

Abstract
Pancreatic ductal adenocarcinoma (PDAC) constitutes 90% of pancreatic cancers. PDAC is a complex and devastating disease with only 1%–3% survival rate in five years after the second stage. Treatment of PDAC is complicated due to the tumor microenvironment, changing cell behaviors to the mesenchymal type, altered drug delivery, and drug resistance. Considering that pancreatic cancer shows early invasion and metastasis, critical research is needed to explore different aspects of the disease, such as elaboration of biomarkers, specific signaling pathways, and gene aberration. In this review, we highlight the biomarkers, the fundamental signaling pathways, and their importance in targeted drug delivery for pancreatic cancers.
Keywords: EGFR, KRAS, PIM, mTOR, NF-κB, PAF, EMT, MMPs, RAGE, MYC, pancreatic cancer stem cells, miRNA, pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is characterized by a series of molecular aberrations.1 Due to the heterogeneity and the complex nature, it is hard to diagnose and treat this malignancy, which has only a 1%–3% survival rate in five years after the second stage.2 In most cases, diagnosis occurs in the later stages, with a well-developed, dense, desmoplastic stroma and metastasis to other organs. The spreading of the disease from the pancreas to multiple distant sites renders major surgery impossible. The complexity of the disease manifests as different, patient-specific, aberrant biochemical pathways and makes the treatment challenging. The dense extracellular matrix (desmoplasia) in PDAC leads to early development of hypoxia, expression of inflammatory cytokines and other extra-cellular components, and epithelial-to-mesenchymal transition (EMT). All of these incriminating factors make drug delivery complicated, resulting in drug resistance and disease relapse.3
Altered gene expression patterns and mutations are frequently observed in PDAC.1 Gene expression microarray analysis has identified the following three main subtypes of PDAC: classical, quasimesenchymal, and exocrine like. The classical PDAC cells (BxPC3 and CaPan-2) have the characteristic epithelial-like genes, while the quasimesenchymal cells (Panc-1 and MiaPaCa-2) express mesenchymal features. Exocrine-like primary tumor cells overexpress digestive enzymes.4 For example, tissue microarray analysis detected the expressions of ABCC3 and TLR2 in AsPC-1, CaPan-1, HPAFII, PSN-1, and SU86.86 pancreatic cancer cell lines. ABCC3 is an ATP-binding cassette mostly observed in the tumor tissues of pancreatic cancer and may be used for cell surface-targeted imaging and delivery of therapeutics.5 Discovery of other biomarkers and aberrant biochemical pathways (contributing to tumorigenicity) has made tremendous progress in recent years. Despite the considerable research and clinical studies, PDAC is still a lethal disease. In this review article, we summarize the biomarkers of PDAC and recent developments of targeting several pathways for treating the disease.
Epidermal Growth Factor Receptor
Epidermal growth factor receptor (EGFR), a transmembrane glycoprotein of the EGFR family, is overexpressed in 40%–70% of patient samples with pancreatic cancer.1,6 The ErbB, also known as the human EGFR-1 (HER-1), belongs to the EGFR family. The glycoprotein EGFR has an intracellular tyrosine kinase domain, a transmembrane domain, and an extracellular domain for ligand binding. Interactions of the tumor growth factor-α and EGF with the extracellular domain lead to dimerization and autophosphorylation of EFGR protein, producing downstream signal transduction Activation of the EGFR kinase stimulates the following two signaling pathways: RAS-RAF-mitogen-activated ERK-activating kinase (MEK)-mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)-PTEN-Akt-mTOR-GSK3 (Fig. 1).7, 8
Figure 1.

Signaling pathways stimulated by the activation of EGFR kinase.
The anticancer drugs erlotinib and gefitinib inhibit the autophosphorylation of EFGR tyrosine kinase by competing with adenosine triphosphate in the intracellular domain.7 The US Food and Drug Administration (FDA) has approved erlotinib as a combination therapy (with gemcitabine) for PDAC. Boeck et al9 evaluated the overexpression of EGFR in tumor tissues treated with erlotinib from 181 phase III randomized patients by immunohistochemistry (49% showed EGFR overexpression). Cardnell et al reported that EMT leads to resistance to EGFR inhibitors and metastatic progression of PDAC.10 Recently, researchers have discovered the role of Na+/H+ exchanger protein NHE1 in promoting EGFR signaling pathway and pancreatic cancer metastasis. The coadministration of cariporide (an NEH1 inhibitor) with erlotinib results in a decreased three-dimensional colony growth and invasion for both classical (BxPC3 and CaPan-2) and quasimesenchymal (Panc-1 and MiaPaCa-2) pancreatic cancer cell lines.11 Anti-EGFR monoclonal antibodies (eg, cetuximab and panitumumab) inhibit receptor dimerization at the extracellular domain. In a recent phase II clinical study, radiotherapy along with cetuximab increased radiosensitivity in locally advanced pancreatic cancer.12
Kirsten Rat Sarcoma Viral Oncogene
Kirsten rat sarcoma viral (KRAS) oncogene is a GTPase protein belonging to the RAS gene family.13 In 1982, the mutated human RAS gene was found to be activated in cancer.14 The KRAS proto-oncogene point mutation occurs in 75%–95% of PDAC.1 The most common mutation is the replacement of glycine with aspartate at position 12 (KrasG12D). KRAS in pancreatic cancer is characterized by the mutation type, allelic ratio, and tumor subtype.15,16 Tumors with high dependency on KRAS might have poor prognosis.4
KRAS oncogene mutation activates the P21 RAS protein and a series of signaling pathways.17 The RAS protein is located on the inner surface of the cell membrane and binds to guanosine triphosphate (GTP)/guanosine diphosphate (GDP). In the presence of RAS mutation, GTPase cannot undergo transition from the GTP (active) form to GDP (inactive) form, and RAS remains in a permanently active state, resulting in a cascade of downstream activation.13 Figure 2 depicts the RAS protein regulation GTP/GDP cycle. Prenylation of the RAS protein increases its capability to interact with cell membrane and endoplasmic reticulum (ER) compartments via the hydrophobic C terminus.18 Farnesyltransferase and geranylgeranyltransferase I, respectively, attach the farnesyl (15 carbon) and geranylgeranyl (20 carbon) isoprenoid lipids to the cysteine residue of RAS protein with the C terminus of CAAX (C: cysteine, A: aliphatic amino acids, and X: usually serine or methionine).14,18 To inhibit the RAS protein, a farnesyltransferase inhibitor for posttranslational prenylation Tipifarnib (R115777) was investigated in conjunction with gemcitabine in a double-blinded phase III clinical study on advanced pancreatic cancer. However, the results did not show any statistically significant clinical benefit over gemcitabine and placebo.19 The lack of increased efficacy may be due to the presence of other RAS isoforms (such as non-farnesylated RAS) or the RAS-independent activity of tipifarnib.17,19
Figure 2.
