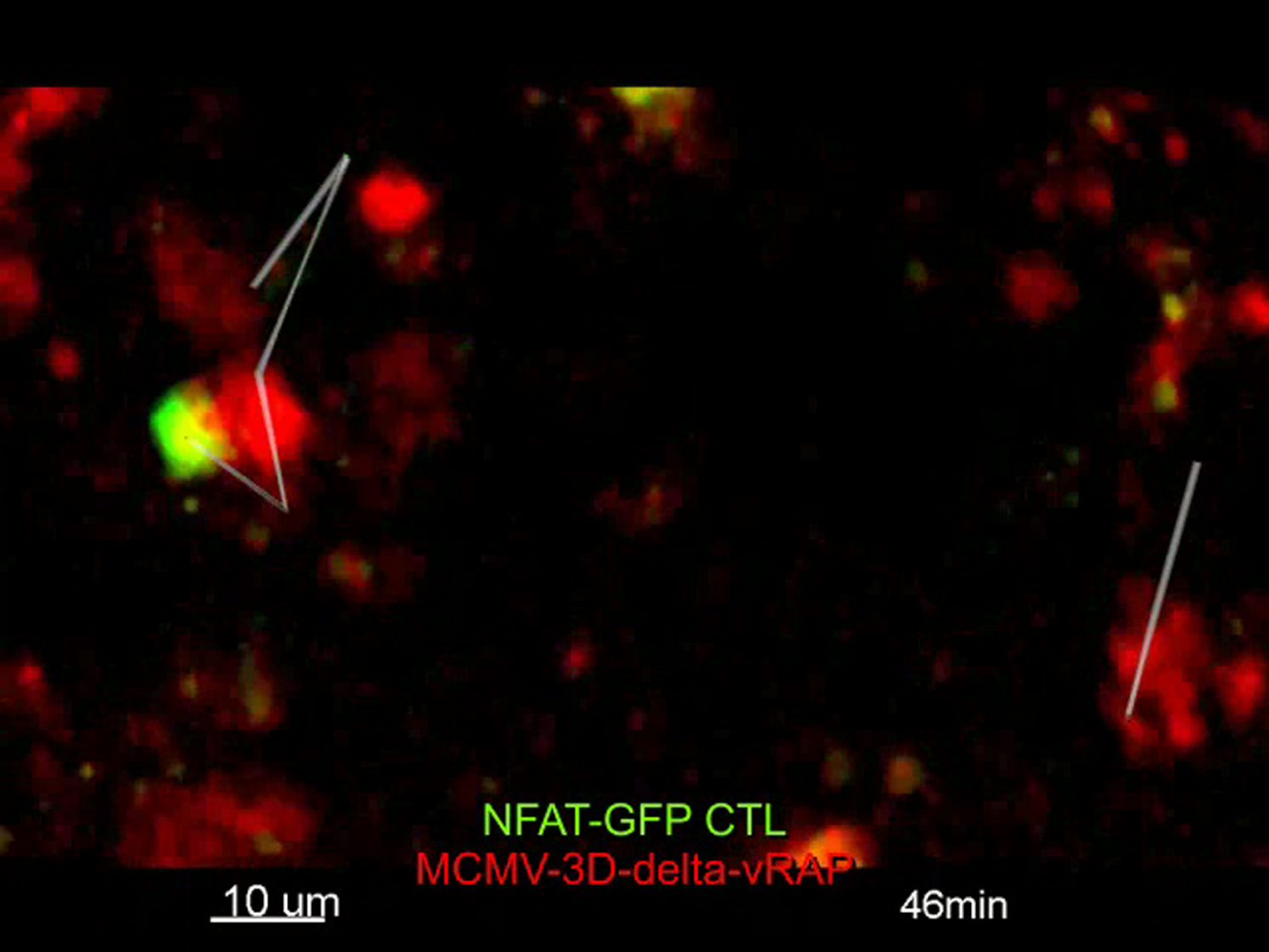
In contrast, in lymph nodes with high CTL densities, most MCMV-3D-ΔvRAP-infected cells were killed within 24 hr after infection (Figure 2G; Movie S1). Together, these findings demonstrate that viral MHC-I immune evasion protects MCMV-3D-infected cells from CTL-mediated killing and that in the absence of viral immune evasion, high CTL densities correlate with local eradication of infected cells.
Next, we characterized migration parameters of CTLs within the first 14–20 hr after infection. Compared to CTLs in mock, MCMV-2D, or MCMV-3D infection, CTLs that attacked MCMV-3D-ΔvRAP-infected cells showed significantly reduced track speeds (Figure 3A). Furthermore, CTLs showed very low motility coefficients, a measure for CTL displacement over time, according to the random-walk-hypothesis framework (Beltman et al., 2009). Also, CTL turning angles were increased during local confinement to regions with infected cells (Figures 3B–3D). These findings demonstrate that CTLs migrate at low velocities and stay confined in small tissue volumes while recognizing virus-infected cells and that viral immune evasion significantly alters CTL migration behavior and killing of virus-infected cells.
Figure 3.

Cognate Antigen Presentation and Viral Immune Evasion Determine the Migration Behavior of CTLs while Attacking Virus-Infected Cells
(A–C) One day after infection, two-photon microscopy was used to quantify OT-I CTL track speed (A), motility coefficient, (B) and turning angles (C) in movies with intact MCMV-infected cells. Dots represent median values from all tracks per movie, and red bars represent IQRs. Kruskal-Wallis and Dunn’s tests were used for comparing multiple groups. Data were pooled from two to six independent experiments per condition. ∗p < 0.05; ∗∗p < 0.01; ns, not significant.
(D) OT-I CTL population median track straightness (y axis) was compared to median track speed (x axis) per imaging region (dots represent median track straightness and median track speed for the different viruses per mouse).
CTL priming was performed with SIINFEKL and poly(I:C); see also Figure S1.
Two-Photon Microscopy Reveals that Multiple CTLs Are Needed to Robustly Kill Virus-Infected Cells
MCMV-3D-ΔvRAP-infected targets usually remained intact after contact by a single OT-I CTL (Figures S1A and S1B; Movie S1). In contrast, when numerous CTLs interacted simultaneously or serially with MCMV-3D-ΔvRAP-infected cells, we frequently observed target cell disruption (Figure 4A; Movie S2). The killing process of the virus-infected cells took some time, and morphological changes were observed over 10–40 min, long before the target cells finally disappeared. Typically, we observed initial morphological disturbances followed by the formation of blebs and later by the shedding of larger portions, resembling apoptotic bodies, of the mCherry-labeled cell body (Figure 4A [lower panel], Figure 4B).
Figure 4.

Two-Photon Microscopy Allows for Real-Time Visualization of CTL-Mediated Killing of Virus-Infected Cells
(A) Upper panel: two-photon microscopy revealed disruption of mCherry+ infected cells (red) and contact events by OT-I CTLs (green) 14 hr after MCMV-3D-ΔvRAP infection of mice harboring FP-CTLs (elapsed time displayed). Scale bar represents 20 μm. Lower panel: example of mCherry+ cell-body morphology during disruption (CTLs not shown). Dots represent center spots of original cell and large fragments. Scale bar represents 5 μm.
(B) Example of four CTLs (red, orange, green, and blue tracks) that interacted with one virus-infected target (red).
(C) Instantaneous speed of the four CTLs shown in (B).
(D) Number of OT-I CTL contacts (>1 min) with MCMV-3D-ΔvRAP-infected target cells that survived (intact) or were killed during the observation period of 1–3 hr. Dots represent target cells. ∗∗∗p < 0.001.
(E) Cumulative CTL-contact duration per target cell. Dots represent target cells. ∗∗∗p < 0.001.
(F) Duration of individual CTL-contact events. Dots represent CTL contacts. ns, not significant.
(D–F) Red bars represent the median with IQR, and data were pooled from nine movies from four independent experiments. A Mann-Whitney test was used for comparing intact and killed target cells.
