

Abstract
Adenoviruses represent the most widely used viral-vectored platform for vaccine design, showing a great potential in the fight against intracellular infectious diseases to which either there is a lack of effective vaccines or the traditional vaccination strategy is suboptimal. The extensive understanding of the molecular biology of adenoviruses has made the new technologies and reagents available to efficient generation of adenoviral-vectored vaccines for both preclinical and clinical evaluation. The novel adenoviral vectors including nonhuman adenoviral vectors have emerged to be the further improved vectors for vaccine design. In this review, we discuss the latest adenoviral technologies and their utilization in vaccine development. We particularly focus on the application of adenoviral-vectored vaccines in mucosal immunization strategies against mucosal pathogens including Mycobacterium tuberculosis, flu virus, and human immunodeficiency virus.
Introduction
Since their discovery nearly half a century ago, adenoviruses have quickly become attractive vectors for vaccine development. Adenoviruses originally showed tremendous promise as gene replacement therapy vectors due to their ability to be easily genetically modified, their excellent safety profiles, broad tissue tropism, and ability to drive robust, sustained transgene expression.1 Their efficacy in clinical settings was unfortunately short-lived due to the robust host innate and adaptive immune responses against adenoviral and transgene products shortly after their administration. The robust immunogenicity of adenoviruses, combined with their favorable safety profiles, however, prompted a rapid transition of adenoviruses from being the gene therapeutic vector to the widely used vaccine platform to-date.2
Adenoviruses are efficacious vaccine vectors against diseases in which traditional vaccine development strategies have proven ineffective.3 These include, but not limited to, diseases such as tuberculosis (TB) and the human immunodeficiency virus (HIV).4 Adenoviral-based vaccines also have the potential to eventually replace existing vaccine platforms which are either financially and/or technically challenging to generate (such as the seasonal influenza vaccine), and less efficacious platforms which have to be administered multiple times in order to engender protective immunity (such as the rabies vaccine).4
In this review we discuss the most often used adenoviral-based vaccine technologies and their pre-clinical and clinical development in the fight against mucosal pathogens. We focus on three mucosal pathogens, Mycobacterium tuberculosis, influenza, and HIV, given the high global prevalence, unmet vaccine needs, and well-documented effort in preclinical and clinical vaccine development using adenoviral technology for these pathogens.
Advantages of Adenoviruses as Platforms for Vaccine Design
Adenoviruses possess the features which make them favorable platforms for vaccine design. Firstly, recombinant adenoviral vectors have excellent safety records which are apparent by their initial use as vectors for gene therapy in humans.5 Combined with their broad tissue tropism, adenoviral vectors have been widely explored for developing vaccines desired to be delivered via the respiratory mucosal route against mucosal pathogens such as M. tuberculosis and influenza.6,7 Secondly, adenoviruses are highly immunogenic, capable of driving robust, long-lasting immune responses to vector-encoded antigens. Adenoviruses are particularly effective in inducing potent CD8, and to a lesser extent, CD4 T-cell responses.4,8 This makes them attractive platforms for the vaccines against intracellular pathogens to which cellular immune responses are indispensable for protection.4 The intense interest in using adenoviral vectors for vaccine design is not restricted to only using the human serotype 5 adenovirus (AdHu5) or other human serotypes. Nonhuman adenoviruses are being increasingly explored for vaccine development. The following sections discuss the tools and procedures available for the generation of such vectors.
Molecular Biology of Adenoviruses
Adenoviruses are species-specific with multiple serotypes in each species. Adenoviruses are further classified into different subgroups based on genetic homology between serotypes.9 Currently 57 different human adenovirus serotypes have been identified, being classified into 7 subgroups.9,10 Of these, AdHu5, a group C adenovirus, is both the most common serotype to infect humans and the most widely used serotype for recombinant vaccine design owing to its superior immunogenicity in comparison to other human adenovirus serotypes.2,11 However, other human adenovirus serotypes and nonhuman adenoviruses have increasingly been used for vaccine design due to the global prevalence of preexisting anti-AdHu5 immunity in humans which has been clinically seen to negatively impact vaccine efficacy.12–14
Adenoviruses are double-stranded DNA viruses with linear genomes ranging between 34 and 43kbp in size. Both linear strands of the viral genome code for viral polypeptides thereby compensating for the viruses’ relatively compact genome size. The genome encodes five early gene transcription units (dictated as E1a, E1b, E2, E3, and E4) and a single late transcription unit that is subdivided into L1 through L5 (Figure 1). The early transcription units play critical roles in viral DNA replication and evasion of host immunosurveillance while the late transcription units primarily encode viral structural components.10 Readers are directed to ref. 10 for a detailed review on the molecular biology of adenoviruses.
Figure 1.
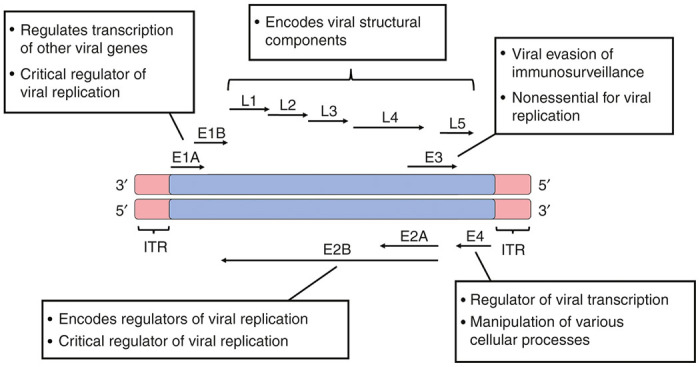
Organization and function(s) of the adenoviral genome. The organization, orientation, and broad function(s) of the adenoviral transcriptional units are depicted. Knowledge of the entire adenoviral genome is critical in the downstream construction of adenoviral-vectored vaccines.
Generation of Recombinant Adenoviruses
Recombinant adenoviral vaccines are generated through the insertion of a transgene cassette into the adenoviral backbone. Transgene cassettes are constructed to express one or more foreign antigens-of-interest under the control of a given promoter (usually the cytomegalovirus (CMV) promoter due to its ability in driving robust and sustained transgene expression). Depending on numerous factors such as the size, composition, or complexity and the expression pattern of the chosen promoter, different promoters may be utilized.15–17 Readers are directed to refs. 15–17 for other commonly used promoters.
Adenoviruses are able to efficiently package 105% of their original genome in viable virion before becoming unstable.18 This allows for up to a 2,000-bp transgene cassette to be inserted into the adenoviral backbone. As regulatory elements within transgene cassettes (such as the promoter) can encompass over 1,000 bp in size, this can limit the size and number of heterologous antigens. As such, the adenoviral genome must be manipulated to accommodate larger transgene cassettes. The majority of recombinant adenoviral vaccine vectors are known as first-generation vectors as they are deficient in the E1 transcription unit. Deletion of E1, which is critical in viral replication, not only bolsters the safety of adenoviral vectors, but further enhances vector capacity allowing for accommodation of transgene cassettes up to 5,000 bp in size.18,19 A majority of first generation vectors are also deficient in E3, further increasing the potential size of transgene cassettes to 7,500 bp in size.18,19 The deletion of multiple transcription units provides several advantages. For instance, it enhances vector capacity for generating multivalent vaccines against complex pathogens. Secondly, it reduces the number of viral-encoded antigens, resulting in a diminished magnitude of antivector immunity.