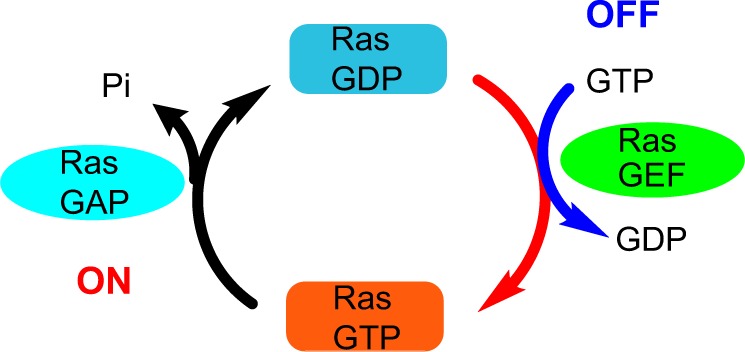
RAS protein regulation through the GTP/GDP cycle.
Pao et al20 investigated KRAS mutation and the development of drug resistance to monotherapy by EGFR inhibitors (erlotinib and gefitinib) in non-small lung carcinoma. Cotreatment of locally advanced pancreatic cancer with erlotinib and gemcitabine did not significantly increase survival, even though 60 % of the patients harbored EGFR expression.21 Moreover, in a phase II clinical study, treatment with gemcitabine along with cetuximab (an anti-EGFR monoclonal antibody) was not more effective than gemcitabine alone.22 Lee et al23 suggested an alternate mechanism for EGFR signaling in KRAS-mutated pancreatic cancer cells that does not follow the canonical MAPK pathway. Moreover, inactivation of Akt might happen as a result of treatment with an EGFR inhibitor, such as erlotinib.24 Three major signaling pathways, PI3K-3-phosphoinositide-dependent protein kinase-1-Akt, Raf-Mek-Erk, and Ral-GEFs, are affected by the KRAS oncogene in PDAC.4,25,26 The PI3K-3-phosphoinositide-dependent protein kinase-1 downstream pathway is mostly dominant in Kras-driven PDAC.26,27 Inhibition of tumor growth was demonstrated by blocking and deletion of Pdk-1 in the PI3K pathway in a Kras-engineered mouse model.26 Collins et al28 reported a mouse model of on and off Kras oncogene that developed metastatic PDAC. Inhibition of the MEK-ERK pathway using AZD-6224 in combination with glycosphingolipid synthesis inhibitor 1-Phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) induced apoptosis in human pancreatic cancer.29 Recently, Lindberg et al ruled out the effect of EGFR and HER-2 signaling pathways on the growth of patient-derived PDAC xenograft (PDX) using mice bearing wild-type and mutant Kras alleles. Coadministration of panitumumab (anti-EGFR antibody) and trastuzumab (anti-HRE2 antibody) synergistically enhanced the anticancer effect of trametinib (an MEK inhibitor) in PDX mouse models.1 Khvalevsky et al developed the biodegradable polymer matrix Local Drug EluteR (LODER) to encapsulate Kras G12D siRNA. LODER drug eluter inhibited tumor growth by decreasing the Kras expression in an orthotopic mouse model of PDAC.30
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases of the metzincin superfamily and degrade extracellular matrix. Therefore, they have a significant role in tissue remodeling and tumor progression in pancreatic cancer. Overexpression of MMP-1 (collagenase), MMP-2 (gelatinases-A), MMP-7 (matrilysin), MMP-9 (gelatinase-B), MMP-10, MMP-11 (stromelysin), and MMP-13 (collagenase) is observed in pancreatic cancer.31–33
Since MMPs play a key role in altering cell behavior, inhibition of MMPs is an attractive approach for anticancer therapy. Small synthetic metalloproteinase inhibitors showed promising results in preclinical studies but failed in phase III clinical trials due to lack of specificity.34 We recently reported MMP-9-triggered release of the anticancer drug gemcitabine from liposomes. The liposomes presented a layer of polyethylene glycol on the surface for long circulation and accumulation in the tumor by the enhanced permeation and retention effect. At the tumor site, the enhanced concentration of the reducing agent glutathione reductively removed the polyethylene glycol layer and exposed the substrate peptides toward MMP-9-mediated hydrolysis. The loss of liposomal structural integrity led to the rapid release of encapsulated gemcitabine and reduction in xenograft pancreatic tumor volume in mice.35 Munshi et al36 reported that increased collagen leads to the overexpression of MMP-14 (MT1-MMP) in the desmoplastic regions of pancreatic cancer, causing tumor progression and gemcitabine resistance. Srivastava et al37 reported the inhibition of MMP-2, -7, -9, and -12 by epigallocatechin-3-gallate (extracted from green tea) in vitro and xenograft mouse model of pancreatic cancer.
Receptor for Advanced Glycation Endproducts
The membrane-associated receptor for advanced glycation endproducts (RAGE) belongs to immunoglobulin-like receptor family. RAGE is present in normal cells, such as epithelial cells, neurons, smooth muscle cells, and hepatocytes. The expression is upregulated in cancers and diverse types of diseases, including diabetes, Alzheimer’s, osteoarthritis, and cardiovascular.38 Several signaling cascades (eg, PI3K-Akt, MAPK, and small GTPase) are activated upon stimulation by binding of the ligands S100P, S100A4, and S100A6 to the RAGE. Overexpression of the ligands S100P and S100A6 are reported in pancreatic cancer. In a recent study, the administration of 5-methyl cromolyn (an S100 inhibitor) resulted in the reduction of tumor growth and metastasis in an orthotopic mouse model of PDAC.39
Nuclear Factor Kappa B
The nuclear transcription factor kappa B (NF-κB) belongs to the Rel/NF-κB protein and has significant roles in targeting genes for encoding cytokines, cell growth, cell molecule adhesion, apoptosis, and inflammatory responses.40 Overactivation of NF-κB pathway is observed in 70% of pancreatic cancer cell lines.41 In most cases, the noncanonical NF-κB pathway overexpression is present in PDAC.42 Small molecule inhibitors for NF-κB have not yet progressed to the clinical trials. However, several researchers have studied the inhibitory effects of curcumin (extracted from turmeric) on the expression of NF-κB using in vitro and in vivo models of pancreatic cancer.43,44 Kurzrock et al45 reported the inhibition of NF-κB and reduced toxicity for advanced pancreatic cancer patients treated with 8 g of oral curcumin (phase II clinical trial).