(G) Duration of OT-I CTL contact with MCMV-2D-, MCMV-3D-, and MCMV-3D-ΔvRAP-infected cells. Here, data from CTLs not in contact with infected cells are shown (independent analysis of dataset used in D–F). ∗p < 0.05; ns, not significant.
(H) Percentage of CTLs that showed target-cell contact (data from G).
(I) Time from first observed CTL contact to death of MCMV-3D-ΔvRAP-infected cells. Target-cell death was defined as complete target disintegration, as shown in (A) and (B) (relative frequencies and binned data from 76 killed infected cells from four independent experiments are shown).
CTL priming was performed with SIINFEKL and poly(I:C) in (A)–(H). In (I), data with MVA-OVA priming was also added; see also Figures S2, S3, and S4.
The cognate interaction between CTLs and their targets rarely led to stable synapses with complete arrest of CTLs. Instead, while contacting their targets, CTLs remained motile with an instantaneous velocity of 2–4 μm/min for periods of approximately 10–15 min (Figure 4C) before regaining velocity. This type of interaction between targets and CTLs resembled the formation of migratory “kinapses” observed between T cells and dendritic cells presenting antigen with intermediate or low affinity during T cell priming (Dustin, 2008). Throughout the manuscript, we will use the term “kinapse” to describe this type of migratory interaction between CTLs and virus-infected cells during target-cell killing.
Direct observation of CTL-target-cell interaction behavior and quantification of target-cell fate revealed that killed virus-infected cells experienced a median of 3.5 distinct CTL contacts, whereas surviving cells were rarely targeted (0 median contacts; Figure 4D). Killed targets encountered a cumulative median contact time of 50 min (Figure 4E). Individual contacts between CTLs and surviving targets lasted 8.5 min (Figure 4F), and individual contacts between CTLs and killed targets lasted for approximately 9.0 min (median; Figure 4F). These observations indicate that successful killing of infected cells is not simply determined by the duration of individual CTL contacts.
When comparing the different MCMV strains, we observed that 1% and 8% of CTLs established contacts with MCMV-2D- and MCMV-3D-infected cells, respectively, whereas 38% of the CTLs contacted MCMV-3D-ΔvRAP infected cells (Figures 4G and 4H). Furthermore, we found that virus-infected cells were usually disrupted within 20–60 min (but rarely within 10 min) after the first observed CTL contact (Figure 4I). Together, these findings indicate that induction and execution of pro-apoptotic signaling cascades in virus-infected cells does not ensue instantaneously after a single CTL contact but that the accumulation of multiple CTL contacts determines the fate of virus-infected cells.
Different Virus-Infection Models Reveal Low Average CTL PCKRs
To study CTL-mediated killing in another infection model, we generated two reporter strains of the poxvirus MVA (Kremer et al., 2012). Injection of MVA-mCherry into the footpad led to infection of CD169+ subcapsular sinus macrophages in the draining lymph node (data not shown). OT-I CTLs killed MVA-OVA-mCherry- but not MVA-mCherry-infected cells (Figure 5A; Movie S3). As observed during MCMV-3D-ΔvRAP infection, CTLs within the MVA-OVA-mCherry-infected lymph nodes showed reduced track speed (Figure 5B), antigen-triggered dynamic contacts (Figure 5C), and disruption of targets that depended on CTL density at the site of infection (Figure 5D).
Figure 5.

CTLs Show Low Average PCKRs in Both Poxvirus and Cytomegalovirus Infection Models
Experimental setup for two-photon imaging of MVA-infected lymph nodes: transfer of FP-OT-I (day 0), expansion with SIINFEKL plus poly(I:C) (day 1), infection with MVA or MVA-OVA (day 6), and two-photon imaging (day 7).
(A) One day after footpad infection with MVA or MVA-OVA (107 PFU), two-photon microscopy was used for observing OT-I CTLs (green) at the site of MVA-infected mCherry+ cells (red).
(B) Median track speed of the OT-I CTL population in non-infected or MVA- or MVA-OVA-infected lymph nodes. Dots represent the mean of >50 CTLs analyzed per lymph node, and red bars represent the mean ± SD. A t test with Welch’s correction was used for comparing MVA and MVA-OVA. ∗∗p < 0.01.
(C) OT-I CTL contacts with MVA- or MVA-OVA-infected cells were analyzed. Dots represent contact duration per CTL, and red bars represent IQRs. A Mann-Whitney test was used for comparing MVA and MVA-OVA. ∗∗p < 0.01.
(D) One day after infection, the number of OT-I CTLs and MVA-infected cells per imaging region was correlated (green dots, MVA infection; green line, linear regression; red dots, MVA-OVA infection; red line, one-phase exponential decay). Dots represent individual mice (B), cells (C), and lymph nodes (D). Data were pooled from three independent experiments in (B)–(D). Scale bars represent 20 μm.
(E) OT-I CTLs PCKRs were calculated from automated-cell-tracking data for different MCMV and MVA strains. Dots represent movies, and red bars represent IQRs. A Kruskal-Wallis test was used for comparing multiple groups (outliers are not shown but were included in the test). ∗p < 0.05; ns, not significant.
To quantify CTL-mediated killing of the different virus-infected cells, we analyzed the expression kinetics of the mCherry reporter to test whether detection of all infected cells can be expected during the two-photon imaging time windows. In vitro infection revealed stronger and faster mCherry expression by MVA at 4–8 hr after infection (Figures S2A–S2D), whereas after 12 hr, both MVA and MCMV were robustly detectable both in vitro and in the infected lymph node (Figures S3A–S3D; Movie S4). Furthermore, virus-specific CTLs could already recognize their specific virus-encoded peptide at 8 hr after infection (Figures S3D and S3E). Thus, direct observation of single CTLs and single virus-infected cells was feasible from 12–24 hr after infection.
Next, we used two-photon-microscopy-derived datasets from the different virus-infection models to calculate PCKRs of OT-I CTLs, i.e., the theoretical average number of infected cells killed per CTL per day (Elemans et al., 2012). PCKR values reported in the literature are highly variable and were calculated on the basis of indirect assays or on assays relying on adoptively transferred artificial targets (Elemans et al., 2012, Regoes et al., 2007). On the basis of our direct observation of endogenous virus-infected cells killed, here we calculated PCKRs of 4.8 (median, MCMV-3D-ΔvRAP) and 4.2 (median, MVA-OVA-mCherry; Figure 5E). Notably, viral MHC-I immune evasion of MCMV-3D reduced the PCKR to 0.0 (median), thus resembling infection with MCMV-2D (Figure 5E). Together, direct visualization and quantification revealed that the OT-I CTL population showed a limited killing efficiency in different infection models and that viral MHC-I immune evasion dramatically reduced CTL killing efficiency in vivo.
Intralymphatic CTL Transfer and Mathematical Modeling Confirm Low Killing Rates
To determine whether CTLs generated from the endogenous pool of CD8+ T cells show killing efficiencies similar to those of TCR-transgenic CD8+ T cells, we generated CTLs in wild-type or perforin-deficient B6 mice (Prf−/−) by intraperitoneal infection with MCMV-3D. After 6–8 days, MCMV-specific CTLs were isolated by tetramer staining (Altman et al., 1996). Employing our previously developed technique of intralymphatic injection (Braun et al., 2011), we delivered tetramer-enriched (∼60% purity) or tetrameter-sorted (90% purity) CTLs or negatively enriched CTLs (depletion of CD62Lhi and non-CD8+ T cells) into the afferent lymphatic vessel draining toward the popliteal lymph node of MCMV-3D-ΔvRAP-infected mice. One day after intralymphatic transfer, we found that the reduction of virus-infected cell numbers, i.e., killing of targets, was dependent on the local number of B6 CTLs (Figures 6A and 6B). Similar to B6 CTLs, tetramer-sorted Prf−/− CTLs showed the typical migration behavior during target cell attack but were unable to disrupt virus-infected cells (Figure 6C; Movie S4).
Figure 6.