Adenoviral vectors can be molecularly designed to lack all viral genes and only express essential elements for viral replication and packaging. These gutted vectors are recoverable with the use of helper adenoviruses which provide in trans complementation of the viral genes needed to properly package the gutted adenovirus.1,17 Such vectors are not only highly flexible, capable of expressing transgene cassettes up to 36 kbp in size, but due to the lack of viral genes, induce drastically reduced antivector immunity, thus allowing for longer and more efficient transgene expression than the first-generation vectors.20 These vectors also have favorable safety profiles following the respiratory mucosal delivery as shown in large animal models.21 As such, such gutted adenoviral vectors continue to represent promising platforms for gene replacement therapy. However, their application for vaccination is limited due to their reduced immune adjuvant effects,22 the technical difficulty in their large-scale production, and the potential negative effect on quality memory immune responses resulting from prolonged high levels of transgene-encoded immunogens.
There are the two main strategies currently utilized in the design and development of recombinant adenoviral vectors.23 The first involves homologous recombination between the adenoviral genome and a transgene cassette-expressing shuttle plasmid within mammalian systems. The second involves directly cloning a transgene cassette into the adenoviral backbone, bypassing the need for homologous recombination.
Method 1: homologous recombination in mammalian systems
The most widely used method in adenoviral vector development involves homologous recombination between a shuttle plasmid carrying a given transgene cassette and the adenovirus backbone.18 This strategy is relatively straightforward and can be easily performed with a variety of commercially available protocols, reagents, and kits.
Generation of adenoviral vectors is commonly carried out in mammalian cell lines. A shuttle plasmid is constructed to express the 5′ end of the adenoviral genome in which the E1 transcription unit is replaced with a transgene cassette.18,19 This shuttle plasmid is transfected into a mammalian system alongside either a rescue plasmid DNA containing an E1-deleted adenoviral genome which is designed to slightly overlap with the 3′ region of the shuttle plasmid or the wild-type adenoviral genome with cleavages in the E1 region. Removal/cleavage of E1 greatly reduces the risk of generating replication-competent viruses.18 Within the mammalian system, homologous recombination between the shuttle plasmid and the adenoviral vector leads to incorporation of the transgene cassette into the adenoviral backbone (Figure 2). The shuttle plasmid can also be generated in such a way that the E3 transcription unit is replaced with a given transgene cassette.18
Figure 2.
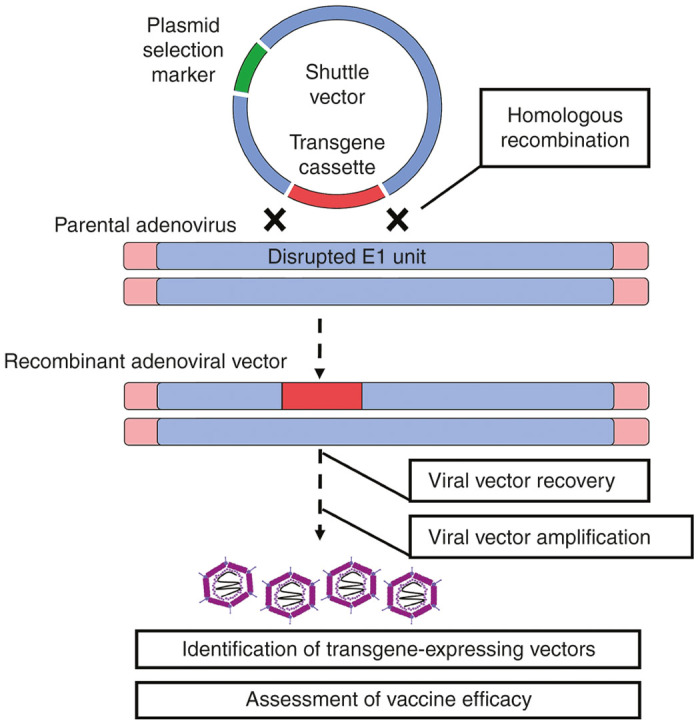
Development of adenoviral-vectored vaccines through homologous recombination. The most commonly used strategy in the generation of E1-deleted human adenoviral vectors (homologous recombination in a viral packaging cell line) is depicted.
As the lack of E1 renders recombinant adenoviral vectors replication-deficient, vector amplification and propagation must be done within cell lines which can provide E1 in trans. The HEK 293 cell line, a modified human embryonic kidney cell line, constitutively expresses E1 from AdHu5 has been the staple workhorse in the generation of recombinant adenoviral vectors for both preclinical and clinical applications.24 The 293 cell line, however, is not without its shortcomings. Firstly, the E1 protein expressed by this cell line is from AdHu5. As such, this cell line cannot fully complement the recovery of all non-AdHu5 adenoviruses. This is particularly evident for group B adenoviruses (such as AdHu35) and certain nonhuman adenoviruses shown poorer recovery yields when propagated in 293 cells.25 This, however, can be overcome by two methods: (i) transcomplementing the E1 protein specific to the adenovirus through transfecting the cell line with an additional vector expressing the adenovirus-unique E1 protein, and (ii) using a different cell line. Readers are directed to ref. 24 for a comprehensive review on the various available adenoviral producer cell lines.
Furthermore, propagation of E1-deficient adenoviral vectors on the 293 cell line can lead to a small degree of contamination with replication-competent adenovirus, which may be one of the questions to address when preparing such vaccines for human applications.24,26 The main cause of replication-competent adenovirus (which is particularity more prevalent for AdHu5 vectors) is due to significant homology between the regions flanking E1 expressed by the cell line and the majority of Ad vectors developed via homologous recombination.26 The commercially available Per.C6 cell line is an alternative cell line for the clinical development of adenoviral vectors as it reduces the risk of replication-competent adenovirus formation significantly.24
Homologous recombination in mammalian systems is not highly efficient which can lead to generation of recombinant viral vectors which lack transgene cassette expression. As such, multiple rounds of viral purification are required to recover and amplify recombinant vectors which properly express the encoded transgene. This can be partly circumvented through implementation of selection strategies (such as the use of β-galactosidase or florescent markers27,28), but are not ideal for development of clinical-grade vectors. Issues relating to ineffective recombination have also been addressed through utilization of bacterial systems (such as the pAdEasy system for AdHu5 vector development) but this strategy is technically more challenging and also more restricted in terms of the adenovirus serotypes available.29–31 Readers are directed to refs. 29–31 for detailed reviews and protocols of homologous recombination in bacterial systems for adenoviral vector development.
Method 2: direct molecular cloning of the adenoviral genome
Despite the widely used homologous recombination method, reconstruction of the entire adenoviral genome into a plasmid represents an alternative strategy which can address some of the issues associated with homologous recombination.
This strategy involves the molecular cloning of the entire adenoviral genome into a plasmid vector.32 Vectors generated this way can be altered in vitro, allowing for direct cloning of transgene cassettes into the adenoviral backbone. Although this strategy is technically more challenging as it requires access to a wider array of molecular biology reagents and techniques, it has multiple advantages. Firstly, transcription units can be easily omitted during vector construction and replaced with cloning (linker) regions. This allows users to develop recombinant vectors which can accommodate multiple transgene cassettes of varying sizes (as per user’s discretion).33 Secondly, it is a one-step straightforward way to rescue the virus without relying on the chances of homologous recombination. Thirdly, this strategy has significant clinical implications as the molecular cloning of the entire adenoviral genome into a plasmid ensures that potential infectious contaminants which may be present in an original adenoviral preparation used for homologous recombination method are completely eliminated.27 This is particularly useful in the development of nonhuman adenoviruses which are originally isolated from tissues that may harbor unknown or undetectable pathogens. Detailed protocols outlining this method are available in refs. 27,32,33. Figure 3 outlines this strategy. Readers are directed to ref. 33 for an up-to-date and in-depth protocol in the generation of both recombinant human and chimpanzee adenoviral vectors.
