

When covalently linked to an acyl carrier protein (ACP) and loaded with acyl substrate-mimics, some 4′-phosphopantetheine prosthetic group arms swing freely, whereas others stick to the protein surface, suggesting a possible mode of interaction with enzyme domains during polyketide biosynthesis.
Keywords: acyl carrier protein, mycolactone, NMR spectroscopy, 4′-phosphopantetheine, type I polyketide synthase
Abstract
Type I modular polyketide synthases (PKSs) produce polyketide natural products by passing a growing acyl substrate chain between a series of enzyme domains housed within a gigantic multifunctional polypeptide assembly. Throughout each round of chain extension and modification reactions, the substrate stays covalently linked to an acyl carrier protein (ACP) domain. In the present study we report on the solution structure and dynamics of an ACP domain excised from MLSA2, module 9 of the PKS system that constructs the macrolactone ring of the toxin mycolactone, cause of the tropical disease Buruli ulcer. After modification of apo ACP with 4′-phosphopantetheine (Ppant) to create the holo form, 15N nuclear spin relaxation and paramagnetic relaxation enhancement (PRE) experiments suggest that the prosthetic group swings freely. The minimal chemical shift perturbations displayed by Ppant-attached C3 and C4 acyl chains imply that these substrate-mimics remain exposed to solvent at the end of a flexible Ppant arm. By contrast, hexanoyl and octanoyl chains yield much larger chemical shift perturbations, indicating that they interact with the surface of the domain. The solution structure of octanoyl-ACP shows the Ppant arm bending to allow the acyl chain to nestle into a nonpolar pocket, whereas the prosthetic group itself remains largely solvent exposed. Although the highly reduced octanoyl group is not a natural substrate for the ACP from MLSA2, similar presentation modes would permit partner enzyme domains to recognize an acyl group while it is bound to the surface of its carrier protein, allowing simultaneous interactions with both the substrate and the ACP.
INTRODUCTION
Type I modular polyketide synthases (PKSs) are large, multi-domain complexes responsible for generating natural products with a spectrum of medically important activities, including antibiotic, anticancer, antifungal, antitumour and immunosuppressive properties [1]. Like type I fatty acid synthases (FASs), these systems consist of a series of covalently linked enzymes that extend a polyketide substrate by two carbon atoms and modify the functionality of the newly added building block via reactions at the β-ketone site. In FAS systems the substrate is cycled repeatedly between a single set of enzymes to produce long, saturated acyl chains, but in the majority of modular PKS systems each extension step is carried out in sequence by a distinct set or ‘module’ of enzymes.
PKS extension modules comprise three domains essential for constructing the product chain [2]: a small (∼10 kDa) acyl carrier protein (ACP) to which the polyketide substrate is tethered via thioester linkage to a 4′-phosphopantetheine (Ppant) prosthetic group; an acyltransferase (AT), which selects an appropriate extender unit (commonly malonate or methylmalonate as their coenzyme A thioesters) for loading on to the ACP; and a ketosynthase (KS), which accepts a polyketide chain from a previous module and attaches the new extender unit by catalysing a decarboxylative condensation reaction (Figure 1A). To introduce chemical diversity into the polyketide product, modules can contain additional enzyme domains [2]: a ketoreductase (KR) that reduces the β-ketone group to an alcohol and may also epimerize the adjacent α-centre; a dehydratase (DH), which eliminates the β-hydroxy to form an α-β double bond; and an enoyl reductase (ER) that reduces the resulting alkene, producing a saturated β-methylene group (Figure 1A). The terminal module of a PKS system normally contains a thioesterase (TE) domain, which promotes release and often cyclization of the substrate. The length and functionality of the final product is therefore defined by the number, order and domain composition of modules within the system [1].
Figure 1. Reaction scheme and module organization for the mycolactone PKS system.

(A) Catalytic cycle for domains from the MLSA2 module. (B) Module organization for the three subunits of the mycolactone PKS system (MLSA1, MLSA2 and MLSB). The product of MLSA2 is shown attached to its carrier protein domain. The structure of mycolactone is colour coded to match the subunits responsible for synthesizing each segment. DH domains predicted to be inactive are shaded black.
The intuitive, linear, assembly-line nature of modular PKS and similarly configured non-ribosomal peptide synthetase (NRPS) systems make them attractive targets for combinatorial biosynthesis and synthetic biology strategies [3,4]. Although numerous new compounds have been generated by such approaches, engineered PKS multi-enzyme complexes often display reduced activity or result in undesirable product mixtures [5,6]. A major limitation in overcoming such deficiencies is a poor understanding of the interactions that occur within and between modules. The role of the ACP is central to this question, since it must present covalently tethered substrates to the active sites of each enzymatic domain within its module, as well as a KS or a TE in the subsequent module [7,8]. However, whether the ACP plays an active role in this process or merely restricts free diffusion of the substrate has not yet been fully established for type I PKS systems [9,10].
Transitions between different module configurations may restrict the access of an ACP to a subset of partner domains during different phases of the reaction cycle [10], but from the point of view of the carrier protein three potential mechanisms for identifying the appropriate active site can be envisaged: (i) substrate-mediated modifications of ACP structure to promote enzyme-specific protein–protein interactions; (ii) recognition of a combination of the ACP surface and substrate chemistry; or (iii) pure recognition of substrate chemistry by the catalytic domain in the absence of specific interactions with the ACP. Identifying which of these mechanisms is used to regulate interactions between substrate-loaded ACPs and the various catalytic domains will have important consequences for rational engineering of PKS assembly lines to produce novel polyketide scaffolds.
Type II PKS systems consist of discrete domains, so their acylated ACP domains can only interact with partner enzymes in trans. Carrier proteins of this sort typically protect complex substrate chains from contact with the solvent by sequestering them inside a hydrophobic pouch to minimize premature hydrolysis and unwanted side reactions [9,11]. In the case of the Streptomyces coelicolor FAS ACP, solution structures for a complete set of C6 reaction intermediates demonstrate that although the global fold of the domain is retained, the conformations of the thioester, the Ppant moiety and nearby regions of the protein are subtly different [12]. These structural changes could assist in selecting the correct active site for the next step in the catalytic cycle. However, the type I FAS and PKS ACP domains studied to date do not appear to bury their substrates [13–16], so the same mode of conformational programming may not apply. Similarly, the ‘switchblade’ [17] or ‘chain-flipping’ [11] paradigms required to explain how a partner enzyme can gain access to a reduced acyl chain that is sequestered inside the hydrophobic core of a type II ACP domain may not be relevant to type I systems if ACP-tethered substrates are continuously on display, sheltered by the quaternary architecture of the surrounding module. Current investigations of substrate-loaded NRPS aryl and peptidyl carrier protein domains from the Type I yersiniabactin [18] and the Type II pyoluteorin [19] systems are consistent with a more delicate mechanism: the distribution of charge across the surface of the domain is modulated by interactions with both the Ppant arm and the substrate in ways that could favour productive encounters with specific partners.
Recent cryo-electron microscopy (cryo-EM) studies on the fifth chain-extension module from the pikromycin PKS (Pik module 5) suggest that positioning of the ACP within a type I module may be driven by the identity of the substrate attached to the Ppant arm [20,21]. ACP domains modified with substrate mimics were found to dock on to the appropriate enzymatic domain for the ensuing reaction within the module's catalytic cycle, although predominantly at distances too remote for the substrate to access the relevant active sites [22]. Further, loading a polyketide chain that mimicked the module's final product caused the ACP to protrude from the base of the module at a position thought to be suitable for downstream transfer of the substrate to a subsequent module or TE. Of the three mechanisms for ACP partner selection suggested above, these results favour the second, as in most cases both protein–protein interfaces and correct substrate chemistry appear to be necessary for progression through the catalytic cycle [10,20,21].
The PKS responsible for production of the Mycobacterium ulcerans toxin mycolactone, the molecular cause of Buruli ulcer [23], presents a particularly attractive system in which to study the intricacies of modular PKS systems. Despite significant variations in the length and chemistry of the substrate encountered at each step, the mycolactone synthases display a remarkably high sequence identity (>95%) between equivalent domains across 16 chain extension modules found in three protein chains: MLSA1, MLSA2 and MLSB (Figure 1B) [24,25]. Such a high degree of similarity between modules suggests either that enzymatic domains from the mycolactone PKS are less discriminating (and may therefore be good candidates for combinatorial biosynthesis and synthetic biology), or that the few residues which do vary are specific for particular features of natural substrates.
As a first step towards the biophysical characterization of interactions between carrier protein and partner enzyme domains from a canonical type I PKS module that contains a complete reductive loop and a chain-releasing TE domain, we report here studies of the solution structure and dynamics of mACP9, the ACP domain from MLSA2, module 9 of the PKS system that constructs the macrolactone ring of mycolactone (Figure 1B) [25]. We demonstrate that different substrate-mimics interact with the surface of the ACP in distinct ways that could play a role in the recognition of partner domains.