Intralymphatic CTL Transfer and Mathematical Modeling Show Low Killing Rates
Protocol for intralymphatic transfer: MCMV-3D-ΔvRAP footpad infection (0 hr), intralymphatic injection of different types of CTLs (∼4 hr), and two-photon imaging of single time points (∼24 hr).
(A) One day after infection and intralymphatic delivery of M45-tetramer-enriched CTLs (red) or M45-, M38-, and M139-tetramer-sorted CTLs (blue), the number of MCMV-3D-ΔvRAP-infected cells and CTLs per imaging region was counted from images of single time points.
(B) Same as (A) either without cell transfer or after injection of negatively enriched CTLs (magnetic depletion of CD62Lhi and non-CD8+ T cells).
(C) Same as (A) after injection of perforin-deficient tetramer-sorted CTLs.
(A–C) Data pooled from seven independent experiments (dots represent lymph nodes, and lines represent exponential decay). CTL priming was performed with intraperitoneal infection of CTL-donor mice with MCMV-3D.
(D) Mathematical modeling calculating the kinetics of infected cell numbers in a standard imaging region at the infected site of the lymph node cortical sinus, depending on the number of CTLs present (plot of infected cell numbers over time).
(E) The number of infected cells killed per T cell per day (PCKR) for the different CTL populations was calculated by the mathematical model from the raw data (median and 95% confidence interval) shown in (A)–(C). See Supplemental Experimental Procedures for details on the mathematical model.
To quantify CTL killing efficiencies after intralymphatic injection, we used a mathematical model to describe the killing of virus-infected cells at different CTL densities 0–24 hr after infection (Supplemental Experimental Procedures; Figure 6D). This model makes no assumptions on CTL killing mechanisms and has been developed in accordance with published modeling approaches (reviewed in Elemans et al., 2012, and Regoes et al., 2007). Furthermore, this model calculates average PCKR values for the entire CTL population, and the results obtained can be directly compared to the counting approach described in Figure 5. Applying this simple mathematical model, we calculated PCKRs from 2.0 to 10.4 for tetramer-enriched, tetramer-sorted, or negatively selected polyclonal CTLs (Figure 6E). In contrast, Prf−/− CTLs showed a PCKR close to 0 (Figure 6E). Thus, PCKRs obtained by intralymphatic transfer of in-vivo-primed CTLs and mathematical modeling were in agreement with the PCKRs obtained by real-time two-photon imaging, together arguing for a limited average PCKR for CTLs attacking virus-infected cells.
Low In Vivo CTL-Mediated Killing Rates in the Dermis
Because CTL killing efficiency might be different in a non-lymphoid organ, we next studied CTL killing in the dermis of T-cell-deficient mice (Cd3e−/−; Rag2−/−). These animals were reconstituted with FP-OT-I or FP-CD8+ T cells. Intraperitoneal virus infection was then used to prime and expand T cells (Figure S4A). In vivo two-photon microscopy showed that 1 day after secondary s.c. ear infection with MCMV-3D-ΔvRAP, but not after infection with MCMV-3D, OT-I CTLs migrating around infected dermal fibroblasts were found to exhibit the typical dynamic scanning behavior characterized by low track speed and low motility coefficients (Figures S4B–S4D). Likewise, mice reconstituted with polyclonal CD8+ T cells and primed intraperitoneally with MCMV-3D displayed CTLs characteristically scanning infected cells in the dermis, as well as local eradication of MCMV-3D-ΔvRAP-infected targets (Figures S4E–S4G; Movie S5). In the skin, we observed median PCKRs of 16.0 and 12.5 for OT-I and polyclonal CTLs, respectively (Figure S4H). Thus, two-photon in vivo imaging of the infected skin supports the conclusion that CTLs show limited killing efficiency when attacking virus-infected stromal cells.
Calcium Signaling in Virus-Infected Cells Reveals Heterogeneity of Single CTL-Contact Events
It is currently unclear whether all CTLs specific to the same epitope show the same killing rate or whether some CTLs kill more efficiently than others. Thus, we first tested whether all CTLs that are in contact with infected cells show signs of activation. Using OT-I CTLs expressing retrovirally transduced nuclear factor of activated T cells (NFAT)-GFP (Aramburu et al., 1998) and H2B-mOrange reporter constructs, and assuming that cognate recognition results in nuclear NFAT translocation within 1–3 min (Marangoni et al., 2013), we addressed whether CTLs in contact with infected cells can readily recognize the target. After MCMV-2D or -3D infection, the NFAT-GFP fluorescent signals remained in the CTL cytoplasm, which is in agreement with the absence of killing of these virus variants. In contrast, after MCMV-3D-ΔvRAP infection, 80% of CTLs in contact with infected cells—but also some CTLs not in contact with targets—showed nuclear NFAT-GFP (Figures S5A–S5D; Movie S6; data not shown). Thus, CTLs were usually activated during target-cell contact. However, because CTLs frequently retained the NFAT signal in the nucleus after target disengagement (Marangoni et al., 2013), NFAT signaling did not allow us to distinguish between CTLs with different killing rates.
Perforin pores in the plasma membrane have been suggested to trigger a transient calcium (Ca2+) flux in the target cell (Keefe et al., 2005). To gain further insights into the functional heterogeneity of CTLs, we therefore qualitatively and quantitatively analyzed CTL-induced Ca2+ influx in infected targets. To study Ca2+ fluxes in virus-infected cells, we replaced the luciferase-encoding sequence of MCMV-3D and MCMV-3D-ΔvRAP with a sequence encoding the ultra-sensitive Ca2+ sensor GCaMP6s (Chen et al., 2013), resulting in the reporter viruses MCMV-3D-Ca and MCMV-3D-ΔvRAP-Ca, respectively (Figures S6A–S6C). After footpad infection of non-immunized B6 mice with MCMV-3D-Ca or MCMV-3D-ΔvRAP-Ca, most infected cells showed spontaneous short Ca2+ fluxes that lasted on average 6 s (median; Figures 7A–7D; Movie S7). After infection with MCMV-3D-ΔvRAP-Ca, OT-I CTLs dynamically interacted with and triggered long-lasting Ca2+ fluxes in infected targets (Figures 7E–7G; Movie S7). CTL-induced Ca2+ fluxes lasted for 80 s (median; interquartile range [IQR] = 40–240 s; Figures 7E–7G) and started 480 s (median) after CTL encounter (IQR = 60–820 s; Figure 7H). Although CTLs intensively contacted infected cells, 40% failed to induce a long-lasting Ca2+ flux (Figure 7I), and only 10% of CTLs induced three or four death-associated Ca2+ fluxes (Figure 7J). Perforin-deficient CTLs failed to elicit long-lasting Ca2+ fluxes and cell death (Figure 7K; Figure S6D). Together, these findings reveal that individual CTLs and CTL contacts targeting virus-infected cells show strong functional heterogeneity.
Figure 7.

CTLs Trigger Ca2+ Fluxes in Virus-Infected Cells and Cooperate during Killing
(A) One day after infection with MCMV-expressing Ca2+ sensor GCaMP6s (MCMV-3D-ΔvRAP-Ca), two-photon microscopy was used to record GCaMP6s intensity (green) in virus-infected cells (red).
(B) Kinetics of GCaMP6s intensity in an infected cell imaged at 0.2 Hz.
(C) Ca2+ flux (defined as GCaMP6sbright event) duration in virus-infected cells in the absence of specific CTLs (dots represent cells, and red bars represent IQRs; n = 98 infected cells from three mice).
(D) Kinetics of Ca2+ fluxes in one infected cell imaged for 10 min at 0.07 Hz.
(E) In lymph nodes with MVA-OVA-primed CTLs present, a CFP+ OT-I CTL (blue; dotted line) contacted a MCMV-3D-ΔvRAP-Ca-infected cell (long flux defined as GCaMP6sbright event lasting >30 s).
(F) Kinetics of a long-lasting Ca2+ flux of an infected cell imaged for 10 min at 0.05 Hz. The green line represents the locally weighted scatterplot smoothing curve.