Mammalian Target of Rapamycin
Mammalian target of rapamycin (mTOR) is a serine/threonine-associated PI3K signaling pathway responsible for cell proliferation, growth, and survival. The mTOR pathway is deregulated in several cancers, including PDAC.46 Moreover, the mTOR pathway activation has been observed in pancreatic cancer stem cells (PCSCs).47 The FDA has approved an mTOR inhibitor (Afinitor) to treat subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) and renal cancer carcinoma. Morran et al48 demonstrated that the FDA-approved mTOR inhibitor rapamycin along with gemcitabine decreased the tumor size and proliferation in a PTEN-deficient, mouse strain with KRasG21D mutation (KC model) of pancreatic cancer. Coadministration of rapamycin and the PI3K inhibitor LY294002 impeded the proliferation and growth of PCSCs by blocking the PI3K-mTOR signaling pathway.49
Proto-oncogene Serine/Treonine-protein Kinase
Proto-oncogene serine/threonine-protein kinase (PIM) proteins belong to serine/threonine kinase family and are upregulated in several tumors, eg, sarcoma, hepatocellular cancer, prostate cancer, and PDAC. Specifically, PIM1 and PIM3 are overexpressed in pancreatic cancer.50 Moreover, hypoxia and Kras oncogene regulate the PIM proteins. The PIM regulates several signaling pathways in cell cycle regulation and apoptosis. The shRNA-mediated knockdown of PIM1 in MIAPaCa-2 and Capan-1 pancreatic cancer cell lines revealed that the PIM1 protein plays a significant role in anchorage-dependent and anchorage-independent growth, invasion, and radioresistance for pancreatic cancer cells.51
V-MYC Avian Myelocytomatosis Viral Oncogene
V-MYC avian myelocytomatosis viral oncogene (MYC) protein is a transcriptional factor and has an important role in genetic and epigenetic regulations of PDAC.52 The over-expression of Myc was observed in 32% of primary and 29% of metastatic pancreatic tumors.53 Myc accelerates metabolism and proliferation of tumor cells and angiogenesis.52 Zhou et al54 demonstrated the expression of Myc in multipotent progenitor cells during differentiation into the exocrine and endocrine cells in pancreatic organogenesis. Impeding the c-Myc in pdx1+ multipotent progenitor cells resulted in altered differentiation and reduced proliferation of exocrine and endocrine pancreatic cells in a mouse model.55 Several signaling pathways, such as PI3K-Akt, RAS-MAPK, cyclin-dependent kinase 2, and NF-κB, have a role in posttranslational alteration of Myc.52
Platelet-Activating Factor
Platelet-activating factor (PAF) is involved in the phospholipid-regulating MAPK signaling pathway. PAF overexpression in pancreatic cancer leads to cell proliferation and tumorigenesis. Jun et al56 demonstrated that the PAF ectopic activation of the MAPK signaling occurred via the activation of LAM TOR3 pathway, causing neoplasia in pancreatic cancer.
Cell Surface Antigen CD109
CD109, a glycophosphatidylinositol-anchored glycoprotein, was recognized as a cell surface antigen on some normal hematopoietic and metpoietic tumor cells.57 CD109 engages in the EGF signaling in SK-MG-1 glioblastoma cells.58 The cell surface glycoprotein CD109 was identified in BxPC3 cells from primary pancreatic cancer. CD109 glycoprotein is expressed in the BxPC3, MIACaPa-2, and Panc-1 cell lines. Also, CD109 overexpression was observed in PDAC. The expression of CD109 was evaluated in normal pancreatic tissues and PDAC samples by cell-surface capture technique and immunohistochemistry.59
PCSC Biomarkers
Cancer stem cells (CSCs) are inherently immortalized, can self-renew, asymmetrically divide, and differentiate into stem cells. Discovery of PCSCs was first reported in 2007.60 PCSCs contribute to tumor progression, metastasis, and resistance to common chemotherapy.61 Although several cell surface markers, such as CD133, C-Met, aldehyde dehydrogenase 1 (ALDH1), and “side population cells and the triplet combination CD44+CD24+ESA+,”62 have been reported, none is unique to the PCSCs (Table 1).61 Molejon et al63,64 reported the high expression of the cell surface marker CD44 in recurrent PDAC. Maeda et al65 reported that high expression of the cell surface marker CD133 reduced patient survival to 2.1 months, in contrast to 23.5 months when the marker is not expressed at a high level. Pancreatic cancer cells characterized by CD44+CD24+ESA+ on their surface showed resistance to gemcitabine and radiotherapy.66 Hong et al67 reported that CD44 has a key role in gemcitabine resistance in PCSCs.
Table 1.
Surface markers of pancreatic cancer stem cells.
| CELL SURFACE MARKER | PERCENTAGE | CHARACTERISTICS |
|---|---|---|
| CD133 | 1.09–3.21% | Tumorigenicity and metastasis |
| C-Met | 2–16% | Tumor growth and metastasis |
| ALDH-1 | 16% | Tumorigenicity and tumor-initiation |
| CD44+CD24+ESA+ | 0.2–0.8% | Tumorigenicity and self-renewal |
Various cellular signaling pathways, such as Notch, Wnt, and hedgehog, can facilitate the formation of stem cells in pancreatic cancer.68 Sonic hedgehog is usually overexpressed in pancreatic tissues and PCSCs. Deregulation of sonic hedgehog pathway causes pathogenesis and desmoplasia in PDAC.69 Cyclopamine, IPI-269609, and GDC-0449 are hedgehog inhibitors. Cyclopamine inhibits PCSCs and reduces endothelial-to-mesenchymal transition and metastasis in vitro and in vivo.69 Moreover, expression of the cell surface biomarkers CD133 and CD44 decreases in gemcitabine-resistant cells after cyclopamine therapy.70 Feldmann et al71 reported that in an orthotopic xenograft model of PDAC, treatment with cyclopamine and gemcitabine decreased the expression of ALDH, resulting in reduced invasion of PDAC.