MATERIALS AND METHODS
Expression and purification of apo mACP9
The mACP9 sequence from the mlsA2 gene from Mycobacterium ulcerans (Uniprot: Q6MZA5; residues 2050–2140) cloned into pET28 (EMD Millipore) was transformed into competent Escherichia coli Tuner (DE3) cells (EMD Millipore). His6-tagged mACP9 was expressed at 15°C for 16 h in 1 litre of LB or M9 medium, prepared according to standard protocols [26], with 30 μg/ml kanamycin (Sigma) for selection and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma) for induction. Isotopically labelled samples were prepared by supplementing M9 with 15N ammonium chloride (Sigma) and 13C6-D-glucose (Cambridge Isotope Laboratories). Cells were harvested at a 600 nm absorbance of 0.6, resuspended in lysis buffer (50 mM sodium hydrogen phosphate, 300 mM sodium chloride, pH 8.0) with 5 mM imidazole, 2.5 units/ml benzonase nuclease (EMD Millipore) and Sigmafast EDTA-free protease inhibitor cocktail (Sigma) and lysed using an Emulsiflex C5 homogeniser (Glen Creston).
The clarified lysate was passed through Ni-NTA resin (Qiagen), washed twice with lysis buffer containing 30 mM imidazole and eluted with lysis buffer containing 300 mM imidazole. The eluted protein was exchanged into phosphate buffer (50 mM sodium hydrogen phosphate, 150 mM sodium chloride, pH 7.5) and the His6-tag was cleaved using restriction grade thrombin (EMD Millipore). The released mACP9 was further purified by size exclusion chromatography using an Äkta Purifier 10 system and a Superdex 75 10/300 column (GE Healthcare) in phosphate buffer. The sample was concentrated using 5000 MWCO Vivaspin 20 columns (Sartorius Stedim). The expression and purification was monitored by SDS/PAGE (NuPAGE) 4–12% Bis-Tris gels (Life Technologies) stained with InstantBlue (Expedeon). The identity of the sample was confirmed by electrospray injection mass spectrometry (ESI MS; Supplementary Table S1 and Supplementary Figure S1).
Preparation of covalently modified forms of mACP9
Co-expression with Sfp to make 15N-labelled holo mACP9
The mACP9/pET28 plasmid and a pSU2718 plasmid [27] with the Sfp gene cloned between the NdeI and SalI sites were transformed into competent E. coli Tuner (DE3) cells (EMD Millipore). Apart from addition of 34 μg/ml chloramphenicol (Sigma) to growth media for selection of the pSU2718 plasmid, expression and purification of the holo protein was identical with that of the apo protein described above. The extent of modification of mACP9 was monitored by ESI MS (Supplementary Figure S2), and was complete in all cases taken forward for further study.
Sfp-based addition of 4′-phosphopantetheine and acyl-phosphopantetheine groups
In vitro loading reactions were performed on 500 μM apo ACP9 samples by incubation with 5 μM recombinant Sfp [28], 2 mM coenzyme A or its derivatives malonyl, butyryl, 2-butenoyl, β-hydroxybutyryl, acetoacetyl, hexanoyl or octanoyl CoA (all Sigma) in pH 7.5 phosphate buffer with 10 mM magnesium chloride at 20°C for 1 h. 10 mM DTT was added when handling coenzyme A and holo mACP9 to prevent disulfide bond formation between exposed thiol groups. Samples were subjected to size exclusion chromatography as described above prior to further analysis. In each case, the identity and extent of modification was monitored by ESI MS (Supplementary Figures S3–S9).
Methanthiosulfonate-based modification of the 4′-phosphopantetheine thiol
(1-Oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methane-thiosulfonate (MTSL) or (1-acetoxy-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methanethiosulfonate (ATSL) (Toronto Research Chemicals), both dissolved in DMSO, were added to holo mACP9 in a 10-fold molar excess at less than 1% of the sample volume, then incubated at 20°C for 16 h. n-Propyl methanethiosufonate (PMTS) oil (Toronto Research Chemicals) was added directly to holo mACP9 at a 20-fold molar excess and at less than 0.2% of the final sample volume. All samples were incubated at 20°C for 16 h and then subjected to size exclusion chromatography as described above prior to further analysis. Full labelling was confirmed by ESI MS (Supplementary Figures S10–S12).
NMR experiments for assignment and distance restraints
All samples for NMR spectroscopy were prepared at concentrations of 200–800 μM in phosphate buffer supplemented with 10% D2O (Sigma) and 0.0025% 3,3,3-trimethylsilylpropionate (Sigma) in 5 mm Ultra-Imperial grade NMR tubes (Wilmad) to a final volume of 600 μL. 10 mM DTT was added to holo samples. [1H,15N]-HSQC, 15N-TOCSY-HSQC, 15N-NOESY-HSQC, HNCA, HNCOCA, HNCACB and CBCA(CO)NH spectra were recorded at 283 K on a Bruker DRX500 spectrometer equipped with a z-shielded gradient triple resonance probe, using standard procedures [29]. 13C-NOESY-HSQC spectra were recorded at 283 K on a Bruker Avance DRX800 spectrometer equipped with a 5 mm TXI CryoProbe. All NMR spectra were processed using the Azara package (www.ccpn.ac.uk/azara), then analysed and assigned using CcpNmr Analysis software [30]. To compare resonance positions in [1H,15N]-HSQC spectra of different mACP9 species, average chemical shift differences were determined using the formula Δδav={0.5(ΔδH)2 + 0.1(ΔδN)2}0.5 [31]. The threshold for a significant shift change (0.042 ppm) was calculated as twice the S.D. of the differences in all data sets remaining after eliminating outliers with differences greater than two S.D. from the initial mean.
Determination of solution structures for apo and octanoyl-mACP9
All structures of apo mACP9 were calculated from extended templates by simulated annealing using ARIA 2.3 [32], with manual screening of ambiguous restraints. Backbone φ and ψ dihedral angle restraints were determined from chemical shifts using the DANGLE program [33]. NOE distance restraints generated by the resonance assignment process and dihedral angle restraints were fed as input. Nine iterations were performed, each determining 20 structures, except for the final round, in which 100 were calculated, followed by refinement in explicit solvent for the 20 lowest energy structures, all of which were selected for the final ensemble, which contains no distance violations >0.5 Å (1 Å=0.1 nm) and includes >97% of residues in the ‘most favoured’ and ‘allowed’ regions of the Ramachandran plot. The atomic coordinates of the final ensemble for apo mACP9 were deposited in the Protein Data Bank under ID code 5HVC; the corresponding NMR assignments were deposited in the Biological Magnetic Resonance Data Bank under accession code 30007.
Structures of octanoyl-mACP9 were calculated following the method described above for the apo form. Ser2096 was replaced with a modified serine residue in which an octanoyl-phosphopantetheine group replaced the H atom. Topology files for the modified serine residue were created using the programs ACPYPE and ANTECHAMBER [34–36]. The atomic coordinates of the final ensemble for octanoyl-mACP9 were deposited in the Protein Data Bank under ID code 5HV8; the corresponding NMR assignments were deposited in the Biological Magnetic Resonance Data Bank under accession code 30006.
atom. Topology files for the modified serine residue were created using the programs ACPYPE and ANTECHAMBER [34–36]. The atomic coordinates of the final ensemble for octanoyl-mACP9 were deposited in the Protein Data Bank under ID code 5HV8; the corresponding NMR assignments were deposited in the Biological Magnetic Resonance Data Bank under accession code 30006.
15N nuclear spin relaxation experiments
15N nuclear spin relaxation experiments were recorded using standard procedures [29] at 283K on a Bruker DRX500 spectrometer. 15N T1 delays (ms): 10, 50, 100, 150, 250, 400, 550, 700, 850, 1000. 15N T2 delays (ms): 14.4, 28.8, 43.2, 57.6, 72.0, 86.4, 100.8, 155.2. The heteronuclear NOE reference and saturation experiments were carried out in duplicate to allow an estimation of the error. An initial τc estimate was obtained from the R2/R1 ratios for each residue [37]; the same procedure was used to make site specific estimates of the local rotational correlation time τeff. The relaxation parameters were analysed with version 4 of the Modelfree program [38] using the strategy described by Mandel et al. [39]. HN─N bond vectors from the solution structure of apo mACP9 were used for anisotropic diffusion tensor modelling of relaxation data for both the apo and holo forms.