(G) Duration of Ca2+ fluxes of infected cells that were not contacted, were contacted but stayed intact, or were contacted and killed. Dots represent cells, and red bars represent IQRs. A Kruskal-Wallis test was used for comparing multiple groups. Data were pooled from four experiments from six different mice for a total of 307 flux events analyzed. ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant.
(H) Time interval between CTL contact and subsequent long-lasting Ca2+ flux (n = 79 events from four experiments analyzed; red bars represent IQRs).
(I) Percentages of CTL contacts that were followed by a long-lasting Ca2+ flux (n = 128 CTLs). Data were pooled from four experiments.
(J) Percentages of different numbers of long-lasting Ca2+ fluxes that followed a CTL contact event (n = 77 CTLs). Data were pooled from four experiments.
(K) Percentage of long-lasting Ca2+ fluxes that followed contacts of Prf−/− CTLs (n = 51 contacts pooled from three experiments).
(L) Probability of target-cell death for infected cells contacted by 0–14 CTLs. Hypothetical values for the no-cooperativity null hypothesis (open dots) and observed data (red squares) are provided. In total, 660 infected cells were analyzed, and data were pooled from >12 independent experiments.
(M) p values for comparing data derived from the null hypothesis and observed data (0.05 significance level indicated by dotted line); see also Figures S5, S6, and S7.
Cytotoxic T Lymphocytes Cooperate during Killing of Virus-Infected Cells
Because single CTL contacts frequently failed to trigger long-lasting Ca2+ influx or death of target cells, we next addressed whether CTLs cooperate while killing virus-infected cells. In all of the above-described imaging models, virus-infected cells were disrupted more frequently when they were contacted by several rather than single CTLs. To test for CTL cooperativity, we choose a null hypothesis that assumed that every CTL contact is an independent event and that previous contacts do not influence the target cell’s death probability. This Bernoulli-series null hypothesis states that the probability p(n) that a target cell will die after n interactions is given by the term p(n) = 1 – (1 – p(1))n. Data from 660 individual MCMV-3D-ΔvRAP-infected target cells were grouped according to the total number of CTL contacts observed per target cell. Targets without any observed CTL interactions died with a probability of p(0) = 0.01, whereas targets with a single or two CTL contacts died with a probability of p(1) = 0.15 or p(2) = 0.28, respectively. In the case of three or five CTL contacts, the probability that an infected cell would die was significantly higher than predicted from the null hypothesis of independent interaction outcomes (Figures 7L and 7M). Thus, CTLs were able to cooperate to kill virus-infected cells by increasing the probability of target cell death after multiple CTL encounters.
Discussion
In the present study, we visualized how CTLs killed virus-infected cells in vivo. We used MCMV and MVA reporter constructs that, after s.c. injection, infected draining lymph node stromal cells and subcapsular sinus macrophages, respectively. Because all reporter viruses encoded mCherry, we could clearly identify single infected cells and use time-lapse ex vivo and in vivo two-photon microscopy to determine their fates after they were contacted by effector CD8+ T cells.
Two-photon imaging revealed that in most situations, migrating CTLs do not come to a complete arrest to establish a static synapse with their target. Stable and static synapses have been described in lymph nodes (1) in vivo between antigen-presenting cells and T cells during defined stages of T cell priming and (2) in vitro on coated surfaces between CTLs and targets (Mempel et al., 2004, Ritter et al., 2015). As described for static synapses, the formation of dynamic kinapses observed in the present study also relied on the cognate interaction between TCR and MHC-I-presented antigen, because kinapses do not form between OT-I CTLs and cells infected with MCMV-2D (no cognate antigen) or MCMV-3D (low surface MHC-I expression). Among other factors, the formation of static immunological synapses has been shown to depend on integrin activation, which allows firm adhesion and complete arrest of migrating cells (Liu et al., 2009). Interestingly, treatment of mice with neutralizing antibodies against β1- and β2-integrin or injection of CTLs lacking the TCR adaptor protein ADAP, involved in TCR signaling leading to integrin activation, did not show differences in the slow but dynamic CTL migration behavior during the attack on infected cells or during the faster migration when CTLs were not contacting infected target cells (Figure S7). Therefore, integrin-independent formation of dynamic kinapses might represent a mechanism that not only allows CTLs to deliver the content of their cytotoxic granules but also keeps them in a motile state to successfully search for further targets.
By recruiting effector cells to the site of infection, chemokine receptors such as CXCR3 and CCR5 have been shown to contribute to CTL function (Hickman et al., 2015, Kastenmüller et al., 2013). In the present study, we observed no significant difference between wild-type and CXCR3- or CCR5-deficient CTLs regarding killing efficiencies in the intralymphatic transfer model (data not shown). These findings indicate that the mode of CTL delivery, the strain of virus investigated, and the cell type infected might affect the differential requirement for chemokine receptors for CTL recruitment. In our study, CTL attack on virus-infected cells resembled the behavior of NK cells attacking tumor cells (Deguine and Bousso, 2013).
In addition to assessing cell disruption on the basis of changes in cell morphology, we used the genetic Ca2+ sensor GCaMP6s expressed in the target cell to better understand CTL-mediated killing. Earlier in vitro studies have suggested that perforin pores in the plasma membrane of target cells cause a transient Ca2+ influx that is sensed by the cells as a sign to repair plasma-membrane damage (Keefe et al., 2005, Thiery et al., 2011). In virus-infected cells, expression of GCaMP6s proved a sensitive and reliable indicator of Ca2+ fluctuations. In the absence of natural killer cells and CTLs, virus-infected cells showed spontaneous short Ca2+ fluxes (data not shown), whereas CTL-induced fluxes could last for several minutes and reliably indicated subsequent cell death. Our study revealed that, on average, 8 min elapsed between CTL contact and Ca2+ influx, whereas subsequent cell disruption occurred in most situations between 20 and 120 min after the first CTL contact. In one study, in vivo imaging revealed killing of lymphoid cells within 10–20 min of CTL contact (Mempel et al., 2006), whereas other studies failed to observe rapid killing of virus-infected keratinocytes, malaria-infected hepatocytes, or tumor cells (Breart et al., 2008, Cockburn et al., 2013, Hickman et al., 2013). Notably, when CTLs attack peptide-pulsed B cells, additional B cell surface molecules (such as SLAMF7) could strengthen the CTL attachment and affect CTL behavior during killing. Together, the specific tropism of the pathogen and the targeted cell type essentially contribute to the outcome of CTL-mediated immune reactions.
The control of virus infection is critically determined by the quality of the CTLs generated (Plotkin, 2013, Varadarajan et al., 2011). CTLs can be heterogeneous with regard to the expression of effector molecules as well as the expression of co-stimulatory or inhibitory surface molecules (Jenkins et al., 2008, Newell et al., 2012). The data shown here also reveal that CTLs are heterogeneous with regard to nuclear translocation of the transcription factor NFAT after cognate antigen recognition. Furthermore, CTLs are highly diverse with regard to their ability to induce death-associated Ca2+ influx in target cells; for example, in movies with an average duration of 66 min, 40% of the CTLs were unable to trigger Ca2+ influx, whereas 10% induced three or four Ca2+ fluxes. This functional CTL heterogeneity helps to explain the observed overall low average killing rates.
The local presence of antigen-specific regulatory T cells can affect CTL killing efficiency (Mempel et al., 2006). The presence of immune-suppressive cells might also explain why intralymphatically delivered tetramer-enriched cells kill slightly less efficiently than very pure tetramer-sorted CTLs.