Figure 3.
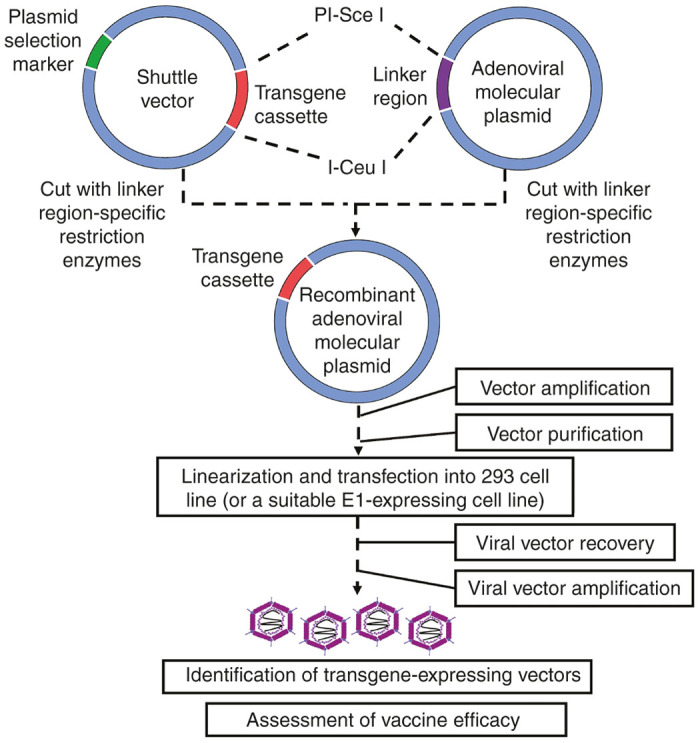
Development of adenoviral-vectored vaccines through a direct-cloning strategy. Generation of adenoviral vectors through direct in vitro molecular cloning of the entire adenovirus is depicted. This represents an alternative strategy in the generation of adenoviral vectors, bypassing the need for homologous recombination.
Molecular cloning of the entire adenoviral genome into a plasmid requires a complete and accurate sequence map of the original viral genome as the location and rarity of restriction sites are critical in vector construction. This strategy is technically challenging due to the large size of the adenoviral genome and the limited number of genome-wide unique restriction sites to be used for vector reconstruction.32 Circumvention of this issue involves cloning the entire adenoviral genome in a step-by-step approach through exploitation of restriction sites which are exclusive to given sections of the adenoviral genome. Adenoviral transcription units which are omitted during vector reconstruction may be replaced by linker sequences that are used for the insertion of transgene cassettes. These linker sites are composed of rare restriction cut sites which are not found within the adenoviral genome. Cut sites such as PI-Sce I and I-Ceu I have been traditionally used due to their absence in a variety of human and nonhuman adenoviruses.33
Following construction of the adenoviral plasmid, a shuttle vector is engineered to express a given transgene cassette with similar restriction sites as those found within the linker sequences. The shuttle vector can be amplified and the transgene cassette subsequently be excised from the shuttle vector and directly ligated into the adenoviral plasmid using standard molecular cloning techniques.33 The recombinant adenoviral plasmid can subsequently be transformed into a suitable bacterial cell line and amplified, and then inserted into a viral packaging cell line for recovery.
Production of Clinical-Grade Recombinant Adenovirus-Based Vaccines
Generation of vectored vaccines that meet the quality and quantity demands for clinical trials can be met with multiple logistical and financial roadblocks. These include the feasibility in vector scaling, availability of proper equipment and facilities, and optimized/standardized Good Manufacturing Practices protocols. Due to the extensive clinical history of adenoviral-vectored therapies (ranging from gene therapy to modern vaccine trials), the ability to scale-up and purify human or nonhuman adenoviral-based vectors from preclinical to clinical studies has become a much more standardized practice compared with other viral vectors.34 This is made possible by the wide availability of quality-controlled cell lines which generate high titre viral batches and of highly scalable clinical-grade purification strategies (such as ion-exchange or size-exclusion chromatography) which allow for generation of high purity and quality vaccine stocks.35,36 The strategies in generating such clinical-grade vectors are well described in refs. 34–36.
Preclinical and Clinical Development of Adenovirus-Based Vaccines Against Tuberculosis
The development of vaccines against TB remains a daunting task with the only clinically approved vaccine, Bacillus Calmette Guerin, failing to protect against the pulmonary form of the disease. With over nine million new cases of active disease and over 1½ million deaths every year, there has been a dire need for the development of improved TB vaccine platforms.37
It is well characterized that protection against pulmonary TB requires the establishment of long-lasting adaptive cellular immune responses at the respiratory mucosa.7,38 Preclinical and human epidemiological studies have shown the importance of type-1 helper CD4+ T cell (Th1) and CD8+ T-cell responses in anti-TB immunity through the production of type-1 cytokines such as interferon γ (IFNγ) and tumor necrosis factor alpha (TNFα), and their cytotoxic effector functions.39 Importantly, such immune responses must be established at the primary site of infection, the lungs, to be protective. As immunization route determines the anatomical location of vaccine-specific T cells, numerous studies have shown that immunization via the respiratory mucosal route is desirable for effective anti-TB immunity.40–44 Collectively these findings suggest that the success of future TB vaccines relies on their ability to induce long-lasting multifunctional T-cell responses situated at the respiratory mucosa before or shortly after M. tuberculosis exposure.
Adenoviruses are one of the most widely used viral-vectored platforms for TB vaccine development. This is not only due to their ability to induce robust T-cell responses, but also due to their natural tropism to the respiratory mucosa, which when combined with their established safety profiles, makes them highly amenable for respiratory mucosal vaccination. A number of human adenovirus serotypes, as well as two recently developed recombinant chimpanzee adenovirus-based TB vaccines are currently under evaluation in both preclinical and clinical settings.
AdHu5-based TB vaccines
AdHu5 is the most immunogenic and utilized serotype for TB vaccines.11 One of the best-characterized AdHu5-based vaccine candidates is a first-generation (E1/E3-deficient) recombinant AdHu5 vector expressing an immunodominant M. tuberculosis antigen 85A under the control of the cytomegalovirus promoter (referred to as AdHu5Ag85A).40,41,45
AdHu5Ag85A has been extensively characterized as a respiratory mucosal vaccine in multiple animal models, ranging from murine models for basic immunogenicity and protective efficacy studies to nonhuman primate models which represent the most clinically relevant model for assessing TB vaccines.41,44,46,47 Multiple studies have collectively shown that respiratory mucosal immunization with AdHu5Ag85A is safe, even in immune-compromised hosts, highlighting the potential use of such first-generation vectors in immunocompromised populations.48
Murine studies have shown that respiratory mucosal immunization with AdHu5Ag85A was immunogenic, inducing robust cytotoxic IFNγ+TNFα+CD8+ T cell, and to a lesser extent CD4+ T-cell responses directly at the respiratory mucosa.41,43 The effector memory CD8+ T cells induced by this strategy persisted at the respiratory mucosa and in an antigen-dependent manner for many months.43 Alongside murine protections studies, guinea pig, bovine, and a recently published nonhuman primate studies have shown significantly greater bacterial control and survival of animals immunized via the respiratory mucosal route with AdHu5Ag85A.41,46,47,49 The potential of AdHu5Ag85A has been further supported by a recently completed phase-1 clinical trial where intramuscular administration of AdHu5Ag85A induced polyfunctional CD4+ and CD8+ T-cell responses particularly in Bacillus Calmette Guerin+ human volunteers.50 A second clinical trial assessing the safety and immunogenicity of AdHu5Ag85A delivered by inhaled aerosol is to be launched soon in Canada.