Paramagnetic relaxation enhancement experiments
1HN T2 experiments [40] were recorded with delays (ms): 0.002, 8, 16 (reference); 0.002, 8, 16, 24 (MTSL-labelled). The reference sample was prepared by reducing the paramagnetic MTSL with a 5-fold molar excess of ascorbic acid (Sigma) from a 200 mM stock solution, added directly to the MTSL-labelled mACP9 NMR sample and incubated at 25°C for 2 h.
Circular dichroism spectroscopy
Thermal denaturation profiles were obtained by monitoring the molar ellipticity [θ] at 220 nm on an Aviv Model 410 circular dichroism spectrometer. Samples of mACP9 species were prepared at 0.1 mg/ml in 20 mM sodium hydrogen phosphate, 50 mM sodium fluoride, pH 7.5. [θ] was recorded at 1°C increments ranging from 20°C to 95°C. The unfolded state percentage was calculated using the formula F(T)={([θ]max − [θ]T)/([θ]max − [θ]min)} × 100, where [θ]max is the maximum observed value of [θ], [θ]min is the minimum observed value and [θ]T is the [θ] recorded at temperature, T. The melting temperature Tm was estimated from the inflexion point of the normalized melting curves. Reported Tm values are the mean for three repeat experiments.
RESULTS
Expression and purification of mACP9 species
mACP9, a protein fragment spanning residues 2050–2140 of the MLSA2 subunit, was expressed with an N-terminal His6-tag, as described in Materials and Methods. Following purification using nickel affinity chromatography, the fusion tag was removed by thrombin cleavage, leaving four non-native amino acid residues originating from the expression vector (GSHM-) at the N-terminus of the construct. Analytical size exclusion chromatography indicated that apo mACP9 is monomeric in solution (Supplementary Figure S13). Phosphopantetheinylation of mACP9 was carried out in vivo by co-expression with Sfp, a broad specificity phosphopantetheinyl transferase [28], resulting in full conversion from the apo to the holo form according to ESI MS (Supplementary Figure S2).
Solution structure of apo mACP9
The [1H,15N]-HSQC spectrum of apo mACP9 displayed well-resolved, dispersed signals indicative of a structured protein (Figure 2). Eighty-three of the 84 expected backbone amide resonances were assigned; no signal was detected for His2077, which is located in a flexible surface exposed loop. Single resonances were observed for each backbone and side-chain amide site, suggesting that the domain adopts a unique conformation in solution.
Figure 2. NMR spectroscopy.
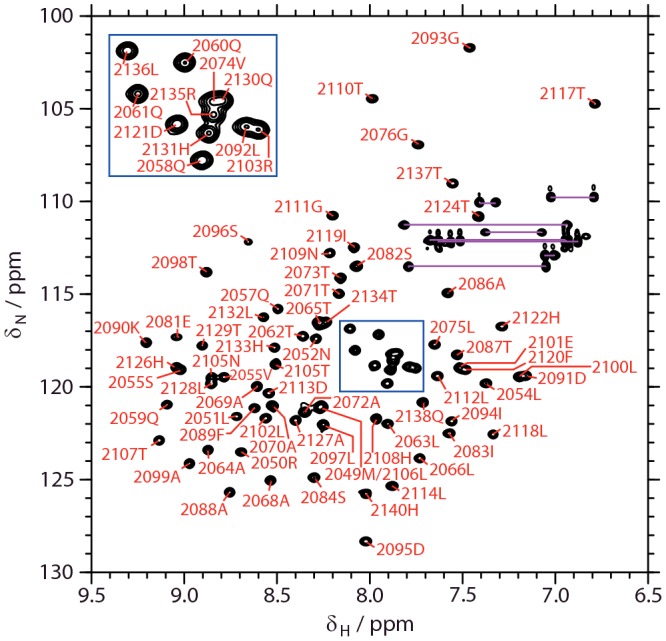
[1H,15N]-HSQC spectrum of apo mACP9, showing residue assignments for backbone amide sites. Pairs of resonances from side-chain amide sites are connected using magenta lines. Assignments for closely spaced signals are displayed in the inset panel.
A total of 2953 NOE-derived distance restraints, of which 1424 were unambiguously assigned, were used to calculate the solution structure of apo mACP9. After water refinement, the final ensemble comprising the 20 lowest energy structures (Figure 3A) has a backbone coordinate RMSD of 0.6 Å for residues 2050–2140, reducing to 0.4 Å over the sections of regular secondary structure; further statistics are summarized in Table 1. The compact, right-hand twisted helical bundle (Figure 3B) is typical of carrier proteins from PKS, FAS and NRPS systems [9]. The three main α helices (α1: 2056–2075; α2: 2096–2110 and α3: 2125–2137) are interspersed with two short helical turns (α2′: 2089–2092 and α3′: 2118–2121). The main helices span the long axis of the domain, with α1 antiparallel to α2 and α3, whereas the shorter turns are approximately perpendicular to the long axis. Ser2096, the attachment point for Ppant modification, is positioned at the opposite end of the domain to the two termini. The side chain of Ser2096 projects out towards the solvent at the beginning of helix α2.
Figure 3. Solution structure of apo mACP9.
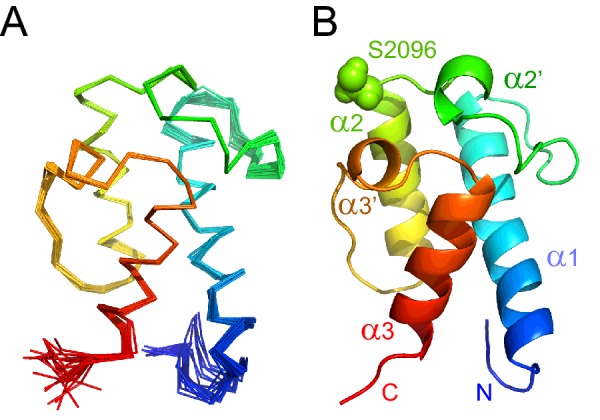
(A) Backbone overlay for the final ensemble of 20 lowest energy structures, coloured from blue at the N-terminus to red at the C-terminus (5HVC). (B) Ribbon representation of the lowest energy structure.
Table 1. Restraints and statistics for the solution structures of apo and octanoyl-mACP9.
| apo mACP9 | Octanoyl-mACP9 | |
|---|---|---|
| NOE-based distance restraints | ||
| Intra-residue, sequential | 705 | 406 |
| Medium range (2 ≤ | i − j | ≤ 5) | 244 | 280 |
| Long range (| i − j | > 5) | 183 | 330 |
| Ambiguous | 1707 | 454 |
| Total | 2839 | 1949 |
| Other restraints | ||
| Hydrogen bond restraints | 0 | 0 |
| φ + ψ dihedral angles restraints | 167 | 167 |
| Coordinate precision* | ||
| Backbone RMSD (Å) | 0.61±0.11 | 0.96±0.19 |
| Heavy atom RMSD (Å) | 1.17±0.18 | 1.46±0.24 |
| Consistency (structure compared with restraints) | ||
| RMSD (Å) from experimental distance restraints | 0.032±0.002 | 0.036±0.006 |
| RMSD (°) from experimental dihedral angle restraints | 1.97±0.08 | 1.12±0.08 |
| Ramachandran plot | ||
| Most favoured | 89.3% | 91.1% |
| Allowed regions | 8.1% | 8.0% |
| Generously allowed regions | 1.2% | 0.7% |
| Disallowed regions | 1.4% | 0.3% |
| WHATIF structure Z-scores | ||
| First generation packing quality | 1.09±0.63 | 1.03±0.59 |
| Second generation packing quality | 6.36±1.72 | 5.63±1.61 |
| Ramachandran plot appearance | −3.60±0.25 | −3.31±0.32 |
| χ1/χ2 rotamer normality | −4.04±0.35 | −5.38±0.51 |
| Backbone conformation | −0.15±0.42 | −0.10±0.02 |
*Coordinate precision, Ramachandran statistics and Z-scores were determined between residues 2050 and 2140.
The core of the calculated structure contains closely packed hydrophobic side chains, with no evidence for the interior cavities observed in type II ACP structures [9]. The exterior surface is predominately composed of hydrophilic side chains, the only exception being a small nonpolar patch adjacent to the attachment serine, produced by Phe2120 from turn α3′ and two solvent-exposed leucine side chains from helix α2 (Leu2097 and Leu2100). The nonpolar surface of the α2 ‘recognition helix’ is a common distinction observed between type I and type II ACPs, for which the equivalent region, proposed to be critical for interactions with partner enzymes, is rich in acidic residues [9]. In the mACP9 structure, helix α2 contains only one acidic residue, Glu2101; the majority of charged residues, which are predominantly basic, are located on a face created by α2, α3′ and α3, whereas the opposing α1 face is relatively uncharged (Supplementary Figure S14).