Two-photon microscopy allowed us to analyze the fate of single infected cells with regard to their CTL-contact history. This approach revealed that CTLs exhibited cooperativity in killing targets when more than two CTLs contacted the infected cell. The underlying mechanisms for this observation are unclear but could be explained by the differential expression levels of granzymes and perforin (and potentially other molecules) in individual CTLs (Jenkins et al., 2008). Two CTL contacts did not result in detectable signs of cooperativity, maybe because the effect of two CTL contacts is too small to be revealed in the assay setup used. In general, if we assume the existence of CTLs with a high or low killing capacity, it is plausible that multiple contacts increase the probability that a fully armed CTL actually disintegrates the infected cell. Alternatively or additionally, it seems possible that the accumulation of sub-lethal pro-death signals delivered by different single CTLs might lead to faster apoptosis of the target. Together, our data show that CTL cooperativity helps to combat viral infections.
The average killing efficacy of individual CTLs crucially affects the outcome of infectious diseases relying on CTL-mediated killing as a main control mechanism. There is currently no consensus on even the order of magnitude of in vivo CTL killing rates in either experimental models or human diseases (Elemans et al., 2014, Elemans et al., 2012, Hickman et al., 2013). Estimated killing rates of mouse and human CTLs vary considerably, and it is unknown how much the different viruses studied, the vaccination protocols applied to CTL generation, or the killing assays used affect the killing estimates (Garcia et al., 2015). Further complicating matters, the definition of killing rates often relies on different rate constants, not taking into account the number of CTLs that might be responsible for the killing (Ganusov and De Boer, 2008).
We therefore chose to calculate the average number of virus-infected cells killed per CTL per 24 hr as a simple measure of the CTL-mediated killing to allow direct comparison between different models. We determined killing rates either by directly observing disintegration of infected cells over time by a defined CTL population size or by determining the ratio of CTLs and infected cells at given time points after infection. The PCKRs calculated by these approaches give an average value that describes the killing efficiency of the whole CTL population (and not of individual CTLs) without assumptions about CTL cooperation or the percentage of non-active CTLs. Strikingly, our data show a consistent range of 2–16 infected cells killed per CTL per day, irrespective of the investigated epitopes recognized by the CTL, the method of calculation (counting or mathematical modeling), the type of cell infected (macrophages or stromal cells), or the virus used (MVA or MCMV-3D-ΔvRAP).
Because two-photon imaging cannot detect killing events that might occur before imaging is started, the PCKR values from direct imaging-based counting might slightly underestimate killing efficiencies. However, killing of “non-visible” cells does not affect our mathematical model, because the time point of killing is not important in this experimental setup. Both direct counting and mathematical modeling might have limitations, but all PCKR estimates obtained here (from different viruses infecting different cells, different organs, and by different methods) are consistent in that they range from 2 to 16 target cells killed per CTL per day.
It remains unknown for how long a single CTL can maintain a certain killing rate, and it will be interesting to measure whether single CTL killing rates increase or decrease after sequential target-cell encounter.
All together, our data indicate that the average in vivo CTL killing capacity is rather limited and not in agreement with the assumption that all CTLs act as “rapid” and “serial” killers that destroy hundreds or thousands of targets per day (Elemans et al., 2012). Therefore, protective CTL immunity might require substantially higher CTL densities than previously thought. This can possibly explain the failure of some CTL-based vaccines (Plotkin, 2013, Yewdell, 2010) or the development of chronic viral infections (West et al., 2011), in which functional CTLs are initially present but fail to eradicate all virus-infected cells.
Experimental Procedures
MCMV Strains
All MCMV strains were derived from the pSM3fr Smith strain (cloned as a bacterial artificial chromosome) and were produced and titrated in vitro according to standard techniques (Marquardt et al., 2011). GCaMP6s-expressing MCMV strains were designed with plasmid pGP-CMV-GCaMP6s (Chen et al., 2013) obtained from Addgene (plasmid 40753). See Supplemental Experimental Procedures for details.
MVA Strains
MVA-mCherry and MVA-OVA-mCherry were generated by homologous recombination using the plasmid vectors pIIIdHR-P7.5 and pLW-73 according to standard procedures. See Supplemental Experimental Procedures for details.
Mice
C57BL/6 (B6) mice were bred at the central animal facility at Hannover Medical School under specific-pathogen-free conditions or purchased from Charles River or the Jackson Laboratory (JAX). Female or male mice were used at the age of 6–18 weeks. The following mutant B6 mouse strains were used: C57BL/6-Tg(CAG-EGFP) (JAX 003291) mice expressing GFP under an artificial CMV-actin-globin promoter (here named GFP), C57BL/6-Tg(CAG-eCFP) mice expressing CFP under the beta-actin promoter (JAX 004218), TCR-transgenic OT-I CD45.1+ mice (derived from C57BL/6-Tg(TcraTcrb)1100Mjb/J; JAX 003831), F1-cross between GFP and OT-I CD45.1+ (named FP-OT-I), Adap−/− mice (C57BL/6J Fyb × B6.PL-Thy1a/CyJ × B6-Tg(TcraTcrb)1100Mjb), Cd3e−/− mice (C57BL/6-CD3etm1Mal/Orl), Prf1−/− mice (JAX 002407), and Rag2−/− mice (JAX 008449). All experiments were conducted in accordance with the local animal welfare regulations reviewed by the institutional review board and the Lower Saxony State Office for consumer protection and food safety (LAVES).
Generation of OT-I CTLs In Vivo
CD8+ T cells from OT-I mice, containing 105 CD8+ TCR Vα5+ OT-I cells, were transferred into recipient mice. OT-I CTL priming was performed by different regimes. See Supplemental Experimental Procedures for details.
Intralymphatic Injection of CTLs
Two to six hours after MCMV-3D-ΔvRAP infection, 2.5 × 104 to 1 × 105 CTLs diluted in 5 μl PBS were injected in 90 s into the afferent lymph vessel draining toward the popliteal lymph node, as described before (Braun et al., 2011).
Two-Photon Microscopy
Ex vivo two-photon imaging was performed with custom-build chambers for imaging explanted lymph nodes, as described in Halle et al. (2009). In Figure S4, the skin at the site of infection in the ear of anesthetized mice was imaged directly in vivo. See Supplemental Experimental Procedures for details.
Calculation of Average CTL Killing Rates
We calculated the number of infected cells killed per T cell per day on the basis of the observed time course of the number of killed virus-infected cells and the average number of CTLs in two-photon microscopy movies. Data from independent experiments were pooled, and average killing rates were reported. Alternatively, we used the single-time-point imaging data from the intralymphatic injection together with the mathematical model to calculate average CTL killing rates and 95% confidence intervals. See Supplemental Experimental Procedures for details.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 4. When comparing two groups, we calculated p values with the nonparametric Mann-Whitney test. To compare multiple groups, we used the Kruskal-Wallis test with Dunn’s test. All data were pooled from multiple independent experiments. See Supplemental Experimental Procedures for details.
Author Contributions
S.H., F.R.S., X.Z., K.H., J.B., K. Wagner, Y.B., R.M., A. Braun., K. Werth, and R.A. performed the experiments; K.A.K., A. Busche., A.M., M.G., V.H., M.K., G.S., and M.M. designed and produced the different viral constructs and virus strains; A.U., H.K., and M.M.-H. designed and applied the mathematical models; and S.H. and R.F. designed the experiments and wrote the manuscript.
Acknowledgments
This study was supported by DFG grant SFB 900-B1 to R.F. and M.M., ERC advanced grant 322645-LYMPHATICS-HOMING to R.F, the Niedersachsen Research Network on Neuroinfectiology, N-RENNT to R.F., a Lichtenberg stipend to F.R.S., and a Boehringer-Ingelheim stipend to A. Braun. A.U. and M.M.-H. were supported by the German Federal Ministry of Education and Research (BMBF) within the Measures for the Establishment of Systems Medicine (eMed) project Systems Immunology and Image Mining in Translational Biomarker (SYSIMIT). H.K. and M.M.-H. were supported by the Human Frontier Science Program. We thank Mathias Herberg for excellent animal handling and Immo Prinz, Andreas Krueger, and Olga Kristina Halle for reading and commenting on the manuscript. We would like to acknowledge the expert assistance of the Cell Sorting Core Facility of Hannover Medical School.