The Notch signaling pathway has important roles in cellular differentiation, apoptosis, stem cell regeneration, EMT, drug resistance, and tumorigenesis.72 The Notch signaling pathway proteins are overexpressed in pancreatic cancer cells and PCSCs.68,73 Notch acts as the tumor suppressor in the skin and small cell carcinomas but as an oncogenic protein in pancreatic cancer.73 Notch signaling pathway is activated through γ-secretase. The γ-secretase inhibitor MRK-003 can be used to block the Notch signaling pathway in pancreatic cancer. Coadministration of MRK-003 and gemcitabine resulted in reduced tumor size in PDAC xenograft model.74
Another cell surface biomarker of CSCs is the tyrosine kinase C-Met. Cabozantinib, a C-Met inhibitor, impedes sphere formation and escalates apoptosis via downregulation of C-Met, CD 133, and SOX2 in PCSCs.75 Cotreatment of XL184 (a C-Met inhibitor) with gemcitabine or XL184 alone reduced cell proliferation and growth of PCSC in NOD-SCID mice.76 Recent research by Singh et al77 reported that PAK4 (p-21 activated kinase 4, serine/threonine kinase family) activates the STAT3 signaling pathway, resulting in a stemness phenotype. In addition, they demonstrated that the PAK4 overexpression in PCSCs compared to the non-CSCs is associated with chemotherapy resistance and sphere formation.
Epithelial-to-Mesenchymal Transition
Through the process of EMT, epithelial cells lose their normal characteristics, such as apical–basal polarity, cell–cell tight junctions, and transition to spindle-like, motile, and invasive mesenchymal cells.78 EMTs can be of the following three types: Type I (embryogenesis), Type II (wound healing and organ fibrosis), and Type III (cancer).79 In addition to embryogenesis and wound healing,80 EMT plays pivotal roles in metastasis and drug resistance in pancreatic and other cancers.81 Due to the EMT in pancreatic cancer, epithelial cells downregulate E-cadherin and upregulate vimentin, N-cadherin, and fibronectin.78 For pancreatic cancer patients with EMT in the primary tumor, 75% showed metastasis to the lungs and liver.82 Tumor microenvironmental factors, such as hypoxia, inflammatory cytokines, extracellular components, and mechanical characters contribute to EMT progression.3 The inflammatory cytokines’ transforming growth factor-β (TGFβ), tumor necrosis factor-α, interleukin-1, and interleukin-6 cause progression of EMT in PDAC.3 The TGFβ signaling pathway can act either as a tumor suppressor or as a tumor promoter, depending on the stage of PDAC.83 The TGFβ signaling pathway leads to apoptosis in the early phases of the tumor but in later stages contributes to tumor progression and invasion via EMT.84
The TGFβ signaling pathway upregulates TWIST1, SNAIL1, and SNAIL2 transcription factors.3 TGFβ pathway inhibitors, such as trabedersen (AP12009) and galunisertib (LY2157299), decreased metastasis and invasion in animal model studies and clinical trials.85,86 In contrast, the TGFβ inhibitors SB431542 and galunisertib showed opposite effects when the Panc-1 cells and normal fibroblasts (VI-38) were cocultured in a three-dimensional collagen gel. The Panc-1 cells showed rapid invasion, changes in morphology, and EMT after treatment with TGFβ inhibitors. It is possible that the secreted hepatocyte growth factor from the fibroblasts and the cancer cells (in response to TGFβ inhibitors) cause invasion and cell proliferation of the Panc-1 cells into the collagen gel.87
MicroRNAs in Pancreatic Cancer
MicroRNAs (miRNAs) are small, single-stranded, noncoding, 20–25 nucleotide RNA sequences with regulatory effects on gene expressions and in several physiological and pathological processes.88 miRNAs behave as tumor suppressors and oncogenes in pancreatic adenocarcinoma. Overexpression of the oncogene miRNAs (oncomir) increases in tumor progression, while tumor suppressors inhibit cell proliferation and induce apoptosis.89 The miRNAs are expressed selectively in the tumor tissues90 and inactivate the tumor suppressor genes p53, p16, and SMAD4 in pancreatic cancer.91
The miR-21 is upregulated in pancreatic cell lines and tissue and decreases survival rate significantly.92 The miR-21 overexpression is reported as the lesion initiator, causing tumor progression in a KRAS (G12D) mouse model.92 The miR-155 is also upregulated in pancreatic cancer and contributes to tumor progression. Knockdown of miR-155 downregulates EGFR, KRAS, and MT1-MMP expressions, leading to inhibition of cell proliferation.93 The upregulation of miR-221 in pancreatic cancer leads to distant metastasis and unresectable tumors.94
Point mutation of p53 is present in 50%–70% of human pancreatic cancers.95 The p53 facilitates transcription of a vast number of miRNAs. Stress signaling in cells induced by hypoxia and starvation upregulates p53 and activates the expressions of several genes, such as miR-107, -34a/b/c, and miR-34. The expressed miRNAs modulate apoptosis and inhibit hypoxia in PDAC.91,96 Mutation of p53 mediates transcription of miR-130b and miR-155, modifies the expressions of the corresponding target genes (ZEB1 and ZNF652), and leads to cell proliferation and invasion in several cancers.97,98 In addition, p53 mutation impairs maturation of miR-145 and miR-16-1 causing cell proliferation, invasion, and migration in PDAC.91
Hypermethylation of the DNA regions coding for the miRNAs suppresses their expressions. For example, the miRNA-124 genes in pancreatic cancer tissues are silenced via hypermethylation, resulting in cell proliferation, invasion, metastasis, and decreased survival rates. Silencing of miRNA- 124 occurs via downregulation of Rac1, proceeding to the inactivation of the MKK4-JNK-c-Jun pathway.99 miRNA-200a and miR-205 are downregulated during EMT in PDAC in response to TGFβ. Expression of the miR-200 family mediates regulation of the E-cadherin and suppresses transcriptions of ZEB1 and SIP1.100 Wellner et al101 demonstrated that ZEB1 inhibits the expression of the miRNA-200 family and regulates the activation of EMT in PCSCs. The downand upregulated miRNAs in pancreatic cancer are summarized in Tables 2 and 3.