Holo and acyl-loaded forms of mACP9
Backbone assignments for the apo form were transferred to experiments recorded on a holo mACP9 sample and verified using NOE connections. As noted for the apo form above, the number of signals detected was consistent with population of a single conformational state. Two additional peaks in the [1H,15N]-HSQC spectrum of holo mACP9 were identified as amide signals from N4 and N8 in the Ppant arm (Supplementary Figure S15). The profile of average 1HN/15N chemical shift differences shown in Figure 4A demonstrates that the majority of backbone amide sites experience only minor perturbations in their electronic environment (<0.03 ppm), suggesting that the structure of mACP9 changes little between the apo and holo forms. Significant differences are observed for residues adjacent to the attachment serine (Ile2094-Asn2104) and for sites towards the N-terminus of the nearby α3′ turn (Thr2117-Asp2121).
Figure 4. Chemical shift differences between modified forms of mACP9.

Underneath a schematic defining the boundaries of α-helices in the structure of apo mACP9, average chemical shift differences, Δδav, are plotted as a function of residue number. Differences are shown between holo mACP9 and the (A) apo; (B) malonyl; (C) acetoacetyl; (D) hydroxybutyryl; (E) buten-2-oyl; (F) butyryl; (G) hexanoyl; and (H) octanoyl forms. Ser2096, the point of attachment for the prosthetic group, is indicated with an asterisk.
In type II ACP domains, the α3′ turn can undergo a minor structural rearrangement upon conversion to the holo form [9], but analysis of 15N- and 13C-separated NOESY spectra recorded on both apo and holo states of mACP9 revealed no significant differences, even for residues that display Δδav values >0.05 ppm, indicating that the backbone conformations of the two species remain very similar. In a comparison of apo and holo forms of the type I ACP from module 6 of the DEBS system, similar patterns of chemical shift perturbation were proposed to be a consequence of proximity to the attachment site, resulting from transient interactions with the dynamic Ppant moiety [13]. Interestingly, for mACP9 the magnitude of shift change does not have a simple relationship to distance from the attachment serine. For example, in the apo mACP9 structure the backbone amide proton (HN) of Thr2117 is 10.5 Å from the side-chain O atom of Ser2096 and possesses a Δδav of 0.16 ppm, whereas Lys2090 HN, which is 10.1 Å away, shows a Δδav of only 0.01 ppm. Enhanced chemical shift perturbations in the α3′ turn could indicate that the Ppant arm has a preferred orientation towards this region. Alternatively, the backbone amide sites of residues 2117–2121 are all close to the side chain of Phe2120, which projects out from α3′ towards the attachment serine. A subtle change in side-chain χ1 rotamer populations caused by intermittent contacts with the prosthetic group could alter the ring current contribution to the chemical shifts of nearby sites sufficiently to account for the observed shift differences.
Additional derivatives were prepared by in vitro incubation of apo mACP9 with Sfp and various acyl coenzyme A thioesters. In this way malonyl, acetoacetyl, β-hydroxybutyryl, 2-butenoyl and butyryl groups were loaded on to holo mACP9 to mimic the various chemistries that occur during the reaction cycle of the MLSA2 module for a C4 chain (Figure 1A; Supplementary Figure S16). Comparison of the [1H,15N]-HSQC spectra of loaded mACP9 species with that of the holo form reveals that short acyl chains have little or no effect on the structure of the domain. Polar malonyl, acetoacetyl and β-hydroxybutyryl modifications yield only minor chemical shift differences (Figures 4B–4D), but when nonpolar 2-butenoyl and butyryl groups are attached to the prosthetic group small but significant (>0.06 ppm) perturbations are observed for sites in the α3′ turn (Figures 4E and 4F).
By contrast, longer, completely reduced hexanonyl and octanoyl chains cause much larger (>0.10 ppm) shift changes at backbone sites through the length of helix α2 to the end of the α3′ turn (Figures 4G and 4H). A 12C/14N-filtered 13C-separated NOESY-HSQC experiment on a sample in which the ACP domain was uniformly 13C/15N-labelled but the Ppant and octanoyl groups remained unlabelled revealed 26 NOE connections between the protein and the acyl chain. Together with 1949 protein–protein distance restraints, these were used to determine the solution structure of octanoyl-mACP9. The final ensemble has a backbone RMSD of 0.8 Å for residues 2054–2140 and the lowest energy structure is very close to that obtained for the apo state of mACP9 (backbone RMSD 1.1 Å). As Figure 5A shows, the Ppant moiety remains somewhat disordered (with a heavy atom RMSD of 3.4 Å), whereas the octanoyl group (heavy atom RMSD 1.3 Å) becomes more ordered towards the tip, which inserts into a small nonpolar pocket between helix α2 and the α2′ and α3′ turns.
Figure 5. Solution structures of octanoyl-ACP species.

Ribbon representations, coloured from blue at the N-terminus to red at the C-terminus, for (A) octanoyl-mACP9 (5HV8); (B) octanoyl-actACP (2KGC); and (C) octanoyl-ACP from the S. coelicolor FAS (2KOS). Stick representations of pantoate, β-alanine, cysteamine and octanoyl sections are coloured orange, red, magenta and blue, respectively.
Experiments following the change in mean residue ellipticity at 222 nm as a function of temperature demonstrated that apo, holo, hexanoyl- and octanoyl-mACP9 species all undergo two-state unfolding transitions, with melting temperatures of 326 K, 332 K, 331 K and 330 K respectively (Supplementary Figure S17). These results confirm earlier work on the type II FAS ACP from P. falciparum, which showed that priming with Ppant enhanced the thermal stability of the domain [41]. For mACP9, it is interesting that further modification with hexanoyl and octanoyl groups had minimal effects, rather than causing the melting temperature to increase. This suggests that the bound state captured in the solution structure of octanoyl-mACP9 is formed only transiently, with a lifetime long enough to allow for magnetization transfer effects when the substrate chain is close to the protein surface, but leaving the undocked state sufficiently populated that the free energy difference between the folded and denatured states of the domain is hardly affected.
In summary, the attachment of C3 and C4 chains has only a modest effect on the behaviour of the Ppant group, especially if the acyl groups are polar, whereas longer nonpolar C6 and C8 chains exceed a threshold for more significant, but still transient, interactions with a hydrophobic patch on the surface of the ACP domain.
15N nuclear spin relaxation studies
To investigate the solution state dynamics of mACP9, nuclear spin relaxation properties were measured for 15N-labelled amide sites in both the apo and holo forms and analysed using the Lipari–Szabo model-free approach (Figure 6; Supplementary Figure S18). The data fitted best to an axially symmetric diffusion tensor model with a Dpar/Dper ratio of 1.30 and overall rotational correlation times of 10.3 ns and 10.4 ns for the apo and holo states, respectively, consistent with a monomeric, globular 90 amino acid domain at 283 K. In both the apo and holo states, backbone sites were found to be predominantly rigid (mean order parameter S2 of 0.87±0.10 for both), with the same five residues possessing more dynamic S2 values (<0.70): Met2050 at the N-terminus; Leu2106 in α2; Leu2114 in the loop between α2 and α3′ and Gln2138 and His2140 at the C-terminus. These similar relaxation properties indicate that no functionally relevant changes in backbone dynamics are induced on conversion of apo mACP9 to the holo form.
Figure 6. 15N relaxation parameters for holo mACP9.

Underneath a schematic defining the boundaries of α-helices in the structure of apo mACP9, NMR parameters for backbone and prosthetic group amide sites in the holo state are plotted as a function of residue number for: (A) the 15N longitudinal relaxation rate, R1; (B) the 15N transverse relaxation rate, R2; (C) the {1H}-15N NOE ratio (I′/I0, where I′ is the intensity when the 1H spectrum has been saturated and I0 is the intensity in the reference spectrum); and (D) the Lipari–Szabo the order parameter, S2. Bars shaded in grey correspond to values for the N8 (left) and N4 (right) sites of the prosthetic group.
In vivo phosphopantetheinylation allowed relaxation parameters to be measured for the two 15N-labelled amide sites in the Ppant arm. Both nuclei displayed properties indicating elevated levels of local motion relative to the rest of the protein (see Table 2). For example, the heteronuclear NOE ratios for N4 (−0.76) and N8 (−0.13) possessed the opposite sign to the average value for holo protein backbone sites (+0.72±0.13). 15N R1 and R2 values for the Ppant sites were used to estimate effective local rotational correlation times (τeff) of 3.7 ns and 4.1 ns for N4 and N8, respectively [37]. The relaxation properties of the Ppant sites were also analysed using the Lipari–Szabo model-free approach together with data from the rest of the holo state. As no structural data was available for the prosthetic group, an isotropic rotational diffusion model was assumed, which fit the data best using an overall correlation time of 10.9 ns and S2 order parameter values of 0.07 and 0.13 for N4 and N8 respectively. These low S2 values demonstrate that the Ppant moiety reorients much more rapidly than the rest of the protein and that the arm becomes significantly more dynamic as it extends outward from its point of attachment at the side chain of Ser2096. This behaviour is consistent with freely swinging motion in the prosthetic group, a conclusion supported by the observation that HN4 and HN8 display only weak, intra-Ppant NOE connections in the 15N-separated NOESY spectrum of holo mACP9. Our in vitro strategy for preparing acyl-loaded mACP9 samples relied on reactions between the apo protein and unlabelled coenzyme A derivatives; as a consequence, we did not have the opportunity to measure the relaxation properties of Ppant amide sites in acyl-ACP species.