Despite their efficacy as vaccine vectors, such (and other) AdHu5-based vaccines are not without their inherent limitations. Pre-existing immunity to the adenoviral backbone may dampen the potency of adenoviral-based vaccines.13 Such preexisting AdHu5 immunity is particularly prevalent in the TB endemic regions, which has been seen to limit the efficacy AdHu5-based vaccines.14,51,52 Although utilization of higher vaccine doses can overcome this limitation, this situation may limit its respiratory mucosal application in humans where the smallest safe effective doses are desired. Such considerations have prompted the development of adenovirus-vectored vaccines based on rarer, less prevalent human serotypes or nonhuman adenoviral species.
Human serotype 35 adenovirus-based TB vaccines
One approach designed to circumvent preexisting anti-AdHu5 immunity is to use rare human serotypes that humans have low global preexisting immunity against.53 Human adenovirus serotype 35 (AdHu35), a group B adenovirus, represents such a vector which is currently being assessed as a platform for TB vaccine design.25
rAd35-TBS is currently the most advanced recombinant AdHu35-based TB vaccine. Based on an E1/E3-deleted AdHu35 vector, this vaccine takes advantage of the genetic plasticity of adenoviral vectors and expresses three different M. tuberculosis antigens under the control of the cytomegalovirus promoter.25,54 Similar to AdHu5Ag85A, this vaccine has also been evaluated as a respiratory mucosal vaccine.55 The studies in nonhuman primate models showed its favorable safety profile and its ability to elicit long-lived antigen-specific immune responses within the respiratory mucosa. Both CD4+ and CD8+ T cells elicited by vaccination were polyfunctional, secreting IFN-γ+, TNF-α+, and IL-2+, with CD8+ T cells possessing cytotoxic activity, as measured by granzyme B staining.55,56 Hokey et al. further studied rAd35-TBS following multiple homologous boost immunizations showing repeated aerosol vaccinations to be well tolerated and elicit the persisting polyfunctional antigen-specific T cells in the lung. It is important to note that in the majority of these studies, rAd35-TBS had to be administered repeatedly, indicative of the poor immunogenicity of the AdHu35 vector, relative to the AdHu5 vector. As a result, following intranasal vaccination it induced only a moderate level of protection in a murine model54 and provided no protection in aerosol-vaccinated nonhuman primate.55
rAd35-TBS has also been assessed clinically in phase-1 clinical trials for safety and immunogenicity in Bacillus Calmette Guerin-immunized humans. These studies show that intramuscular administration of this vaccine was safe in both infants and HIV− and HIV+ adults and induced polyfunctional CD4+ and CD8+ T-cell immune responses.57–61 Again, such immune responses were only measurable following repeated high-dose administrations of the vaccine.
Recent work has shown the importance of considering the effect of TB vaccines on the innate immune responses as they can have drastic impact on the immunogenicity and efficacy of vaccines. In this regard, the AdHu35 vector was found to induce type-1 interferons which in turn suppressed T-cell activation,62,63 providing a mechanism for the poor immunogenicity of AdHu35 vectors. Another independent study compared AdHu5 with VSV-based TB vaccines in respiratory mucosal boost vaccination. This study shows that the AdHu5-vectored vaccine-induced little type-1 IFN responses and conferred significantly enhanced protection in comparison to the VSV vaccine, which induced high levels of type-1 IFN responses, resulting in heightened IL-10 production and decreased anti-TB activities in infected APCs.64 Type-1 IFNs are also employed by M. tuberculosis to dampen the Th1 immunity.65,66 Collectively, these findings indicate the importance of careful selection of viral vectors for TB vaccine development and the detrimental activities of type-1 IFNs in anti-TB immunity. Readers are directed to ref. 11 for an excellent overview of the immunogenicity of a variety of human and nonhuman adenoviral vectors.
Chimpanzee adenovirus-based TB vaccines
With advancements in the molecular tools and developmental strategies available to generating nonhuman adenoviruses, chimpanzee adenoviruses have emerged as attractive platforms for vaccine design.33,67 The growing popularity of chimpanzee adenoviruses as vaccine vectors is associated with their ability to bypass the negative impact of preexisting antihuman adenovirus immunity while inducing immune responses that are similar to or more potent than those elicited by their human adenoviral counterparts.11,12,51
Currently two chimpanzee adenovirus-based TB vaccines including AdCh68Ag85A and ChAdOx1.85A are under evaluation, both designed to express an M. tuberculosis antigen Ag85A.68,69
AdCh68Ag85A, which was developed based on chimpanzee adenovirus serotype 68, expresses the same transgene cassette as AdHu5Ag85A and has been recently assessed for its anti-TB efficacy in the murine model. The study shows that respiratory mucosal immunization with this vaccine was safe, and similar to its AdHu5 counterpart, induced minimal type-1 IFN responses.69 AdCh68Ag85A induced significantly greater and more persisting antigen-specific T-cell responses within the lungs than AdHu5Ag85A. Although similar levels of protection were seen following immunization with AdHu5Ag85A, protection from M. tuberculosis-induced lung pathology varied between the two vaccines. The lungs from animals vaccinated with AdCh68Ag85A showed less pathology as indicated by a drastic reduction in the formation of granulomatous regions (a hallmark of pulmonary TB infection) in comparison to AdHu5Ag85A-vaccinated animals. Furthermore, when assessed in the context of preexisting anti-AdHu5 immunity, the protective efficacy of AdCh68Ag85A was maintained. This finding is particularly important as preexisting anti-AdHu5 immunity is globally prevalent and represents a potential roadblock to the clinical success of AdHu5-vectored vaccines.
A recent study assessing another chimpanzee adenoviral-vectored TB vaccine, ChAdOx1.85A, shows that although respiratory mucosal immunization with ChAdOx1.85A failed to significantly enhance anti-TB protection in Bacillus Calmette Guerin-primed animals, and enhanced protection when combined in a booster regimen with MVA85A.68
Preclinical and Clinical Development of Adenovirus-Based Vaccines Against Influenza
Seasonal epidemics of influenza remain a huge challenge to public health worldwide. It is estimated that epidemics of influenza result in three to five million severe cases and 250,000−500,000 deaths globally each year. Vaccination has been the primary prophylactic strategy against influenza infection.
Optimal protection against influenza viral infection requires both humoral and cellular-mediated immunity. Antibodies specific for the best-known antigenic determinants in influenza virus, namely hemagglutinin (HA), neuraminidase (NA), and/or matrix proteins, are critically required for blocking virus attachment to the host cell, preventing the viral release, and interfering with virus assembly, respectively.70 T cells specific for conserved influenza viral components are required for the clearance of virally infected cells. Therefore, ideal influenza vaccines are expected to induce both neutralizing antibodies and T cellular-mediated immunity.