Table 2.
Downregulated miRNA genes in pancreatic cancer.
Table 3.
Upregulated miRNA genes in pancreatic cancer.
Treatment of Pancreatic Cancer
Diagnostic staging of pancreatic cancer is the key to the treatment of the disease. Computed tomography is routinely used to determine the tumor stage and the resectability. Tumor characteristics, such as size, vascularity, lymphatic node, locations, and degree of metastasis, ascertain the success of the surgery.102 The carbohydrate antigen 19-9 in body fluids is a biomarker for diagnosis, prognosis, and determining chemotherapy response for pancreatic adenocarcinoma, albeit not specific for the disease.103,104 Table 4 shows the validated serum biomarkers for pancreatic cancer.103 Chemotherapy and radiation still are the primary treatment for advanced pancreatic cancer. Surgical removal of the tumor followed by six months of gemcitabine treatment increased the median patient survival to 22.8 months and the one-year survival to 70%.105 Locally advanced pancreatic cancer is treated with an initial chemotherapy and subsequent 5-fluorouracil chemoradiation.106 Several clinical trials also show promising results (Table 5). In the CONKO-001 randomized, multicenter trial, patients with completely resectable tumors were treated for six months with gemcitabine (following surgery). The gemcitabine treatment showed the median disease-free survival of 13.4% (confidence interval 95%) compared to 6.7% (confidence interval 95%) for the nontreated group.105 Clinical trials also suggest that the combination of gemcitabine and fluorouracil derivatives (CAP/S-1) improve the one-year survival rates compared to monotherapy with the drugs.107 Gemcitabine, FOLFIRINOX (FOL: folinic acid, F: 5-fluorouracil, IRIN: irinotecan hydrochloride, OX: oxaliplatin) and gemcitabine plus nab-paclitaxel are the suggested treatments for metastatic pancreatic adenocarcinoma.108–110
Table 4.
Serum biomarkers in pancreatic adenocarcinoma.
Table 5.
Selected phase III clinical trials for the treatment of advanced pancreatic cancer.
| TREATMENTS | MEDIAN SURVIVAL (MONTHS) | OBJECTIVE RESPONSE | BEST TREATMENT TOXICITIES GRADE (III/IV) | REFERENCE |
|---|---|---|---|---|
| Gemcitabine vs Gemcitabine/nab-paclitaxel | 8.5 vs 6.7 | 23% vs 7% | Fatigue, neutropenia, peripheral neuropathy | 108 |
| FOLFIRINOX vs Gemcitabine | 11.1 vs 6.8 | 31.6% vs 9.4% | Fatigue, neutropenia, diarrhea | 110 |
| Gemcitabine vs Fluorouracil | 5.65 vs 4.41 | 5.4% vs 1% | neutropenia | 109 |
Conclusion
PDAC is a sequence of complex deviations at the molecular levels.111 Cell signaling pathway alterations, pancreatic stem cells, and EMT led to resistance to conventional chemotherapy. In this review, we summarize the primary molecular changes, biomarkers, and small molecule inhibitors for blocking different pathways of PDAC. Although several inhibitors are reported for most of the molecular aberrations, extensive efforts need to be made to bring the research to the clinics. Targeted delivery reduces toxicity and enhances the efficacy of the anticancer drugs.112 The knowledge of biomarkers and small molecule inhibitors is expected to promote further research and development of targeted therapies, alleviating the severe side effects of pancreatic cancer therapy and increasing the survival rates.
Abbreviations
- PDAC
pancreatic ductal adenocarcinoma
- EGFR
epidermal growth factor receptor
- K-RAS
Kristen rat sarcoma viral
- MAPK
mitogen-activated protein kinase
- PIM
protooncogene serine/threonine-protein kinase
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor kappa B
- PAF
platelet-activating factor
- MMPs
matrix metalloproteinases
- MYC
V-MYC avian myelocytomatosis viral oncogene
- RAGE
receptor for advanced glycation endproducts
- PCSCs
pancreatic cancer stem cells
- miRNA
microRNAs
- EMT
epithelial-to-mesenchymal-transition
- TNFα
tumor necrosis factor-α mentioned on page 6, line 4, column 1 (SMAD), SMAD family member
- ErbB
epidermal growth factor receptor-1
- MEK
mitogen-activated ERK-activating kinase
- PI3K
phosphoinositide 3-kinase
- PTEN
tumor suppressor phosphatase and tensin homolog
- Akt
serine/threonine-specific protein kinase
- GSK3
glycogen synthase kinase
- Pdk1
3-phosphoinositide-dependent protein kinase-1
- TP53
tumor suppressor protein 53
- TGFβ
tumor necrosis factor-β
- SHH
sonic hedgehog
Footnotes
ACADEMIC EDITOR: Barbara Guinn, Editor in Chief
PEER REVIEW: Five peer reviewers contributed to the peer review report. reviewers’ reports totaled 568 words, excluding any confidential comments to the academic editor.
FUNDING: a part of the summarized research was supported by NSF grant DMR 1306154 and NIH grant 1 R01 GM 114080 to SM. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert single-blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Analyzed the data: FK. Wrote the first draft of the manuscript: FK. Contributed to the writing of the manuscript: SM. Agree with manuscript results and conclusions: FK and SM. Jointly developed the structure and arguments for the paper: FK and SM. Made critical revisions and approved final version: FK and SM. Both author reviewed and approved of the final manuscript.
REFERENCES
- 1.Lindberg JM, Newhook TE, Adair SJ, et al. Co-treatment with panitumumab and trastuzumab augments response to the MEK inhibitor trametinib in a patient-derived xenograft model of pancreatic cancer. Neoplasia. 2014;16(7):562–571. doi: 10.1016/j.neo.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Cancer Society, Pancreatic Cancer (2015) 2015. http://www.cancer.org/cancer/pancreaticcancer/index.