Table 2. Dynamics parameters derived from 15N nuclear spin relaxation experiments.
| State | Species | τeff (ns)* | NOE ratio |
|---|---|---|---|
| apo | Backbone average | 10.3 | 0.68 |
| holo | Backbone average | 10.5 | 0.72 |
| holo | N4 | 3.7 | –0.76 |
| holo | N8 | 4.1 | –0.13 |
| ATSL, major | Backbone average | 10.5 | 0.72 |
| ATSL, major | N4 | 6.3 | 0.00 |
| ATSL, major | N8 | 6.6 | 0.24 |
| ATSL, minor | Backbone average | 10.6 | 0.79 |
| ATSL, minor | N4 | 5.3 | 0.00 |
| ATSL, minor | N8 | 5.4 | 0.20 |
*Determined using the equation τeff=[(6R2/R1) − 7]0.5/(2ωN) [37].
Paramagnetic relaxation enhancement studies
The chemical shift perturbations observed on conversion of mACP9 from the apo to the holo form could be interpreted as evidence that the Ppant group prefers to populate conformations that are oriented towards the α3′ turn, similar perhaps to those captured in the ensemble of structures for octanoyl-mACP9. By contrast, our 15N nuclear spin relaxation measurements suggest that the prosthetic group is highly flexible, implying that the arm samples multiple conformations in an isotropic fashion. To investigate this issue further, the Ppant arm of holo mACP9 was modified with the paramagnetic spin label reagent MTSL. Two reference samples were also prepared: one modified with ATSL, with the nitroxyl moiety replaced by an acetyl group (Supplementary Figure S16); and one in which the nitroxyl radical of MTSL was reduced to a diamagnetic hydroxy group via treatment with ascorbate. The ascorbate-reduced reference sample showed only minor chemical shift perturbations compared with non-MTSL-labelled holo mACP9 (Figure 7B), confirming that any interactions between the MTSL label and mACP9 must be similar to those observed for C4-loaded species.
Figure 7. Average shift difference and PRE parameters for acyl-mACP9 species.

Underneath a schematic defining the boundaries of α-helices in the structure of apo mACP9, properties of disulfide-linked substrate mimics are plotted as a function of residue number. Average chemical shift differences, Δδav, are shown between holo mACP9 and (A) the apo form; (B) the reduced MTSL form; (D) the ATSL minor form (doubled peaks only); (E) the ATSL major form; and (F) the PMTS form. Panel (C) shows the PRE effects (defined as R2,MTSL – R2,ref) for MTSL-mACP9. Cartoons of the apo mACP9 structure below highlight the Ser2096 modification site in red and amide sites that: (G) show significant shift changes between the apo and holo forms (green); (H) are bleached in the MTSL form (yellow); and (I) exhibit doubled resonances in ATSL-labelled mACP9 (blue). Small cream spheres indicate the nitrogen atoms of proline residues.
Paramagnetic relaxation enhancements (PREs) were quantified as the difference between 1HN transverse relaxation rates in the spin-labelled sample and in the reduced reference sample (R2,MTSL − R2,ref) [40]. In the paramagnetic sample, several resonances exhibited greatly enhanced relaxation, to the extent that accurate R2,MTSL measurements could not be made; to include these sites in Figure 7C, the value of R2,MTSL − R2,ref was set to an arbitrary maximum of 75 Hz. The relaxation enhancement profile is similar to that observed for chemical shift perturbations between the apo and holo forms (Figure 7A), with residues surrounding the attachment serine and around the α3′ turn affected most. As Figure 7H suggests, all amide signals from sites within 12.5 Å of the O atom of Ser2096 were bleached or strongly broadened (including those from N4 and N8 in the Ppant arm). Proline residues lack an amide proton, so they are not probed by this technique.
Modification with disulfide-linked groups
Unexpectedly, labelling of mACP9 with ATSL yielded a [1H,15N]-HSQC spectrum in which several resonances were doubled (Supplementary Figure S19), rendering the sample unsuitable for use as a reference state for PRE experiments. ESI MS revealed that the sample contained a single species at the expected molecular mass (Supplementary Figure S11), suggesting that the two forms detected by NMR correspond to distinct conformational states rather than alternative modification products. None of the doubled peaks coincide with resonances from the apo or holo forms of mACP9, indicating that modification with ATSL creates two significantly different conformations. All resolvable doublets comprise a minor peak with chemical shifts close to the corresponding holo signal (Figure 7D) and a major peak showing a larger shift perturbation (Figure 7E), with an intensity ratio of approximately 1:1.4. In zz-HSQC experiments, no chemical exchange cross-peaks between resolved doublets were detected (results not shown), implying that if the two states are capable of interconverting, this must occur on a timescale slower than 0.5 s. As Figure 7I shows, sites that give rise to doublets include those displaying the strongest PRE effects (Figure 7H), the largest chemical shift differences between the apo and holo forms (Figure 7G), and the most significant perturbations in 2-butenoyl- and butyryl-loaded samples, but affect fewer sites than those changed in an octanoyl-loaded sample. Amide signals from the prosthetic group were also doubled and displayed relaxation properties (τeff values of 6.5 ns and 5.4 ns for the major and minor forms, respectively; Table 2) intermediate between those of the protein backbone (∼10.5 ns) and the more dynamic arm of the holo species (∼3.9 ns). Consistent with these results, the HN4 and HN8 sites in both forms exhibited more intense 1H-1H NOEs than the holo state, although once again only intra-Ppant connections were observed.
Aiming to mimic the length of a butyryl substrate chain, we modified a sample of the holo domain using PMTS. The [1H,15N]-HSQC spectrum of PMTS-modified mACP9 showed a single set of resonances with chemical shift changes (Figure 7F) more extensive than those observed for butyryl-mACP9 (Figure 4F), but smaller than for either of the ATSL-modified forms (Figures 7D and 7E). Together with an absence of peak doubling for the reduced MTSL sample, these observations suggest that multiple ATSL-mACP9 states are a property of the altered nitroxide moiety rather than a consequence of the linkage mode, such as conformational isomerism of the disulfide bond [42]. The most likely explanation is that the ATSL-modified prosthetic group populates two distinct conformers, both of which dock against the surface of the ACP domain, differing perhaps in the orientation of the acetoxy moiety (Supplementary Figure S16). Within the Ppant group, motions are restricted compared with the freedom of movement experienced in the holo state, but a significant degree of flexibility is retained. Although ATSL is not a natural substrate for mACP9, these results provide a further demonstration that longer substrates can interact with the surface of a type I PKS ACP domain. Evidence that the prosthetic group possesses moderately restricted dynamics in both ATSL-loaded conformers is consistent with the picture conveyed by the solution structure of octanoyl-mACP9, in which the Ppant arm shows a degree of conformational heterogeneity that decreases as the acyl group approaches the protein surface.
DISCUSSION
Although a wide variety of carrier protein domains have been studied previously [9], this report on MLSA2 from the mycolactone system is the first to compare the solution structures of both the apo and acyl-loaded forms of an ACP domain excised from a canonical type I PKS module. The only other published atomic resolution structure for an ACP from a type I modular PKS is for the apo domain from module 2 of the DEBS system (eryACP2), which lacks DH and ER domains [43]. EryACP2 shares 48% sequence identity with mACP9 and its overall fold is highly similar, showing a backbone RMSD of 1.3 Å over residues equivalent to Leu2054-His2140 (Supplementary Table S3); the three main helices possess almost identical orientations, whereas the connecting loops and helical turns display a greater degree of variation. In contrast with the clustering of charged side chains towards one face observed for mACP9, the distribution of positive and negative charges is more even across the surface of eryACP2 (Supplementary Figure S14). The mACP9 structure also resembles those of ACP domains from the CalE8 iterative type I PKS (backbone RMSD 1.1 Å), the curacin trans-AT hybrid PKS/NRPS system (1.5 Å), S. coelicolor type II FAS (1.8 Å), the actinorhodin type II PKS (2.0 Å) and rat type I FAS (2.3 Å) (Supplementary Table S3).