Conventional influenza vaccines include inactivated (intramuscular flu vaccine) and live-attenuated (nasal spray vaccine) vaccines which are derived from virulent viruses identified in previous epidemics which have proved to be protective. However, due to its error-prone polymerase, the influenza virus is characterized by its frequent antigenic drift in HA and to a less extent NA which facilitates its evasion from preexisting antibodies and memory T cells induced by prior immunizations.70 As a result, such prediction-based flu vaccination strategy may not always be reliable and the overall protective efficacy is only about 50–60%. For these reasons, there is a need to develop further improved influenza vaccination strategies.
Novel strategies for the generation of influenza vaccines have been proposed, including recombinant proteins, virus-like particles, viral vectors, and DNA-based vaccines. These novel strategies facilitate the availability of candidate vaccines shortly after the genetic sequence of a new dominant influenza virus is determined early in a given epidemics.70 Among these novel strategies for generating influenza vaccines, replication-deficient human and chimpanzee adenovirus vectors are promising platforms.
Human adenovirus-based influenza vaccines
In murine models, human adenovirus-vectored influenza vaccines induced both humoral and cell-mediated immune responses against influenza virus-encoded antigens.71 One well known vaccine, a replication-defective human adenoviral-vectored vaccine expressing H5 from the avian H5N1 influenza virus (H5HA), when administered either intramuscularly or intranasally was seen to induce comparable titers of circulating neutralizing antibodies as compared with an adjuvanted recombinant H5HA protein-based vaccine. Importantly, intranasal or intramuscular immunization with this vaccine induced higher frequencies of IFN-γ+ CD8+ T cells in the spleen, as compared with the protein plus adjuvant equivalent.71 These results suggest that adenoviral-vectored influenza vaccines might confer superior protection over protein-based ones as the former is better in CD8+ T-cell priming. Importantly, intramuscular and intranasal immunization with this adenoviral-vectored vaccine comparably protected against lethal challenge with H5N1, presumably due to the preferential generation of lung resident memory T cells.
In humans, nasal and epicutaneous vaccination with a replication-defective AdHu5 vector encoding the A/PR/8/34 H1N1 influenza virus HA (referred to as AdCMV-PR8.ha) was shown to be safe and immunogenic in a graded-dose phase-1 clinical trial both following primary and booster immunization.72 Immunogenicity of nasal vaccination was demonstrated by the induction of serum hemagglutination-inhibition antibodies. More importantly, nasal vaccination induced significantly higher frequencies of seroconversion compared with subcutaneous vaccination following both primary and booster immunization even though the dose of nasal vaccine was around 10% of the subcutaneous dose.72 These findings suggest that respiratory mucosal immunization with human adenovirus-vectored influenza vaccines may be of superior safety and efficacy, though it is unknown in this study to which extent the T cells were activated following mucosal immunization.
A more recent phase-1 clinical trial, however, evaluated the safety and immunogenicity of a replicating human serotype 4 adenovirus-vectored influenza vaccine expressing the HA from H5N1 (Ad4-H5-Vtn) administered via oral route as a priming vaccine that was followed by parenteral H5N1 boosting vaccination.73 The cumulative frequency of such adverse events as abdominal pain, diarrhoea, and nasal congestion was significantly higher than placebo group, though no serious treatment-related events occurred. Oral Ad4-H5-Vtn priming immunization induced cellular responses in the peripheral blood, as demonstrated by IFN-γ and interleukin-2 (IL-2) enzyme-linked immunospot assay (ELISPOT). However, the haemagglutination-inhibition seroconversion of all dose levels was only 11% following three repeated doses of the priming vaccine, as compared with that of 7% in placebo group. Following H5N1 boosting vaccination, 80% of prime-vaccinated subjects had seroconversion, which was significantly higher than that of 36% in placebo group. Preexisting anti-AdHu4 immunity dampened Ad4-H5-Vtn-induced cellular responses against H5 antigen, as well as seroconversion after boost vaccination, though only in low priming dose groups (107, 108, and 109 viral particles). This study suggests a potential new paradigm where adenovirus vectored priming vaccine may be used in combination with conventional seasonal influenza vaccination to enhance the immunogenicity and possibly the efficacy of the latter.
In addition to these published studies, there are recently completed or ongoing phase-1 clinical trials evaluating replication-deficient AdHu5-vectored (ClinicalTrials.gov Identifier: NCT00755703) and a replicating human Ad4-vectored influenza vaccines (ClinicalTrials.gov Identifier: NCT01806909). Notably, both adenovirus vectors were administered intranasally in both clinical trials, which together with the above published studies, suggest that the respiratory mucosal route is increasingly recognized as the preferred route to deliver human adenovirus-vectored influenza vaccines. However, the potential impact of preexisting anti-human adenoviruses immunity on the vaccine potency remains to be fully appreciated in future studies.
Chimpanzee adenovirus-based influenza vaccines
As mentioned previously, the high global prevalence of preexisting anti-human adenovirus immunity adversely impacts the efficacy of human adenoviral-based vaccines.13 As such, chimpanzee adenovirus vectors represent more promising and efficacious platforms for the development of future clinical adenoviral-based influenza vaccines. A chimpanzee serotype 7 adenovirus-vectored vaccine expressing the NP from the H1N1 strain A/PR/8/34 (referred to as AdCh7-NP) has been assessed in a murine model.74 Compared with an AdHu5-vectored vaccine expressing the same NP (AdH5-NP), the AdCh7-NP induced comparable T-cell immune responses against NP, and was as protective against a lethal dose of H1N1 strain A/PR/8/34. In heterosubtype challenge experiments with H5N1 strains, AdCh7-NP was as protective compared with its AdHu5 equivalent.
A recent clinical study assessed the safety and immunogenicity of a replication-deficient chimpanzee adenovirus-vectored influenza vaccine expressing NP and matrix protein 1 (ChAdOx1 NP+M1).75 Intramuscular immunization with ChAdOx1 NP+M1 was safe and immunogenic in terms of both cellular and humoral immunity. Heterologous boosting with a vaccinia virus Ankara (MVA) expressing NP and M1 enhanced T cellular responses to NP+M1 primed by ChAdOx1 NP+M1.
Currently chimpanzee adenovirus-vectored influenza vaccines have been less studied as compared with human adenovirus vectors, though it is generally believed that chimpanzee vectors may be more immunogenic in humans than human adenovirus vectors due to the prevalence of preexisting immunity against the latter. However, due to the huge gap of knowledge on human lung mucosal immune responses induced by chimpanzee versus human adenovirus-vectored influenza vaccines, much more efforts are required to testify this potential superiority of chimpanzee adenovirus-vectored influenza vaccine.
In summary, compared with conventional egg- or cell-based influenza vaccine production, adenovirus-vectored vaccines bypass the time-consuming adaptation of influenza virus to the culture system. The adenovirus-vectored vaccine strategy is also antigen-sparing without the need of producing a large quantity of proteins required for the recombinant protein vaccination strategies. With its documented safety and immunogenicity in humans, adenovirus-vectored influenza vaccination represents a promising novel strategy in future influenza prophylaxis.