Published: February 9, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven movies and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2016.01.010.
Contributor Information
Stephan Halle, Email: halle.stephan@mh-hannover.de.
Reinhold Förster, Email: foerster.reinhold@mh-hannover.de.
Supplemental Information
Mice harboring GFP-expressing OT-I CTL were generated as described in Figure 2. One day after footpad challenge with MCMV-2D (in the absence of cognate SIINFEKL antigen), GFP+ OT-I CTLs (green) migrated fast and unconfined in MCMV-2D (red) infected lymph nodes (part 1). One day after MCMV-3D infection (SIINFEKL expression, but intact viral MHC-I immune evasion), most OT-I CTLs migrated next to the still intact MCMV-3D-infected cells (red; part 2). In contrast, one day after MCMV-3D-ΔvRAP challenge, the same region of the popliteal lymph node was devoid of intact infected cells, and most OT-I CTLs migrate fast (part 3). One day after infection, few individual CTLs could dynamically contact but not rapidly kill MCMV-3D-ΔvRAP infected cells (part 4). One day after footpad challenge with MCMV-3D-ΔvRAP, in lymph nodes with few OT-I CTLs, long contacts and confined migration of effector cells (green) were observed. However, most infected cells stay intact during the observation period. Three examples are shown, representative for >4 independent experiments. In part 5, CFP-OT-I cells (blue) were observed for 3 hr in a MCMV-3D-ΔvRAP-infected region (center spots, blue; SHG, blue). Related to Figure 2.
{kind=link}
In the popliteal lymph node, GFP+ OT-I CTLs (green) attacked MCMV-3D-ΔvRAP infected cells (red), 18 hr after footpad infection. Two different examples are shown (part 1-2). Part 3: Detailed view of one MCMV-3D-ΔvRAP-infected cell that was attacked by multiple CTLs. The CTLs that contacted the target are tracked and shown in red, orange, green and blue tracks. Note that some tissue drift and shaking occurred over time, as can be seen in the lateral views presented next to the surpass view. Related to Figure 4.
{kind=link}
Mice harboring GFP+ OT-I CTLs were generated as described in Figure 5A. One day after footpad injection of MVAmCherry (red, part 1), no cognate antigen is expressed and no stable contacts could be observed. One day after footpad injection of MVA-OVA-mCherry (red), many OT-I CTLs slowly migrated around the infected cells and formed dynamic contacts that lasted for minutes. In situations where only few CTLs are present, MVA-OVAmCherry-infected cells stayed intact (part 2). Over time, at sites with high and increasing CTL density, MVAOVA-mCherry expressing cells were disrupted (part 3). Part 4 shows a lateral view from part 3. Related to Figure 5.
{kind=link}
Time-lapse live cell microscopy was used to record the kinetics of mCherry expression starting at the time of infection until 16 hr after infection in vitro. Cells infected with MCMV-3D-ΔvRAP were observed every 30 min to follow the brightness of MCMV-encoded mCherry (red) over time. Automated cell tracking was used to detect target cells over time (part 1). In parallel experiments, MVA-mCherry-infected cells were imaged and mCherry-expression was recorded every 30 min (part 2). M45-tetramer selected CTLs were transferred by intralymphatic injection. One day following MCMV-3D-ΔvRAP infection, GFP-expressing M45-tetramer-selected wild-type CTLs contacted and killed virus-infected cells (part 3). In contrast, intralymphatically transferred tetramer-sorted CTLs from perforin-deficient donors failed to kill MCMV-3D-ΔvRAP-infected cells (part 4). Flow cytometry-sorted, tetramer-selected Prf1−/− effector CD8+ T cells were labeled and transferred by intralymphatic injection. One day following MCMV-3D-ΔvRAP infection, these effector cells contacted but failed to kill virus-infected cells (part 4). Related to Figure 6.
{kind=link}
One day following MCMV-3D-ΔvRAP infection of the ear dermis of primed mice, polyclonal GFP+ CTL migrated in the ear in regions more than 1000 μm away from the site of infection. These cells showed the typical “search mode” migration of CTLs (lateral region, part 1). Even at day 2 of infection, infected cells stayed intact during imaging when no effector cells are present (part 2). In non-primed mice, CD8+ T cells did not contact or kill virus-infected cells (part 3). In contrast, in MCMV-3D-primed mice one day after secondary MCMV-3D-ΔvRAP infection, CTLs attacked and killed virus-infected cells, showing typical scanning behavior. mCherry+ remnant formation in the upper part of the movie (part 4). A magnified view of a polyclonal CTL attack on MCMV-3D-ΔvRAP-infected cells (parts 5 and 6). Related to Figure 6.
{kind=link}
In a non-infected lymph node, cytoplasmic NFAT-GFP (green) was observed in migrating CTLs (part 1). In MCMV-3D-ΔvRAP-infected lymph nodes, CTLs observed in contact with target cells (red) showed a dense nuclear NFAT-GFP signal (part 2). Related to Figure 7.
{kind=link}
A single Ca2+-sensor-expressing virus-infected cell (red) showed short pulses of GCaMP6s fluorescence (green) in the absence of CTLs (part 1, imaging at 0.125 Hz). Another example of short flux events typically seen in intact infected cells, imaged at 0.05 Hz (part 2). In contrast, when virus-specific CTLs (blue) interacted with the virus-infected cells, we observed bright, long-lasting GCaMP6s signals in the targets (part 3 and 4; imaging at 0.05 Hz). Related to Figure 7.
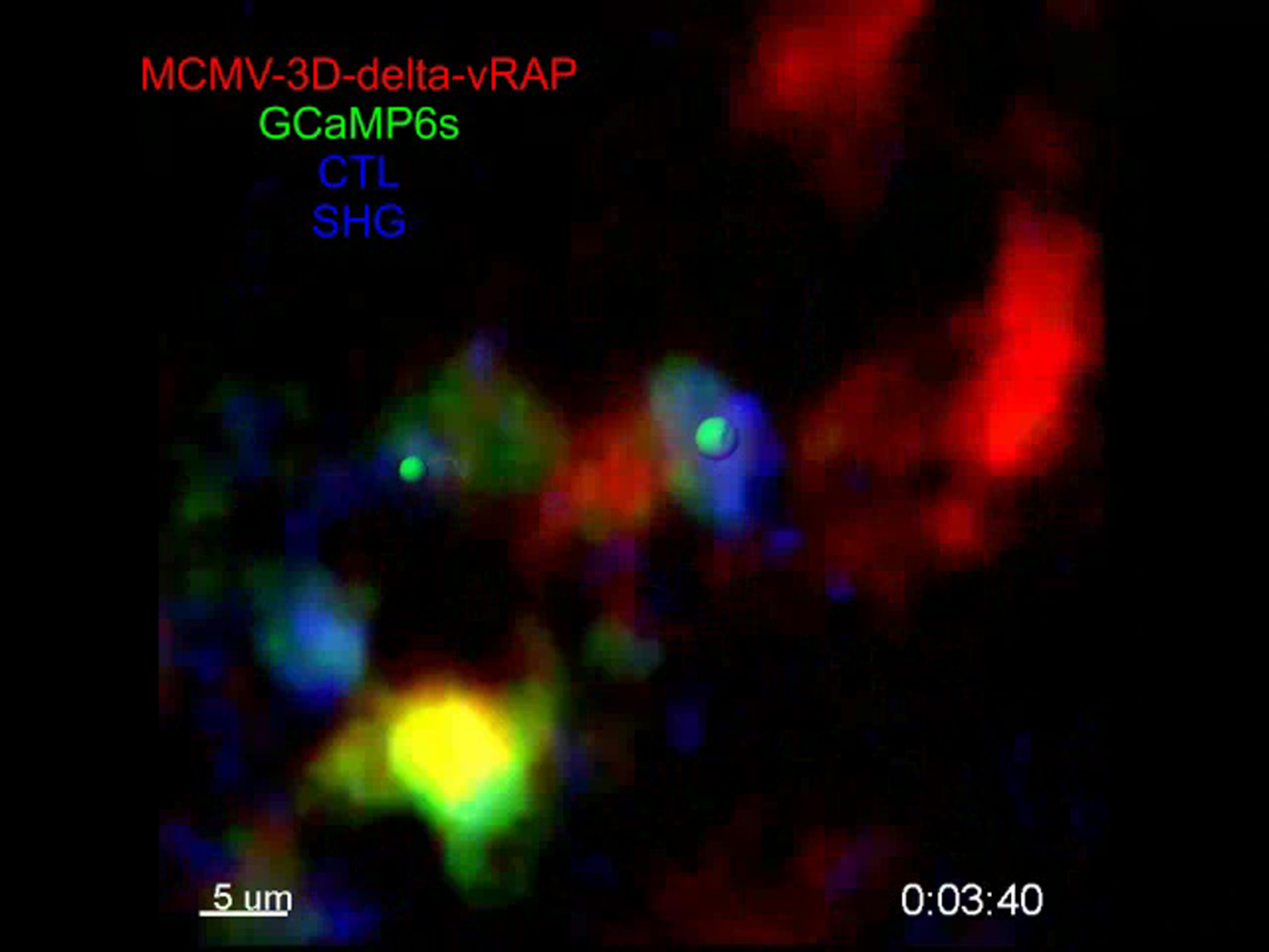{kind=link}
References
- Altman J.D., Moss P.A., Goulder P.J., Barouch D.H., McHeyzer-Williams M.G., Bell J.I., McMichael A.J., Davis M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- Aramburu J., Garcia-Cózar F., Raghavan A., Okamura H., Rao A., Hogan P.G. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Beltman J.B., Marée A.F.M., de Boer R.J. Analysing immune cell migration. Nat. Rev. Immunol. 2009;9:789–798. doi: 10.1038/nri2638. [DOI] [PubMed] [Google Scholar]
- Braun A., Worbs T., Moschovakis G.L., Halle S., Hoffmann K., Bölter J., Münk A., Förster R. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat. Immunol. 2011;12:879–887. doi: 10.1038/ni.2085. [DOI] [PubMed] [Google Scholar]
- Breart B., Lemaître F., Celli S., Bousso P. Two-photon imaging of intratumoral CD8+ T cell cytotoxic activity during adoptive T cell therapy in mice. J. Clin. Invest. 2008;118:1390–1397. doi: 10.1172/JCI34388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn I.A., Amino R., Kelemen R.K., Kuo S.C., Tse S.W., Radtke A., Mac-Daniel L., Ganusov V.V., Zavala F., Ménard R. In vivo imaging of CD8+ T cell-mediated elimination of malaria liver stages. Proc. Natl. Acad. Sci. USA. 2013;110:9090–9095. doi: 10.1073/pnas.1303858110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguine J., Bousso P. Dynamics of NK cell interactions in vivo. Immunol. Rev. 2013;251:154–159. doi: 10.1111/imr.12015. [DOI] [PubMed] [Google Scholar]