Some type II carrier proteins exhibit multiple slowly exchanging structural forms in solution [44–46] although such heterogeneity is apparently not ubiquitous [18,47]. The single sets of resonances observed in our [1H,15N]-HSQC experiments demonstrate that the apo, holo and acyl-loaded forms of mACP9 each appear to adopt a single conformational state on the timescale of milliseconds to seconds monitored by slow regime chemical exchange. The 15N relaxation properties of the apo and holo forms of mACP9 (Figure 6; Supplementary Figure S18) support this conclusion, showing that backbone amide sites are rigid on the sub-nanosecond and the micro- to milli-second timescales, except for close to the N- and C-termini and in the long linker region between helices α1 and α2. It therefore seems unlikely that chemical modification of mACP9 species would manipulate the populations of significantly different pre-existing protein conformations in order to optimize their surface structures for interactions with particular partner enzyme domains [44,48]. Similarly modest degrees of backbone flexibility have been detected in inter-helical regions for the CalE8 [49] and frenolicin type II PKS ACP domains [50].
The Ppant arms of holo ACP species adopt a spectrum of dynamic states, ranging from tightly bound to the carrier protein surface to freely swinging. For example, the prosthetic group of the holo form of an atypical Geobacter metallireducens ACP (GmACP3) is rigid, displaying S2 order parameter values similar to those for structured backbone sites (∼0.8), together with 23 Ppant-protein NOE connections [51]. The Plasmodium falciparum type II FAS ACP (PfACP) represents the centre ground, exhibiting two conformers in slow exchange due to different docked orientations of the Ppant moiety; in both states the prosthetic group is semi-rigid, with S2 values for N4 and N8 in the 0.1–0.5 range, but also 6 NOEs between the arm and the protein surface [46]. The prosthetic group of frenolicin holo ACP is at the high flexibility end of the scale, exhibiting chemical shifts identical with those for free coenzyme A, low S2 values (∼0.1) and no detectable Ppant-protein NOEs [50]. For holo mACP9, Ppant group behaviour is close to this freely swinging extreme, showing no evidence for NOEs to the protein surface and N4 and N8 order parameter values of 0.07 and 0.13, respectively (Table 2), consistent with the arm becoming progressively more dynamic as it protrudes into the solvent.
Interestingly, backbone 1HN/15N chemical shift perturbations appear to be less sensitive probes of protein/prosthetic group interactions than Ppant 15N relaxation parameter measurements or the detection of direct Ppant-protein NOE connections. For mACP9, which possesses a highly dynamic arm, the largest detected Δδav value between the apo and holo forms is ∼0.3 ppm (Figure 4A), whereas the largest corresponding shift change for the firmly bound GmACP3 species is only ∼0.4 ppm [51]. Furthermore, we found that the magnitude of shift changes between apo and holo mACP9 did not depend in a straightforward way on distance from the point of attachment of the prosthetic group.
We therefore modified the Ppant thiol group of holo mACP9 with a nitroxide spin label to determine whether PRE experiments could provide complementary information. As the reduced, diamagnetic form of MTSL-mACP9 shows no 1HN/15N shift perturbations from the holo species larger than 0.1 ppm (Figure 7B), we deduced that the motional properties of the prosthetic group are altered only to a minor extent by addition of the spin label. In [1H,15N]-HSQC spectra of the oxidized, paramagnetic form of MTSL-mACP9, all amide signals from sites close to Ser2096 either disappear or are strongly broadened (Figure 7C). It is not possible to interpret completely bleached signals in fine-grained detail, but we take the clustering of residues that experience substantial intensity reductions (Figure 7H) as an indication that each site is transiently approached by the paramagnetic centre to within 10 Å [40]. This result is consistent with the Ppant arm sampling a wide range of conformations in an isotropic manner, as would be expected for freely swinging motion. Similar conclusions were drawn in recent studies of an MTSL-modified holo peptidyl carrier protein from a teicoplanin-producing NRPS system [47].
In common with the structure of rat FAS ACP [14], apo mACP9 lacks an obvious central cavity, meaning that considerable rearrangement would be required to bury an acyl group in its hydrophobic core, in the style of type II carrier proteins [11]. When the holo form of rat FAS ACP was compared with species loaded with hexanoyl and palmitoyl chains, no 1HN/15N shift changes >0.05 ppm and no acyl-protein NOEs could be detected, leading to the conclusion that this type I domain does not sequester long hydrophobic chains from the solvent [14]. Likewise, no Δδav values >0.13 ppm were found between the holo and propionyl-, malonyl-, butyryl-, crotonyl- or hexanoyl-loaded species of eryACP2 [16]. Derivatization of mACP9 with C3 and C4 acyl chains has a similar outcome, with no Δδav values >0.15 ppm (Figure 4), suggesting that these substrate mimics remain predominantly exposed to the solvent. In contrast, weighted shift changes >0.5 ppm are apparent for both hexanoyl- and octanoyl-mACP9 species, alongside 26 NOE connections between the octanoyl chain and the protein surface.
The ensemble of solution structures determined for octanoyl-mACP9 shows the Ppant arm curling over to allow the acyl chain to nestle into a nonpolar pocket formed between the α2′ and α3′ turns and helix α2, whereas the prosthetic group itself remains solvent exposed (Figure 5A). This behaviour is similar to that observed for the ACP domain from the actinorhodin type II PKS (actACP), with no significant interactions detected between the protein and acetyl, malonyl, 3-oxobutyl or 3,5-dioxohexyl chains, but with large shift changes apparent for nonpolar butyryl, hexanoyl and octanoyl chains, yielding solution structures that showed Ppant and the acyl moieties rigidly docked into a groove between the α3′ turn and helices α2 and α3 (Figure 5B) [45]. For octanoyl-mACP9, the interaction of the acyl chain with the protein surface is clearly more superficial than that for octanoyl-actACP, which in turn does not penetrate as deeply into the hydrophobic core as seen in studies of octanoyl-ACP from the S. coelicolor type II FAS (Figure 5C) [12]. Furthermore, in the ensemble of structures for octanoyl-mACP9, the conformation of the Ppant moiety is relatively uncertain (RMSD 3.4 Å), implying that it retains a degree of flexibility analogous to that measured here for the major and minor states of ATSL-mACP9 (Table 2). This behaviour is different to the more precise definition of the prosthetic group in the octanoyl-actACP ensemble (RMSD 1.7 Å), which is restrained by 50 intra-Ppant, 12 Ppant-protein and 30 acyl-protein NOEs [45].
The NOE connection between a pair of nuclear spins corresponds to an r−6-weighted time and ensemble average over all the inter-nuclear distances sampled during the mixing time of a NOESY experiment [52]. This strong distance dependence rarely complicates the interpretation of NOE-based structures for folded protein domains, but it implies that structures of more dynamic systems may be biased towards conformations in which the two spins are close in space, even if these are populated sparsely and for relatively brief time periods. In this light, the ensemble of solution structures for octanoyl-mACP9 should be regarded as a collection of snapshots of bound conformations that provide no information about potential unbound states in which the acyl chain is fully exposed to solvent.
If a conformation in which a Ppant-tethered octanoyl group is bound to the surface of an ACP domain is highly populated, acyl-loading might be expected to stabilize the structure of the protein compared with the holo state, making it more resistant to thermal denaturation. To our surprise, when we used circular dichroism spectroscopy to monitor the unfolding of apo, holo, hexanoyl- and octanoyl-mACP9 species, acyl group attachment was found to have minimal effects on the melting temperature. By contrast, modification with Ppant increased the melting temperature of apo mACP9 by 6 K (Supplementary Figure S17). Phosphopantetheinylation of the apo state of pfACP has been reported to cause a similar degree of stabilization, attributed to the formation of hydrophobic contacts between the prosthetic group and the protein surface [41]. In our hands, the Ppant group of holo mACP9 appears to swing freely around its attachment point, which suggests that other factors must be responsible for increasing its resistance to denaturation. Further studies will be needed to fully explain the effects of Ppant attachment on ACP domain thermostability, but the results of our NMR and circular dichroism experiments on octanoyl-mACP9 are consistent with its acyl chain making weak transitory interactions with the surface of the carrier protein.
Within the mycolactone PKS system, four other extender module ACP domains display >95% identity with mACP9: those from the third, seventh and eighth modules of MLSA1 and the seventh module of MLSB [25]. When mACP9 is included, this family of domains can be loaded with either malonyl or methylmalonyl extender units, whereas its natural substrate chains range in length from C8 to C20, can either possess or lack an α-branching methyl group, and can sample three of the four possible β-position oxidation states: an alcohol, an alkene or a fully reduced methylene group (Supplementary Figure S20). Compared with such diversity, this work has been able to characterize a limited panel of commercially available substrate-mimics (Supplementary Figure S16). Nevertheless, we have shown that when attached to a type I PKS ACP fragment short polar acyl chains remain exposed to the solvent, whereas longer, more saturated chains can interact (albeit transiently) with patches on the surface of the domain without significantly changing the structure of the protein.