- 3.Jung H-Y, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial–mesenchymal transition in metastasis. Clin Cancer Res. 2015;21(5):962–968. doi: 10.1158/1078-0432.CCR-13-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collisson EA, Sadanndam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morse DL, Balagurunathan Y, Hostetter G, et al. Identification of novel pancreatic adenocarcinoma cell-surface targets by gene expression profiling and tissue microarray. Biochem Pharmacol. 2010;80(5):748–754. doi: 10.1016/j.bcp.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamanaka Y, Friess H, Kobrin M, Buchler M, Beger H, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1992;13(3):565–569. [PubMed] [Google Scholar]
- 7.Philip PA. Targating epidermal growth factor receptor-related signalling pathway in pancreatic cancer. Pancrease. 2015;44:1041–1052. doi: 10.1097/MPA.0000000000000389. [DOI] [PubMed] [Google Scholar]
- 8.Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers. 2011;3(2):1513–1526. doi: 10.3390/cancers3021513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boeck S, Jung A, Laubender R, et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br J Cancer. 2013;108(2):469–476. doi: 10.1038/bjc.2012.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardnell R, Diao L, Wang J, et al. An epithelial-mesenchymal transition (EMT) gene signature to predict resistance to EGFR inhibition and A X L identification as a therapeutic target in head and neck squamous cell carcinoma. J Clin Oncol. 2013:31. abstr6011. [Google Scholar]
- 11.Cardone RA, Greco MR, Zeeberg K, et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor–dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia. 2015;17(2):155–166. doi: 10.1016/j.neo.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rembielak AI, Jain P, Jackson AS, et al. Phase II trial of cetuximab and conformal radiotherapy only in locally advanced pancreatic cancer with concurrent tissue sampling feasibility study. Transl Oncol. 2014;7(1):55–64. doi: 10.1593/tlo.13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan MB, Der CJ, Wang-Gillam A, Cox AD. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1(3):183–198. doi: 10.1016/j.trecan.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox AD, Der CJ. Ras history. Small GTPases. 2010;1(1):2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lennerz JK, Stenzinger A. Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist. 2015;20:e8–e9. doi: 10.1634/theoncologist.2014-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: hopes and realities. Eur J Cancer. 2015;54:75–83. doi: 10.1016/j.ejca.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol. 2016;17(2):110–122. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 19.Van Cutsem E, van de Velde H, Karasek P, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 20.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley RK, Ko AH. Erlotinib in the treatment of advanced pancreatic cancer. Biologics. 2008;2(1):83–95. doi: 10.2147/btt.s1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: southwest oncology group–directed intergroup trial S0205. J Clin Oncol. 2010;28:3605–3610. doi: 10.1200/JCO.2009.25.7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S, Heinrich EL, Lu J, et al. Epidermal growth factor receptor signaling to the mitogen activated protein kinase pathway bypasses Ras in pancreatic cancer cells. Pancreas. 2016;45(2):286–292. doi: 10.1097/MPA.0000000000000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buck E, Eyzaguirre A, Haley JD, Gibson NW, Cagnoni P, Iwata KK. Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol Cancer Ther. 2006;5(8):2051–2059. doi: 10.1158/1535-7163.MCT-06-0007. [DOI] [PubMed] [Google Scholar]
- 25.Feldmann G, Mishra A, Hong S-M, et al. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70(11):4460–4469. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 27.Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-RasG12D. Cancer Discov. 2011;1(2):128–136. doi: 10.1158/2159-8290.CD-10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins MA, Brisset J-C, Zhang Y, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7(12):e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Wei J, Wang N, Ma J-L, Hui PP. The glucosylceramide synthase inhibitor PDMP sensitizes pancreatic cancer cells to MEK/ERK inhibitor AZD-6244. Biochem Biophys Res Commun. 2015;456(3):821–826. doi: 10.1016/j.bbrc.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 30.Khvalevsky EZ, Gabai R, Rachmut IH, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci. 2013;110(51):20723–20728. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloomston M, Zervos EE, Rosemurgy AS., II Matrix metalloproteinases and their role in pancreatic cancer: a review of preclinical studies and clinical trials. Ann Surg Oncol. 2002;9(7):668–674. doi: 10.1007/BF02574483. [DOI] [PubMed] [Google Scholar]
- 32.Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13(12):904–927. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- 33.Shay G, Lynch CC, Fingleton B. Moving targets: emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015;44–46:200–206. doi: 10.1016/j.matbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer—trials and tribulations. Science. 2002;295(5564):2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 35.Kulkarni PS, Haldar MK, Nahire RR, et al. MMP-9 responsive PEG cleavable nanovesicles for efficient delivery of chemotherapeutics to pancreatic cancer. Mol Pharm. 2014;11(7):2390–2399. doi: 10.1021/mp500108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dangi-Garimella S, Krantz SB, Barron MR, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP–mediated expression of HMGA2. Cancer Res. 2011;71(3):1019–1028. doi: 10.1158/0008-5472.CAN-10-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front Biosci. 2007;13:440–452. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 38.Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM. Cellular signalling of the receptor for advanced glycation end products (RAGE) Cell Signal. 2013;25(11):2185–2197. doi: 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Arumugam T, Ramachandran V, Sun D, et al. Designing and developing S100P inhibitor 5-methyl cromolyn for pancreatic cancer therapy. Mol Cancer Ther. 2013;12(5):654–662. doi: 10.1158/1535-7163.MCT-12-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-κB RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5(1):119–127. [PubMed] [Google Scholar]
- 41.Chiao PJ, Ling J, Kras Pten. NF-κB, and inflammation: dangerous liaisons. Cancer Discov. 2011;1(2):103–105. doi: 10.1158/2159-8290.CD-11-0115. [DOI] [PubMed] [Google Scholar]
- 42.Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurzrock R, Li L. Liposome-encapsulated curcumin: in vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis; ASCO Annual Meeting Proceedings; Orlando, FL. 2005; p. 4091. [DOI] [PubMed] [Google Scholar]