- Dustin M.L. T-cell activation through immunological synapses and kinapses. Immunol. Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- Elemans M., Seich Al Basatena N.-K., Asquith B. The efficiency of the human CD8+ T cell response: how should we quantify it, what determines it, and does it matter? PLoS Comput. Biol. 2012;8:e1002381. doi: 10.1371/journal.pcbi.1002381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemans M., Florins A., Willems L., Asquith B. Rates of CTL killing in persistent viral infection in vivo. PLoS Comput. Biol. 2014;10:e1003534. doi: 10.1371/journal.pcbi.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganusov V.V., De Boer R.J. Estimating in vivo death rates of targets due to CD8 T-cell-mediated killing. J. Virol. 2008;82:11749–11757. doi: 10.1128/JVI.01128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V., Richter K., Graw F., Oxenius A., Regoes R.R. Estimating the In Vivo Killing Efficacy of Cytotoxic T Lymphocytes across Different Peptide-MHC Complex Densities. PLoS Comput. Biol. 2015;11:e1004178. doi: 10.1371/journal.pcbi.1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain R.N., Robey E.A., Cahalan M.D. A decade of imaging cellular motility and interaction dynamics in the immune system. Science. 2012;336:1676–1681. doi: 10.1126/science.1221063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold M.C., Munks M.W., Wagner M., McMahon C.W., Kelly A., Kavanagh D.G., Slifka M.K., Koszinowski U.H., Raulet D.H., Hill A.B. Murine cytomegalovirus interference with antigen presentation has little effect on the size or the effector memory phenotype of the CD8 T cell response. J. Immunol. 2004;172:6944–6953. doi: 10.4049/jimmunol.172.11.6944. [DOI] [PubMed] [Google Scholar]
- Halle S., Dujardin H.C., Bakocevic N., Fleige H., Danzer H., Willenzon S., Suezer Y., Hämmerling G., Garbi N., Sutter G. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med. 2009;206:2593–2601. doi: 10.1084/jem.20091472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S.G., Powers C.J., Richards R., Ventura A.B., Ford J.C., Siess D., Axthelm M.K., Nelson J.A., Jarvis M.A., Picker L.J., Früh K. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science. 2010;328:102–106. doi: 10.1126/science.1185350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman H.D., Reynoso G.V., Ngudiankama B.F., Rubin E.J., Magadán J.G., Cush S.S., Gibbs J., Molon B., Bronte V., Bennink J.R., Yewdell J.W. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe. 2013;13:155–168. doi: 10.1016/j.chom.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman H.D., Reynoso G.V., Ngudiankama B.F., Cush S.S., Gibbs J., Bennink J.R., Yewdell J.W. CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity. 2015;42:524–537. doi: 10.1016/j.immuni.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan T., Kadolsky U., Tung S., Seddon B., Yates A. Spatial heterogeneity and peptide availability determine CTL killing efficiency in vivo. PLoS Comput. Biol. 2014;10:e1003805. doi: 10.1371/journal.pcbi.1003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins M.R., Mintern J., La Gruta N.L., Kedzierska K., Doherty P.C., Turner S.J. Cell cycle-related acquisition of cytotoxic mediators defines the progressive differentiation to effector status for virus-specific CD8+ T cells. J. Immunol. 2008;181:3818–3822. doi: 10.4049/jimmunol.181.6.3818. [DOI] [PubMed] [Google Scholar]
- Kastenmüller W., Brandes M., Wang Z., Herz J., Egen J.G., Germain R.N. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity. 2013;38:502–513. doi: 10.1016/j.immuni.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe D., Shi L., Feske S., Massol R., Navarro F., Kirchhausen T., Lieberman J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Kremer M., Volz A., Kreijtz J.H., Fux R., Lehmann M.H., Sutter G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. 2012;890:59–92. doi: 10.1007/978-1-61779-876-4_4. [DOI] [PubMed] [Google Scholar]
- Krmpotic A., Messerle M., Crnkovic-Mertens I., Polic B., Jonjic S., Koszinowski U.H. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 1999;190:1285–1296. doi: 10.1084/jem.190.9.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Bryceson Y.T., Meckel T., Vasiliver-Shamis G., Dustin M.L., Long E.O. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity. 2009;31:99–109. doi: 10.1016/j.immuni.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J.A., Brennan A.J., Whisstock J.C., Voskoboinik I., Trapani J.A. Protecting a serial killer: pathways for perforin trafficking and self-defence ensure sequential target cell death. Trends Immunol. 2012;33:406–412. doi: 10.1016/j.it.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Marangoni F., Murooka T.T., Manzo T., Kim E.Y., Carrizosa E., Elpek N.M., Mempel T.R. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity. 2013;38:237–249. doi: 10.1016/j.immuni.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchingo J.M., Kan A., Sutherland R.M., Duffy K.R., Wellard C.J., Belz G.T., Lew A.M., Dowling M.R., Heinzel S., Hodgkin P.D. T cell signaling. Antigen affinity, costimulation, and cytokine inputs sum linearly to amplify T cell expansion. Science. 2014;346:1123–1127. doi: 10.1126/science.1260044. [DOI] [PubMed] [Google Scholar]
- Marquardt A., Halle S., Seckert C.K., Lemmermann N.A., Veres T.Z., Braun A., Maus U.A., Förster R., Reddehase M.J., Messerle M., Busche A. Single cell detection of latent cytomegalovirus reactivation in host tissue. J. Gen. Virol. 2011;92:1279–1291. doi: 10.1099/vir.0.029827-0. [DOI] [PubMed] [Google Scholar]
- Mempel T.R., Henrickson S.E., Von Andrian U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Mempel T.R., Pittet M.J., Khazaie K., Weninger W., Weissleder R., von Boehmer H., von Andrian U.H. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Newell E.W., Sigal N., Bendall S.C., Nolan G.P., Davis M.M. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin S.A. Complex correlates of protection after vaccination. Clin. Infect. Dis. 2013;56:1458–1465. doi: 10.1093/cid/cit048. [DOI] [PubMed] [Google Scholar]
- Regoes R.R., Yates A., Antia R. Mathematical models of cytotoxic T-lymphocyte killing. Immunol. Cell Biol. 2007;85:274–279. doi: 10.1038/sj.icb.7100053. [DOI] [PubMed] [Google Scholar]