Adenoviral vectors are also useful to developing “universal” or broadly protective flu vaccines, by targeting the conserved region of HA, or internal proteins including NP and matrix proteins. In murine models, a single dose of a candidate universal influenza vaccine based on a replication-deficient AdHu5 vector expressing NP and/or matrix protein 2 (M2) provided rapid protection against subsequent infections with virulent H5N1, H3N2, and H1N1 viruses, and of importance, intranasal immunization provided superior protection over the intramuscular route.76 In another preclinical study, a single dose of replication-defective AdHu5 vector encoding a fusion protein of humanized full-length H5 HA and the ectodomain of the M2 elicited long-lasting and antibody-dependent protection against heterosubtypic viruses (H1N1).77 However, the efficacy clinical trials are critically required to test whether this protection observed in mice can be translated into an universal or broad protection in humans.78
As discussed earlier, the adenovirus vector is featured by its well-documented safety, balanced humoral and cellular immunogenicity, as well as the respiratory mucosal tropism. However, preexisting immunity against human adenoviruses may dampen the immunogenicity of human adenovirus vectors. While increased vaccine doses may overcome the impact of preexisting immunity, this could happen at the cost of safety particularly when the vaccine is delivered to the respiratory tract. Moreover, there is still a lack of solid evidence that the humoral and/or cellular immunogenicity of adenovirus-vectored influenza vaccine correlates with protection in a clinical setting, as was shown in animal models. For these reasons, it is still too early to make a direct comparison between an adenovirus-vectored influenza vaccine and the current seasonal flu vaccine shots.
Preclinical and Clinical Development of Adenovirus-Based Vaccines Against HIV
As a major public health issue worldwide, the HIV/acquired immunodeficiency syndrome (AIDS), has claimed over three million lives globally to date. There were two million new cases of HIV infection with a total of 1.2 million AIDS-related deaths in 2014. Furthermore, as HIV-infected hosts are prone to latent TB reactivation, approximately one-third of AIDS patients succumb to TB worldwide. Although currently both the incidence of HIV infection and AIDS-related deaths per year have decreased compared with those in 2005, the total number of HIV infected people continues to increase. Better prophylactic measures including novel and effective vaccines/vaccination strategies are therefore in urgent need to protect people at high risk of HIV infection.
Based on current understandings of the early events following HIV transmission, it is believed that the viral eradication can only be achieved in the initial 5 to 10 days following HIV transmission. Neutralizing antibodies with broad antigen specificity are critically required to stop HIV from infecting host cells, while cell-mediated immunity including CD8 T cell- and NK cell-mediated killing of infected cells early after HIV transmission forms a second line of host defense to clear the founder virus if neutralizing antibodies fail to do so. Failure to eradicate HIV within the early phase of infection leads to viral spreading and generation of latent viral reservoirs which prove difficult to be cleared.79 For this reason, HIV vaccines are developed to induce the neutralizing antibodies with broad HIV antigen specificity and HIV-specific CD8+ T-cell responses.
The majority of HIV infections occur at the vaginal or rectal mucosal sites. Therefore, it is speculated that vaccination strategies with induced mucosal immune responses may confer superior protection over systemic immunization.80 However, it is still controversial whether systemic neutralizing Abs and/or cell-mediated immune components are competent in eradicating HIV early after mucosal infection, or in other words, it is not clear whether systemic neutralizing Abs and/or cell-mediated immune response are reliable immune correlates in the protection against HIV infection via mucosal routes.
Human adenovirus-based HIV vaccines
Current preclinical evaluation of HIV vaccines relies largely on either the chimpanzee model with HIV-1 infection or the macaque model with simian immunodeficiency virus (SIV) or SIV-HIV hybrid virus (SHIV) infection.81 Adenoviral-vectored HIV vaccines have been assessed in both models. In these studies, these vaccines were used either alone or in combination with protein or DNA vaccines in a prime-boost regimen.
One of the most inspiring preclinical studies was conducted using a replication-deficient AdHu5 vector expressing the SIV gag protein used either as a homologous booster or as a heterologous booster inoculation after priming with a SIV gag DNA vector plus adjuvant. In this study, the AdHu5-vectored vaccine primed CD8+ T cells specific for an immunodominant SIV gag epitope p11CM and attenuated the infection of SHIV as demonstrated by reduced viral load and higher peripheral blood CD4+ T-cell counts.82 Since these promising preclinical studies, multiple clinical trials have been conducted to evaluate the efficacy of novel human adenoviral-vectored HIV vaccines.
The most extensively characterized HIV vaccine is the MERK-developed trivalent AdHu5-based vaccine expressing the HIV antigens gag, pol, and nef (referred to as MRKAd5 HIV-1 gag/pol/nef). Successful phase-1 clinical trials provided the evidence supporting its safety profile and its ability to establish robust humoral and cellular immune responses against the encoded HIV antigens,83 thus forming the basis for its further clinical assessment in the phase-2 efficacy STEP and HVTN503/Phambili trials.84,85
The STEP trial, which was conducted in North and South America, the Caribbean, and Australia, unfortunately showed no efficacy and increased rates of HIV acquisition in immunized males who were either uncircumcised or AdHu5 seropositive prior to vaccination.84 It is important to note that although there was an increase in HIV acquisition in this subcohort, a follow-up study by Duerr et al.86 showed a transient nature of increased HIV infection which waned by 18 months postvaccination. Although the mechanisms still remain unclear for these observations, various studies point to the possibility of either attenuation of innate immunity in AdHu5 seropositive individuals or the transient spikes in the activation and recruitment of anti-AdHu5 CD4 T cells, the main targets of HIV, at the mucosa.14,87 Following the STEP trial’s observations, recruitment for the HVTN503/Phambili trial in South Africa was terminated. Findings from this trial also showed no vaccine-induced protection but instead, it found a nonstatistically significant increase in the rates of HIV acquisition.85
In another attempt carried out in the United States, a DNA prime-rAdHu5 boost vaccine regimen in the subjects at increased risks for HIV-1 infection did not lower the rate of HIV-1 infection or the viral load set point, even though this vaccination strategy primed HIV-specific CD4+ T and CD8+ T cell-mediated IFN-γ and/or IL-2 production in the peripheral blood as well as increased titers of neutralizing antibodies in serum.88
Chimpanzee adenoviral-vectored HIV vaccines
Chimpanzee adenoviral-vectored HIV vaccines have also begun preclinical evaluation, with a recent study further highlighting the potential of such vectors. In this study, chimpanzee serotypes 3 and 63 adenoviruses expressing the HIV gag, pol, and nef antigens were evaluated for T-cell immunogenicity in the murine model. The study shows that parenteral immunization with these vaccines was highly immunogenic, capable of inducing IFN-γ+ T-cell responses to all encoded antigens.89 Follow-up protection studies in comparison to AdHu5 vectors will be helpful to understanding the potential of such chimpanzee adenoviral vectors.
In summary, the lack of efficacy of current immunogenic adenovirus-vectored HIV vaccines highlights the importance of identifying the immune correlates of protection. Also, much more efforts should be made to decipher the relative contribution of systemic versus mucosal immune components in protection as vaginal and rectal mucosa are the primary entry site of HIV.80 Updated knowledge on these issues will be of great value in guiding the development of novel adenovirus-vectored HIV vaccines and/or novel vaccination strategies. Furthermore, as the efficacy trial with human adenoviral-vectored HIV vaccines suggests an increased incidence of HIV infection in some AdHu5-seropositive individuals, it has been concluded that AdHu5 vectors are no longer suitable for HIV vaccine design. In this respect, chimpanzee adenoviral-vectored vaccines may represent an improved approach in future HIV vaccine development due to the very low seroprevalence of chimpanzee adenoviruses in humans. However, it remains to be seen whether the T-cell epitopes potentially shared by both AdHu5 and chimpanzee adenoviruses may still pose a limitation to the use of the latter for HIV vaccine design.90
Concluding Remarks
The continued success of adenoviruses as vaccine vectors is attributed not only to their ability to drive robust and sustained humoral and cellular adaptive immune responses, but to their ability to induce such responses at the mucosal sites of pathogen entry. This makes adenoviral vectors one of the most widely used platforms for the generation of mucosally deliverable vaccines. Recent advances in adenoviral vectorology, viral packaging cell lines, mucosal immunity, and vaccine immunology have expedited the development of adenoviral-vectored vaccines against a number of mucosal pathogens to which there is a lack of effective vaccination strategies. The next few years shall see the much increased clinical knowledge in the potential of mucosal vaccination with adenoviral-vectored vaccines.