- 44.Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-κB–regulated gene products. Cancer Res. 2007;67(8):3853–3861. doi: 10.1158/0008-5472.CAN-06-4257. [DOI] [PubMed] [Google Scholar]
- 45.Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–4499. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 46.Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36(2):124–135. doi: 10.1016/j.tips.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Mueller MT, Hermann PC, Witthauer J, et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137(3):1102–1113. doi: 10.1053/j.gastro.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 48.Morran DC, Wu J, Jamieson NB, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63(9):1481–1489. doi: 10.1136/gutjnl-2013-306202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou J, Wang C, Liu T, et al. Phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway is critical for survival and proliferation of pancreatic cancer stem-like side population cells. Zhonghua Yi Xue Za Zhi. 2008;88(42):2994–2998. [PubMed] [Google Scholar]
- 50.Xu J, Zhang T, Wang T, You L, Zhao Y. PIM kinases: an overview in tumors and recent advances in pancreatic cancer. Future Oncol. 2014;10(5):865–876. doi: 10.2217/fon.13.229. [DOI] [PubMed] [Google Scholar]
- 51.Xu D, Allsop SA, Witherspoon SM, et al. The oncogenic kinase Pim-1 is modulated by K-Ras signaling and mediates transformed growth and radioresistance in human pancreatic ductal adenocarcinoma cells. Carcinogenesis. 2011;32(4):488–495. doi: 10.1093/carcin/bgr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hessmann E, Schneider G, Ellenrieder V, Siveke J. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene. 2015 doi: 10.1038/onc.2015.216. [DOI] [PubMed] [Google Scholar]
- 53.Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Modern Pathol. 2002;15(4):462–469. doi: 10.1038/modpathol.3880547. [DOI] [PubMed] [Google Scholar]
- 54.Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Bonal C, Thorel F, Ait-Lounis A, Reith W, Trumpp A, Herrera PL. Pancreatic inactivation of c-Myc decreases acinar mass and transdifferentiates acinar cells into adipocytes in mice. Gastroenterology. 2009;136(1):309–319. doi: 10.1053/j.gastro.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 56.Jun S, Lee S, Kim H-C, et al. PAF-mediated MAPK signaling hyperactivation via LAMTOR3 induces pancreatic tumorigenesis. Cell Rep. 2013;5(2):314–322. doi: 10.1016/j.celrep.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashimoto M, Ichihara M, Watanabe T, et al. Expression of CD109 in human cancer. Oncogene. 2004;23(20):3716–3720. doi: 10.1038/sj.onc.1207418. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J-M, Murakumo Y, Hagiwara S, et al. CD109 attenuates TGF-β1 signaling and enhances EGF signaling in SK-MG-1 human glioblastoma cells. Biochem Biophys Res Commun. 2015;459(2):252–258. doi: 10.1016/j.bbrc.2015.02.093. [DOI] [PubMed] [Google Scholar]
- 59.Haun RS, Fan C-Y, Mackintosh SG, Zhao H, Tackett AJ. CD109 overexpression in pancreatic cancer identified by cell-surface glycoprotein capture. J Proteomics Bioinform. 2014:S10–S13. doi: 10.4172/jpb.S10-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 61.Zhan H-X, Xu J-W, Wu D, Zhang T-P, Hu SY. Pancreatic cancer stem cells: new insight into a stubborn disease. Cancer Lett. 2015;357(2):429–437. doi: 10.1016/j.canlet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 62.Fitzgerald TL, McCubrey JA. Pancreatic cancer stem cells: association with cell surface markers, prognosis, resistance, metastasis and treatment. Adv Biol Regul. 2014;56:45–50. doi: 10.1016/j.jbior.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Molejon MI, Tellechea JI, Moutardier V, et al. Targeting CD44 as a novel therapeutic approach for treating pancreatic cancer recurrence. Oncoscience. 2015;2(6):572. doi: 10.18632/oncoscience.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Molejon MI, Tellechea JI, Loncle C, et al. Deciphering the cellular source of tumor relapse identifies CD44 as a major therapeutic target in pancreatic adenocarcinoma. Oncotarget. 2015;6(10):7408–7423. doi: 10.18632/oncotarget.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maeda S, Shinchi H, Kurahara H, et al. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br J Cancer. 2008;98(8):1389–1397. doi: 10.1038/sj.bjc.6604307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol. 2008;26(17):2806–2812. doi: 10.1200/JCO.2008.16.6702. [DOI] [PubMed] [Google Scholar]
- 67.Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125(10):2323–2331. doi: 10.1002/ijc.24573. [DOI] [PubMed] [Google Scholar]
- 68.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 69.Xia J, Chen C, Chen Z, Miele L, Sarkar FH, Wang Z. Targeting pancreatic cancer stem cells for cancer therapy. Biochim Biophys Acta. 2012;1826(2):385–399. doi: 10.1016/j.bbcan.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 70.Yao J, An Y, Wie J, et al. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med Wkly. 2011;141:w13208. doi: 10.4414/smw.2011.13208. [DOI] [PubMed] [Google Scholar]
- 71.Feldmann G, Dhara S, Fendrich V, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–2196. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011;31(4):1105–1113. [PubMed] [Google Scholar]
- 73.Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med. 2011;208(10):1931–1935. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mizuma M, Rasheed ZA, Yabuuchi S, et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther. 2012;11(9):1999–2009. doi: 10.1158/1535-7163.MCT-12-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hage C, Rausch V, Giese N, et al. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013;4(5):e627. doi: 10.1038/cddis.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li C, Wu JJ, Hynes M, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218–2227. doi: 10.1053/j.gastro.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 77.Tyagi N, Marimuthu S, Bhardwaj A, et al. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016;370(2):260–267. doi: 10.1016/j.canlet.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dangi-Garimella S, Krantz SB, Shields MA, Grippo PJ, Munshi HG. Epithelial-mesenchymal transition and pancreatic cancer progression. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): Transworld Research Network; 2012. [PubMed] [Google Scholar]
- 79.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 80.Barriere G, Fici P, Gallerani G, Fabbri F, Rigaud M. Epithelial mesenchymal transition: a double-edged sword. Clin Transl Med. 2015;4(1):14. doi: 10.1186/s40169-015-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang J-H, Liu C, Cheng H, et al. Epithelial–mesenchymal transition in pancreatic cancer: Is it a clinica lly significant factor? Biochim Biophys Acta. 2015;1855(1):43–49. doi: 10.1016/j.bbcan.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 82.Rasheed ZA, Yang J, Wang Q, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102(5):340–351. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Javle M, Li Y, Tan D, et al. Biomarkers of TGF-β signaling pathway and prognosis of pancreatic cancer. PLoS One. 2014;9(1) doi: 10.1371/journal.pone.0085942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Drabsch Y, Ten Dijke P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012;31(3–4):553–568. doi: 10.1007/s10555-012-9375-7. [DOI] [PubMed] [Google Scholar]