In the context of an intact PKS module, our observations argue against channelling mechanisms that rely on substantial substrate-mediated structural modifications to optimize the shape of the ACP in such a way that protein–protein interactions with accessible partner domains are promoted, whereas others are prohibited. Alternatively, if the next destination of the ACP is to be decided by random diffusion between a pre-selected subset of partner domains [10] followed by recognition of substrate chemistry alone, this would presumably be favoured by arranging for solvent exposed acyl cargoes to be presented by highly dynamic Ppant arms. Despite an apparent concordance between this mechanism and our results for short polar substrate-mimics, the sub-states identified in cryo-EM experiments on Pik module 5 suggest that both protein–protein interfaces and correct substrate chemistry are required for the ACP to find an appropriate active site [20,21]. Unfortunately, the >7 Å resolution of these cryo-EM studies was not sufficient to localize any Ppant or acyl moieties, so the relative importance of interactions with the ACP surface, the prosthetic group or the substrate itself has not yet been established for any of the detected sub-states.
Although not obtained using a natural substrate, our findings for octanoyl-mACP9 pose the intriguing possibility that a partner domain may be able to recognize an acyl group whereas it is bound to the surface of its carrier protein, allowing simultaneous interactions with both the substrate and the ACP. Subtle variations in the substrate chemistry, ACP-docked conformation and features of the adjacent ACP surface could then favour encounters with the correct partner in order to facilitate progress through the reaction cycle. From this perspective, the apparent flexibility of the Ppant and acyl portions of octanoyl-mACP9 need not be seen as a disadvantage, as a conformational selection mechanism triggered by proximity to the cognate surface of a partner domain could permit the substrate/Ppant/ACP interface to adapt so as to promote productive interactions with the appropriate active site. Similar mechanisms have recently been suggested for substrate-loaded NRPS aryl and peptidyl carrier protein domains from the Type I yersiniabactin [18] and the Type II pyoluteorin [19] systems.
Various novel methods have been used to investigate Ppant behaviour, including the attachment of solvatochromic groups for fluorescence spectroscopy [53], a cyanyl group for vibrational spectroscopy [54] and fluorophenyl [55] or trifluoromethyl [16] substituted acyl groups for 19F NMR spectroscopy. These approaches were primarily designed to sense whether the probe was buried in a nonpolar environment or exposed to solvent, and do not return information about Ppant dynamics or details about any detected site of interaction. Moreover, all three methods require covalent modification of the Ppant arm, making them incompatible with studies of natural acyl-ACP species, as well as potentially perturbing the dynamics and interaction motifs of the prosthetic group. This work has deployed NOE-based structure determination, 1HN/15N chemical shift perturbation, nuclear spin relaxation and PRE as a suite of tools that in combination can report directly on the conformational and dynamic properties of ACP-attached and substrate-loaded Ppant groups. Having demonstrated that the Ppant arm in some circumstances swings freely, but in others is ‘sticky’ enough to interact with the surface of the carrier protein, the logical next step is to characterize the role of prosthetic group dynamics in binary interactions between ACP and partner domain fragments and within an intact PKS module.
Acknowledgments
We are grateful to Professor Kira Weissman for a critical reading of this manuscript; to Dr Annabel Murphy for assistance with the ESI MS data, and to the referees for helpful suggestions about this manuscript.
Abbreviations
- ACP
acyl carrier protein
- AT
acyltransferase
- ATSL
(1-acetoxy-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methanethiosulfonate
- cryo-EM
cryo-electron microscopy
- DH
dehydratase
- ER
enoyl reductase
- ESI MS
electrospray injection mass spectrometry
- FAS
fatty acid synthase
- HSQC
heteronuclear single quantum coherence
- KR
ketoreductase
- KS
ketosynthase
- MTSL
(1-oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methanethiosulfonate
- NRPS
non-ribosomal peptide synthetase
- PKS
polyketide synthase
- PMTS
n-propyl methanethiosufonate
- Ppant
4′-phosphopantetheine
- PRE
paramagnetic relaxation enhancement
- TE
thioesterase
AUTHOR CONTRIBUTION
William Broadhurst designed the research. Steven Vance, Olga Tkachenko and William Broadhurst conceived and designed the experiments. Steven Vance, Olga Tkachenko, Ben Thomas, Mona Bassuni, Daniel Nietlispach and William Broadhurst performed the experiments. Steven Vance, Olga Tkachenko and William Broadhurst analysed the data. Steven Vance, Mona Bassuni, Hui Hong and William Broadhurst contributed reagents/materials/analysis tools. Steven Vance and William Broadhurst wrote the paper.
FUNDING
This work was supported by the Wellcome Trust [grant number 094252/Z/10/Z]. The Yousef Jameel Academic Foundation and the Cambridge Trust are thanked for providing a studentship to M.B.
References
- 1.Staunton J., Weissman K.J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Keatinge-Clay A.T. The structures of type I polyketide synthases. Nat. Prod. Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 3.Weissman K.J., Leadlay P.F. Combinatorial biosynthesis of reduced polyketides. Nat. Rev. Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 4.Kim E., Moore B.S., Yoon Y.J. Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat. Chem. Biol. 2015;11:649–659. doi: 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Floss H.G. Combinatorial biosynthesis: potential and problems. J. Biotechnol. 2006;124:242–257. doi: 10.1016/j.jbiotec.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poust S., Hagen A., Katz L., Keasling J.D. Narrowing the gap between the promise and reality of polyketide synthases as a synthetic biology platform. Curr. Opin. Biotechnol. 2014;30:32–39. doi: 10.1016/j.copbio.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Khosla C., Herschlag D., Cane D.E., Walsh C.T. Assembly line polyketide synthases: mechanistic insights and unsolved problems. Biochemistry. 2014;53:2875–2883. doi: 10.1021/bi500290t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weissman K.J., Müller R. Protein–protein interactions in multienzyme megasynthetases. ChemBioChem. 2008;9:826–48. doi: 10.1002/cbic.200700751. [DOI] [PubMed] [Google Scholar]
- 9.Crosby J., Crump M.P. The structural role of the carrier protein–active controller or passive carrier. Nat. Prod. Rep. 2012;29:1111–1137. doi: 10.1039/c2np20062g. [DOI] [PubMed] [Google Scholar]
- 10.Weissman K.J. The structural biology of biosynthetic megaenzymes. Nat. Chem. Biol. 2015;11:660–670. doi: 10.1038/nchembio.1883. [DOI] [PubMed] [Google Scholar]
- 11.Cronan J.E. The chain-flipping mechanism of ACP (acyl carrier protein)-dependent enzymes appears universal. Biochem. J. 2014;460:157–163. doi: 10.1042/BJ20140239. [DOI] [PubMed] [Google Scholar]
- 12.Płoskoń E., Arthur C.J., Kanari A.L.P., Wattana-amorn P., Williams C., Crosby J., Simpson T.J., Willis C.L., Crump M.P. Recognition of intermediate functionality by acyl carrier protein over a complete cycle of fatty acid biosynthesis. Chem. Biol. 2010;17:776–785. doi: 10.1016/j.chembiol.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Tran L., Broadhurst R.W., Tosin M., Cavalli A., Weissman K.J. Insights into protein–protein and enzyme–substrate interactions in modular polyketide synthases. Chem. Biol. 2010;17:705–716. doi: 10.1016/j.chembiol.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Płoskoń E., Arthur C.J., Evans C., Williams C., Crosby J., Simpson T.J., Crump M.P. A mammalian type I fatty acid synthase acyl carrier protein domain does not sequester acyl chains. J. Chem. Biol. 2008;283:518–528. doi: 10.1074/jbc.M703454200. [DOI] [PubMed] [Google Scholar]
- 15.Wattana-amorn P., Williams C., Płoskoń E., Cox R.J., Arthur C.J., Simpson T.J., Crosby J., Crump M.P. Solution structure of an acyl carrier protein domain from a fungal type I polyketide synthase. Biochemistry. 2010;49:2186–2193. doi: 10.1021/bi902176v. [DOI] [PubMed] [Google Scholar]