Acknowledgments
The work from the authors’ laboratory is supported by funds from the Canadian Institutes of Health Research, The Natural Sciences and Engineering Research Council of Canada, The Canadian Foundation for Innovation, Ontario Government, British Government, Tianjin CanSino Biotechnology, and McMaster University. We also acknowledge the kind provision of Mycobacterium tuberculosis and immunologic reagents by the BEI Resources and NIH Tetramer Core.
References
- Volpers, C and Kochanek, S (2004). Adenoviral vectors for gene transfer and therapy. J Gene Med 6 (suppl. 1): S164–S171. [DOI] [PubMed] [Google Scholar]
- Tatsis, N and Ertl, HC (2004). Adenoviruses as vaccine vectors. Mol Ther 10: 616–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasaro, MO and Ertl, HC (2009). New insights on adenovirus as vaccine vectors. Mol Ther 17: 1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majhen, D, Calderon, H, Chandra, N, Fajardo, CA, Rajan, A, Alemany, R et al. (2014). Adenovirus-based vaccines for fighting infectious diseases and cancer: progress in the field. Hum Gene Ther 25: 301–317. [DOI] [PubMed] [Google Scholar]
- Sheridan, C (2011). Gene therapy finds its niche. Nat Biotechnol 29: 121–128. [DOI] [PubMed] [Google Scholar]
- Lycke, N (2012). Recent progress in mucosal vaccine development: potential and limitations. Nat Rev Immunol 12: 592–605. [DOI] [PubMed] [Google Scholar]
- Lai, R, Afkhami, S, Haddadi, S, Jeyanathan, M and Xing, Z (2015). Mucosal immunity and novel tuberculosis vaccine strategies: route of immunisation-determined T-cell homing to restricted lung mucosal compartments. Eur Respir Rev 24: 356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleman, M, Galea, S, Gavard, F, Merillon, N, Klonjkowski, B, Tartour, E et al. (2011). Antigen encoded by vaccine vectors derived from human adenovirus serotype 5 is preferentially presented to CD8+ T lymphocytes by the CD8α+ dendritic cell subset. Vaccine 29: 5892–5903. [DOI] [PubMed] [Google Scholar]
- Robinson, CM, Singh, G, Henquell, C, Walsh, MP, Peigue-Lafeuille, H, Seto, D et al. (2011). Computational analysis and identification of an emergent human adenovirus pathogen implicated in a respiratory fatality. Virology 409: 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell, MJ and Imperiale, MJ (2004). Biology of adenovirus and its use as a vector for gene therapy. Hum Gene Ther 15: 1022–1033. [DOI] [PubMed] [Google Scholar]
- Colloca, S, Barnes, E, Folgori, A, Ammendola, V, Capone, S, Cirillo, A et al. (2012). Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med 4: 115ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsis, N, Tesema, L, Robinson, ER, Giles-Davis, W, McCoy, K, Gao, GP et al. (2006). Chimpanzee-origin adenovirus vectors as vaccine carriers. Gene Ther 13: 421–429. [DOI] [PubMed] [Google Scholar]
- Zaiss, AK, Machado, HB and Herschman, HR (2009). The influence of innate and pre-existing immunity on adenovirus therapy. J Cell Biochem 108: 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak, DE, Andersen-Nissen, E, Peterson, ER, Sato, A, Hamilton, MK, Borgerding, J et al. (2012). Merck Ad5/HIV induces broad innate immune activation that predicts CD8+ T-cell responses but is attenuated by preexisting Ad5 immunity. Proc Natl Acad Sci USA 109: E3503–E3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouard, D, Alazard-Dany, D and Cosset, FL (2009). Viral vectors: from virology to transgene expression. Br J Pharmacol 157: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton, RJ, McSharry, BP, Armstrong, M, Tomasec, P and Wilkinson, GW (2008). Re-engineering adenovirus vector systems to enable high-throughput analyses of gene function. Biotechniques 45: 659–662, 664. [DOI] [PubMed] [Google Scholar]
- Sakhuja, K, Reddy, PS, Ganesh, S, Cantaniag, F, Pattison, S, Limbach, P et al. (2003). Optimization of the generation and propagation of gutless adenoviral vectors. Hum Gene Ther 14: 243–254. [DOI] [PubMed] [Google Scholar]
- Graham, FL and Prevec, L (1995). Methods for construction of adenovirus vectors. Mol Biotechnol 3: 207–220. [DOI] [PubMed] [Google Scholar]
- Davis, AR, Wivel, NA, Palladino, JL, Tao, L and Wilson, JM (2001). Construction of adenoviral vectors. Mol Biotechnol 18: 63–70. [DOI] [PubMed] [Google Scholar]
- Weaver, EA, Nehete, PN, Buchl, SS, Senac, JS, Palmer, D, Ng, P et al. (2009). Comparison of replication-competent, first generation, and helper-dependent adenoviral vaccines. PLoS One 4: e5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H, Machuca, TN, Yeung, JC, Wu, J, Du, K, Duan, C et al. (2013). Efficient gene delivery to pig airway epithelia and submucosal glands using helper-dependent adenoviral vectors. Mol Ther Nucleic Acids 2: e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruve, DA, Cotter, MJ, Zaiss, AK, White, LR, Liu, Q, Chan, T et al. (2004). Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J Virol 78: 5966–5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthinne, X and Imperiale, MJ (2000). Production of first generation adenovirus vectors: a review. Gene Ther 7: 1707–1714. [DOI] [PubMed] [Google Scholar]
- Kovesdi, I and Hedley, SJ (2010). Adenoviral producer cells. Viruses 2: 1681–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havenga, M, Vogels, R, Zuijdgeest, D, Radosevic, K, Mueller, S, Sieuwerts, M et al. (2006). Novel replication-incompetent adenoviral B-group vectors: high vector stability and yield in PER.C6 cells. J Gen Virol 87: 2135–2143. [DOI] [PubMed] [Google Scholar]
- Murakami, P, Pungor, E, Files, J, Do, L, van Rijnsoever, R, Vogels, R et al. (2002). A single short stretch of homology between adenoviral vector and packaging cell line can give rise to cytopathic effect-inducing, helper-dependent E1-positive particles. Hum Gene Ther 13: 909–920. [DOI] [PubMed] [Google Scholar]
- Gao, G, Zhou, X, Alvira, MR, Tran, P, Marsh, J, Lynd, K et al. (2003). High throughput creation of recombinant adenovirus vectors by direct cloning, green-white selection and I-Sce I-mediated rescue of circular adenovirus plasmids in 293 cells. Gene Ther 10: 1926–1930. [DOI] [PubMed] [Google Scholar]
- Schaack, J, Langer, S and Guo, X (1995). Efficient selection of recombinant adenoviruses by vectors that express beta-galactosidase. J Virol 69: 3920–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier, C, Degryse, E, Gantzer, M, Dieterle, A, Pavirani, A and Mehtali, M (1996). Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol 70: 4805–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, P and Graham, FL (2002). Construction of first-generation adenoviral vectors. Methods Mol Med 69: 389–414. [DOI] [PubMed] [Google Scholar]
- Luo, J, Deng, ZL, Luo, X, Tang, N, Song, WX, Chen, J et al. (2007). A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2: 1236–1247. [DOI] [PubMed] [Google Scholar]