- 85.Schlingensiepen KH, Jaschinski F, Lang SA, et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011;102(6):1193–1200. doi: 10.1111/j.1349-7006.2011.01917.x. [DOI] [PubMed] [Google Scholar]
- 86.Gaspar NJ, Li L, Kapoun AM, et al. Inhibition of transforming growth factor β signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol. 2007;72(1):152–161. doi: 10.1124/mol.106.029025. [DOI] [PubMed] [Google Scholar]
- 87.Oyanagi J, Kojima N, Sato H, et al. Inhibition of transforming growth factor-β signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp Cell Res. 2014;326(2):267–279. doi: 10.1016/j.yexcr.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 88.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6(5):376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 89.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 90.Volinia S, Calin GA, Liu C-G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rachagani S, Macha MA, Heimann N, et al. Clinical implications of miRNAs in the pathogenesis, diagnosis and therapy of pancreatic cancer. Adv Drug Deliv Rev. 2015;81:16–33. doi: 10.1016/j.addr.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J Gastrointest Surg. 2008;12(12):2171–2176. doi: 10.1007/s11605-008-0584-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ali S, Banerjee S, Logna F, et al. Inactivation of Ink4a/Arf leads to deregulated expression of miRNAs in K-Ras transgenic mouse model of pancreatic cancer. J Cell Physiol. 2012;227(10):3373–3380. doi: 10.1002/jcp.24036. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Kawaguchi T, Komatsu S, Ichikawa D, et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br J Cancer. 2013;108(2):361–369. doi: 10.1038/bjc.2012.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morton JP, Timpson P, Karim SA, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci. 2010;107(1):246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brunetti O, Russo A, Scarpa A, et al. MicroRNA in pancreatic adenocarcinoma: predictive/prognostic biomarkers or therapeutic targets? Oncotarget. 2015;6(27):23323–23341. doi: 10.18632/oncotarget.4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial–mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–3295. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neilsen PM, Noll JE, Mattiske S, et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene. 2013;32(24):2992–3000. doi: 10.1038/onc.2012.305. [DOI] [PubMed] [Google Scholar]
- 99.Wang P, Chen L, Zhang J, et al. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene. 2014;33(4):514–524. doi: 10.1038/onc.2012.598. [DOI] [PubMed] [Google Scholar]
- 100.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors, ZEB1 and ZEB2. Genes Dev. 2008 doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 102.Conroy T, Bachet J-B, Ayav A, et al. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur J Cancer. 2016;57:10–22. doi: 10.1016/j.ejca.2015.12.026. [DOI] [PubMed] [Google Scholar]
- 103.Ballehaninna UK, Chamberlain RS. Biomarkers for pancreatic cancer: promising new markers and options beyond CA 19-9. Tumour Biol. 2013;34(6):3279–3292. doi: 10.1007/s13277-013-1033-3. [DOI] [PubMed] [Google Scholar]
- 104.Ballehaninna UK, Chamberlain RS. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: an evidence based appraisal. J Gastrointest Oncol. 2012;3(2):105–119. doi: 10.3978/j.issn.2078-6891.2011.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the conko-001 randomized trial. JAMA. 2013;310(14):1473–1481. doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 106.Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol. 2007;25(3):326–331. doi: 10.1200/JCO.2006.07.5663. [DOI] [PubMed] [Google Scholar]
- 107.Li Q, Yan H, Liu W, Zhen H, Yang Y, Cao B. Efficacy and safety of gemcitabine-fluorouracil combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomized controlled trials. PLoS One. 2014;9(8):e104346. doi: 10.1371/journal.pone.0104346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Burris HA, III, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 110.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 111.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patra CR, Bhattacharya R, Wang E, et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008;68(6):1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 113.Yu S, Lu Z, Liu C, et al. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010;70(14):6015–6025. doi: 10.1158/0008-5472.CAN-09-4531. [DOI] [PubMed] [Google Scholar]
- 114.Jiao LR, Frampton AE, Jacob J, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS One. 2012;7(2):e32068. doi: 10.1371/journal.pone.0032068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31(10):1726–1733. doi: 10.1093/carcin/bgq160. [DOI] [PubMed] [Google Scholar]
- 116.Sureban SM, May R, Lightfoot SA, et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011;71(6):2328–2338. doi: 10.1158/0008-5472.CAN-10-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7(16):2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 118.Tsuda N, Ishiyama S, Li Y, Ioannides CG, Abbruzzese JL, Chang DZ. Synthetic microRNA designed to target glioma-associated antigen 1 transcription factor inhibits division and induces late apoptosis in pancreatic tumor cells. Clin Cancer Res. 2006;12(21):6557–6564. doi: 10.1158/1078-0432.CCR-06-0588. [DOI] [PubMed] [Google Scholar]
- 119.Kent OA, Fox-Talbot K, Halushka MK. RREB1 repressed miR-143/145 modulates KRAS signaling through downregulation of multiple targets. Oncogene. 2013;32(20):2576–2585. doi: 10.1038/onc.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kulshreshtha R, Ferracin M, Wojcik SE, et al. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27(5):1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 2010;126(1):73–80. doi: 10.1002/ijc.24687. [DOI] [PubMed] [Google Scholar]
- 122.Park J-K, Henry JC, Jiang J, et al. miR-132 and miR-212 are increased in pancreatic cancer and target the retinoblastoma tumor suppressor. Biochem Biophys Res Commun. 2011;406(4):518–523. doi: 10.1016/j.bbrc.2011.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harsha HC, Kandasamy K, Ranganathan P, et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009;6(4):e1000046. doi: 10.1371/journal.pmed.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Szajda SD, Waszkiewicz N, Chojnowska S, Zwierz K. Carbohydrate markers of pancreatic cancer. Biochem Soc Trans. 2011;39(1):340–343. doi: 10.1042/BST0390340. [DOI] [PubMed] [Google Scholar]