- 16.Charkoudian L.K., Liu C.W., Capone S., Kapur S., Cane D.E., Togni A., Seebach D., Khosla C. Probing the interactions of an acyl carrier protein domain from the 6-deoxyerythronolide B synthase. Protein Sci. 2011;20:1244–1255. doi: 10.1002/pro.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen C., Haushalter R.W., Lee D.J., Markwick P.R.L., Bruegger J., Caldera-Festin G., Finzel K., Jackson D.R., Ishikawa F., O'Dowd B., et al. Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature. 2014;505:427–431. doi: 10.1038/nature12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodrich A.C., Harden B.J., Frueh D.P. Solution structure of a nonribosomal peptide synthetase carrier protein loaded with its substrate reveals transient, well-defined contacts. J. Am. Chem. Soc. 2015;137:12100–12109. doi: 10.1021/jacs.5b07772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaremko M.J., Lee J.D., Opella S.J., Burkart M.D. Structure and substrate sequestration in the pyoluteorin peptidyl carrier protein PltL. J. Am. Chem. Soc. 2015;137:11546–11549. doi: 10.1021/jacs.5b04525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutta S., Whicher J.R., Hansen D.A., Hale W.A., Chemler J.A., Congdon G.R., Narayan A.R., Håkansson K., Sherman D.H., Smith J.L., Skiniotis G. Structure of a modular polyketide synthase. Nature. 2014;510:512–517. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whicher J.R., Dutta S., Hansen D.A., Hale W.A., Chemler J.A., Dosey A.M., Narayan A.R., Håkansson K., Sherman D.H., Smith J.L., Skiniotis G. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature. 2014;510:560–564. doi: 10.1038/nature13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weissman K.J. Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2015;32:436–453. doi: 10.1039/C4NP00098F. [DOI] [PubMed] [Google Scholar]
- 23.George K.M., Pascopella L., Welty D.M., Small P.L. A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect. Immun. 2000;68:877–883. doi: 10.1128/IAI.68.2.877-883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bali S., Weissman K.J. Ketoreduction in mycolactone biosynthesis: insight into substrate specificity and stereocontrol from studies of discrete ketoreductase domains in vitro. ChemBioChem. 2006;7:1935–1942. doi: 10.1002/cbic.200600285. [DOI] [PubMed] [Google Scholar]
- 25.Stinear T.P., Mve-Obiang A., Small P.L., Frigui W., Pryor M.J., Brosch R., Jenkin G.A., Johnson P.D., Davies J.K., Lee R.E., et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1345–134. doi: 10.1073/pnas.0305877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. 3rd edn. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 27.Martinez E., Bartolomé B., de la Cruz F. pACYC184-derived cloning vectors containing the multiple cloning site and lacX alpha reporter gene of pUC8/9 and pUC18/19 plasmids. Gene. 1998;68:159–162. doi: 10.1016/0378-1119(88)90608-7. [DOI] [PubMed] [Google Scholar]
- 28.Quadri L.E., Weinreb P.H., Lei M., Nakano M.M., Zuber P., Walsh C.T. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 29.Cavanagh J., Fairbrother W.J., Palmer A.G., Skelton N.J. Protein NMR Spectroscopy: Principles and Practice. 2nd edn. San Diego: Academic Press; 2006. [Google Scholar]
- 30.Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 31.Pellecchia M., Sebbel P., Hermanns U., Wuthrich K., Glockshuber R. Pilus chaperone FimC-adhesin FimH interactions mapped by TROSY-NMR. Nat. Struct. Biol. 1999;6:336–339. doi: 10.1038/7573. [DOI] [PubMed] [Google Scholar]
- 32.Bardiaux B., Bernard A., Rieping W., Habeck M., Malliavin T.E., Nilges M. Influence of different assignment conditions on the determination of symmetric homodimeric structures with ARIA. Proteins. 2009;75:569–585. doi: 10.1002/prot.22268. [DOI] [PubMed] [Google Scholar]
- 33.Cheung M.S., Maguire M.L., Stevens T.J., Broadhurst R.W. DANGLE: a Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 2010;202:223–233. doi: 10.1016/j.jmr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Sousa da Silva A.W., Vranken W.F. ACPYPE–antichamber python parser interface. BMC Res. Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 36.Wang J., Wang W., Kollman P.A., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Kay L.E., Torchia D., Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 38.Palmer A., Rance M., Wright P. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance carbon-13 heteronuclear NMR. J. Am. Chem. Soc. 1991;113:4371–4380. doi: 10.1021/ja00012a001. [DOI] [Google Scholar]
- 39.Mandel A.M., Akke M., Palmer A.G. Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 40.Clore G.M., Iwahara J. Theory, practice and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Modak R., Sinha S., Surolia N. Isothermal unfolding studies on the apo and holo forms of Plasmodium falciparum acyl carrier protein: role of the 4′-phosphopantetheine group in the stability of the holo form of the Plasmodium falciparum acyl carrier protein. FEBS J. 2007;274:3313–3326. doi: 10.1111/j.1742-4658.2007.05856.x. [DOI] [PubMed] [Google Scholar]
- 42.Grey M.J., Wang C., Palmer A.G. Disulphide bond isomerisation in basic pancreatic trypsin inhibitor: multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modelling. J. Am. Chem. Soc. 2003;125:14324–14335. doi: 10.1021/ja0367389. [DOI] [PubMed] [Google Scholar]
- 43.Alekseyev V.Y., Liu C.W., Cane D.E., Puglisi J.D., Khosla C. Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 2007;16:2093–2107. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koglin A., Mofid M.R., Löhr F., Schäfer B., Rogov V.V., Blum M.M., Mittag T., Marahiel M.A., Bernhard F., Dötsch V. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 45.Evans S.E., Williams C., Arthur C.J., Płoskoń E., Wattana-amorn P., Cox R.J., Crosby J., Willis C.J., Simpson T.J., Crump M.P. Probing the interactions of early polyketide intermediates with the actinorhodin ACP from S. coelicolor A3(2) J. Mol. Biol. 2009;389:511–528. doi: 10.1016/j.jmb.2009.03.072. [DOI] [PubMed] [Google Scholar]
- 46.Sharma A.K., Sharma S.K., Surolia A., Surolia N., Sarma S.P. Solution structures of conformationally equilibrium forms of holo-acyl carrier protein (PfACP) from Plasmodium falciparum provides insight into the mechanism of activation of ACPs. Biochemistry. 2006;45:6904–6916. doi: 10.1021/bi060368u. [DOI] [PubMed] [Google Scholar]
- 47.Haslinger K., Redfield C., Cryle M. Structure of the terminal PCP domain of the non-ribosomal peptide synthetase in teicoplanin biosynthesis. Proteins. 2015;83:711–721. doi: 10.1002/prot.24758. [DOI] [PubMed] [Google Scholar]
- 48.Tufar P., Rahighi S., Kraas F.I., Kirchner D.K., Löhr F., Henrich E., Köpke J., Dikic I., Güntert P., Marahiel M.A., Dötsch V. Crystal structure of a PCP/Sfp complex reveals the structural basis for carrier protein posttranslational modification. Chem. Biol. 2014;21:552–562. doi: 10.1016/j.chembiol.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Lim J., Kong R., Murugan E., Ho C.L., Liang Z.X., Yang D. Solution structures of the acyl carrier protein domain from the highly reducing type I iterative polyketide synthase CalE8. PloS One. 2011;6:e20549. doi: 10.1371/journal.pone.0020549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Q., Khosla C., Puglisi J.D., Liu C.W. Solution structure and backbone dynamics of the holo form of the frenolicin acyl carrier protein. Biochemistry. 2003;42:4648–4657. doi: 10.1021/bi0274120. [DOI] [PubMed] [Google Scholar]
- 51.Ramelot T.A., Smola M.J., Lee H.W., Ciccosanti C., Hamilton K., Acton T.B., Xiao R., Everett J.K., Prestegard J.H., Montelione G.T., Kennedy M.A. Solution structure of 4′-phosphopantetheine-GmACP3 from Geobacter metallireducens: a specialized acyl carrier protein with atypical structural features and a putative role in lipopolysaccharide biosynthesis. Biochemistry. 2011;50:1442–1453. doi: 10.1021/bi101932s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zagrovic B., van Gunsteren W.F. Comparing atomistic simulation data with NMR experiment: how much can NOEs actually tell us? Proteins. 2006;63:210–218. doi: 10.1002/prot.20872. [DOI] [PubMed] [Google Scholar]
- 53.Beld J., Cang H., Burkart M.D. Visualizing the chain-flipping mechanism in fatty-acid biosynthesis. Angew. Chem. Int. Ed. 2014;53:14456–14461. doi: 10.1002/anie.201408576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson M.N.R., Londergan C.H., Charkoudian L.K. Probing the phosphopantetheine arm conformations of acyl carrier proteins using vibrational spectroscopy. J. Am. Chem. Soc. 2014;136:11240–11243. doi: 10.1021/ja505442h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lloyd R.P. Ph.D. thesis. University of Cambridge; 2014. Structural Studies of Acylated Forms of an Acyl Carrier Protein from Saccharopolyspora erythraea. [Google Scholar]