- Ritter A.T., Asano Y., Stinchcombe J.C., Dieckmann N.M., Chen B.C., Gawden-Bone C., van Engelenburg S., Legant W., Gao L., Davidson M.W. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity. 2015;42:864–876. doi: 10.1016/j.immuni.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J., Keefe D., Boulant S., Boucrot E., Walch M., Martinvalet D., Goping I.S., Bleackley R.C., Kirchhausen T., Lieberman J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011;12:770–777. doi: 10.1038/ni.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan N., Julg B., Yamanaka Y.J., Chen H., Ogunniyi A.O., McAndrew E., Porter L.C., Piechocka-Trocha A., Hill B.J., Douek D.C. A high-throughput single-cell analysis of human CD8+ T cell functions reveals discordance for cytokine secretion and cytolysis. J. Clin. Invest. 2011;121:4322–4331. doi: 10.1172/JCI58653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West E.E., Youngblood B., Tan W.G., Jin H.-T., Araki K., Alexe G., Konieczny B.T., Calpe S., Freeman G.J., Terhorst C. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity. 2011;35:285–298. doi: 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann A., Depoil D., Faroudi M., Valitutti S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA. 2006;103:10985–10990. doi: 10.1073/pnas.0600651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell J.W. Designing CD8+ T cell vaccines: it’s not rocket science (yet) Curr. Opin. Immunol. 2010;22:402–410. doi: 10.1016/j.coi.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Bevan M.J. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H., Thale R., Lucin P., Muranyi W., Flohr T., Hengel H., Farrell H., Rawlinson W., Koszinowski U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mice harboring GFP-expressing OT-I CTL were generated as described in Figure 2. One day after footpad challenge with MCMV-2D (in the absence of cognate SIINFEKL antigen), GFP+ OT-I CTLs (green) migrated fast and unconfined in MCMV-2D (red) infected lymph nodes (part 1). One day after MCMV-3D infection (SIINFEKL expression, but intact viral MHC-I immune evasion), most OT-I CTLs migrated next to the still intact MCMV-3D-infected cells (red; part 2). In contrast, one day after MCMV-3D-ΔvRAP challenge, the same region of the popliteal lymph node was devoid of intact infected cells, and most OT-I CTLs migrate fast (part 3). One day after infection, few individual CTLs could dynamically contact but not rapidly kill MCMV-3D-ΔvRAP infected cells (part 4). One day after footpad challenge with MCMV-3D-ΔvRAP, in lymph nodes with few OT-I CTLs, long contacts and confined migration of effector cells (green) were observed. However, most infected cells stay intact during the observation period. Three examples are shown, representative for >4 independent experiments. In part 5, CFP-OT-I cells (blue) were observed for 3 hr in a MCMV-3D-ΔvRAP-infected region (center spots, blue; SHG, blue). Related to Figure 2.
In the popliteal lymph node, GFP+ OT-I CTLs (green) attacked MCMV-3D-ΔvRAP infected cells (red), 18 hr after footpad infection. Two different examples are shown (part 1-2). Part 3: Detailed view of one MCMV-3D-ΔvRAP-infected cell that was attacked by multiple CTLs. The CTLs that contacted the target are tracked and shown in red, orange, green and blue tracks. Note that some tissue drift and shaking occurred over time, as can be seen in the lateral views presented next to the surpass view. Related to Figure 4.
Mice harboring GFP+ OT-I CTLs were generated as described in Figure 5A. One day after footpad injection of MVAmCherry (red, part 1), no cognate antigen is expressed and no stable contacts could be observed. One day after footpad injection of MVA-OVA-mCherry (red), many OT-I CTLs slowly migrated around the infected cells and formed dynamic contacts that lasted for minutes. In situations where only few CTLs are present, MVA-OVAmCherry-infected cells stayed intact (part 2). Over time, at sites with high and increasing CTL density, MVAOVA-mCherry expressing cells were disrupted (part 3). Part 4 shows a lateral view from part 3. Related to Figure 5.
Time-lapse live cell microscopy was used to record the kinetics of mCherry expression starting at the time of infection until 16 hr after infection in vitro. Cells infected with MCMV-3D-ΔvRAP were observed every 30 min to follow the brightness of MCMV-encoded mCherry (red) over time. Automated cell tracking was used to detect target cells over time (part 1). In parallel experiments, MVA-mCherry-infected cells were imaged and mCherry-expression was recorded every 30 min (part 2). M45-tetramer selected CTLs were transferred by intralymphatic injection. One day following MCMV-3D-ΔvRAP infection, GFP-expressing M45-tetramer-selected wild-type CTLs contacted and killed virus-infected cells (part 3). In contrast, intralymphatically transferred tetramer-sorted CTLs from perforin-deficient donors failed to kill MCMV-3D-ΔvRAP-infected cells (part 4). Flow cytometry-sorted, tetramer-selected Prf1−/− effector CD8+ T cells were labeled and transferred by intralymphatic injection. One day following MCMV-3D-ΔvRAP infection, these effector cells contacted but failed to kill virus-infected cells (part 4). Related to Figure 6.
One day following MCMV-3D-ΔvRAP infection of the ear dermis of primed mice, polyclonal GFP+ CTL migrated in the ear in regions more than 1000 μm away from the site of infection. These cells showed the typical “search mode” migration of CTLs (lateral region, part 1). Even at day 2 of infection, infected cells stayed intact during imaging when no effector cells are present (part 2). In non-primed mice, CD8+ T cells did not contact or kill virus-infected cells (part 3). In contrast, in MCMV-3D-primed mice one day after secondary MCMV-3D-ΔvRAP infection, CTLs attacked and killed virus-infected cells, showing typical scanning behavior. mCherry+ remnant formation in the upper part of the movie (part 4). A magnified view of a polyclonal CTL attack on MCMV-3D-ΔvRAP-infected cells (parts 5 and 6). Related to Figure 6.
In a non-infected lymph node, cytoplasmic NFAT-GFP (green) was observed in migrating CTLs (part 1). In MCMV-3D-ΔvRAP-infected lymph nodes, CTLs observed in contact with target cells (red) showed a dense nuclear NFAT-GFP signal (part 2). Related to Figure 7.
A single Ca2+-sensor-expressing virus-infected cell (red) showed short pulses of GCaMP6s fluorescence (green) in the absence of CTLs (part 1, imaging at 0.125 Hz). Another example of short flux events typically seen in intact infected cells, imaged at 0.05 Hz (part 2). In contrast, when virus-specific CTLs (blue) interacted with the virus-infected cells, we observed bright, long-lasting GCaMP6s signals in the targets (part 3 and 4; imaging at 0.05 Hz). Related to Figure 7.