- Mizuguchi, H and Kay, MA (1998). Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther 9: 2577–2583. [DOI] [PubMed] [Google Scholar]
- Zhou, D, Zhou, X, Bian, A, Li, H, Chen, H, Small, JC et al. (2010). An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nat Protoc 5: 1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallel, H and Kamen, AA (2015). Large-scale adenovirus and poxvirus-vectored vaccine manufacturing to enable clinical trials. Biotechnol J 10: 741–747. [DOI] [PubMed] [Google Scholar]
- Kamen, A and Henry, O (2004). Development and optimization of an adenovirus production process. J Gene Med 6 (suppl. 1): S184–S192. [DOI] [PubMed] [Google Scholar]
- Lusky, M (2005). Good manufacturing practice production of adenoviral vectors for clinical trials. Hum Gene Ther 16: 281–291. [DOI] [PubMed] [Google Scholar]
- WHO (2015). WHO Global Tuberculosis Report 2015. World Health Organization: Geneva. [Google Scholar]
- Cooper, AM (2009). Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27: 393–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Alves, C, Booty, MG, Carpenter, SM, Jayaraman, P, Rothchild, AC and Behar, SM (2014). In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol 12: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes, EK, Sander, C, Ronan, EO, McShane, H, Hill, AV, Beverley, PC et al. (2008). Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J Immunol 181: 4955–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J, Thorson, L, Stokes, RW, Santosuosso, M, Huygen, K, Zganiacz, A et al. (2004). Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol 173: 6357–6365. [DOI] [PubMed] [Google Scholar]
- Horvath, CN, Shaler, CR, Jeyanathan, M, Zganiacz, A and Xing, Z (2012). Mechanisms of delayed anti-tuberculosis protection in the lung of parenteral BCG-vaccinated hosts: a critical role of airway luminal T cells. Mucosal Immunol 5: 420–431. [DOI] [PubMed] [Google Scholar]
- Santosuosso, M, McCormick, S, Zhang, X, Zganiacz, A and Xing, Z (2006). Intranasal boosting with an adenovirus-vectored vaccine markedly enhances protection by parenteral Mycobacterium bovis BCG immunization against pulmonary tuberculosis. Infect Immun 74: 4634–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santosuosso, M, McCormick, S, Roediger, E, Zhang, X, Zganiacz, A, Lichty, BD et al. (2007). Mucosal luminal manipulation of T cell geography switches on protective efficacy by otherwise ineffective parenteral genetic immunization. J Immunol 178: 2387–2395. [DOI] [PubMed] [Google Scholar]
- Huygen, K (2014). The immunodominant T-cell epitopes of the mycolyl-transferases of the antigen 85 complex of M. tuberculosis. Front Immunol 5: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan, M, Shao, Z, Yu, X, Harkness, R, Jiang, R, Li, J et al. (2015). AdHu5Ag85A respiratory mucosal boost immunization enhances protection against pulmonary tuberculosis in BCG-primed non-human primates. PLoS One 10: e0135009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Z, McFarland, CT, Sallenave, JM, Izzo, A, Wang, J and McMurray, DN (2009). Intranasal mucosal boosting with an adenovirus-vectored vaccine markedly enhances the protection of BCG-primed guinea pigs against pulmonary tuberculosis. PLoS One 4: e5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, J, Jeyanathan, M, Shaler, CR, Horvath, C, Damjanovic, D, Zganiacz, A et al. (2010). Respiratory mucosal immunization with adenovirus gene transfer vector induces helper CD4 T cell-independent protective immunity. J Gene Med 12: 693–704. [DOI] [PubMed] [Google Scholar]
- Dean, GS, Clifford, D, Whelan, AO, Tchilian, EZ, Beverley, PC, Salguero, FJ et al. (2015). Protection induced by simultaneous subcutaneous and endobronchial vaccination with BCG/BCG and BCG/adenovirus expressing antigen 85A against Mycobacterium bovis in cattle. PLoS One 10: e0142270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaill, F, Jeyanathan, M, Smieja, M, Medina, MF, Thanthrige-Don, N, Zganiacz, A et al. (2013). A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci Transl Med 5: 205ra134. [DOI] [PubMed] [Google Scholar]
- Dudareva, M, Andrews, L, Gilbert, SC, Bejon, P, Marsh, K, Mwacharo, J et al. (2009). Prevalence of serum neutralizing antibodies against chimpanzee adenovirus 63 and human adenovirus 5 in Kenyan children, in the context of vaccine vector efficacy. Vaccine 27: 3501–3504. [DOI] [PubMed] [Google Scholar]
- Sumida, SM, Truitt, DM, Lemckert, AA, Vogels, R, Custers, JH, Addo, MM et al. (2005). Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol 174: 7179–7185. [DOI] [PubMed] [Google Scholar]
- Thorner, AR, Vogels, R, Kaspers, J, Weverling, GJ, Holterman, L, Lemckert, AA et al. (2006). Age dependence of adenovirus-specific neutralizing antibody titers in individuals from sub-Saharan Africa. J Clin Microbiol 44: 3781–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radosevic, K, Wieland, CW, Rodriguez, A, Weverling, GJ, Mintardjo, R, Gillissen, G et al. (2007). Protective immune responses to a recombinant adenovirus type 35 tuberculosis vaccine in two mouse strains: CD4 and CD8 T-cell epitope mapping and role of gamma interferon. Infect Immun 75: 4105–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah, PA, Bolton, DL, Lackner, AA, Kaushal, D, Aye, PP, Mehra, S et al. (2014). Aerosol vaccination with AERAS-402 elicits robust cellular immune responses in the lungs of rhesus macaques but fails to protect against high-dose Mycobacterium tuberculosis challenge. J Immunol 193: 1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokey, DA, Wachholder, R, Darrah, PA, Bolton, DL, Barouch, DH, Hill, K et al. (2014). A nonhuman primate toxicology and immunogenicity study evaluating aerosol delivery of AERAS-402/Ad35 vaccine: evidence for transient t cell responses in peripheral blood and robust sustained responses in the lungs. Hum Vaccin Immunother 10: 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchyard, GJ, Snowden, MA, Hokey, D, Dheenadhayalan, V, McClain, JB, Douoguih, M et al. (2015). The safety and immunogenicity of an adenovirus type 35-vectored TB vaccine in HIV-infected, BCG-vaccinated adults with CD4(+) T cell counts >350 cells/mm(3). Vaccine 33: 1890–1896. [DOI] [PubMed] [Google Scholar]
- Kagina, BM, Tameris, MD, Geldenhuys, H, Hatherill, M, Abel, B, Hussey, GD et al. 018-402 Clinical Lab study team. (2014). The novel tuberculosis vaccine, AERAS-402, is safe in healthy infants previously vaccinated with BCG, and induces dose-dependent CD4 and CD8T cell responses. Vaccine 32: 5908–5917. [DOI] [PubMed] [Google Scholar]
- Abel, B, Tameris, M, Mansoor, N, Gelderbloem, S, Hughes, J, Abrahams, D et al. (2010). The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4+ and CD8+ T cells in adults. Am J Respir Crit Care Med 181: 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan, S, Harris, SA, Satti, I, Hokey, DA, Dheenadhayalan, V, Stockdale, L et al. (2015). A phase I, open-label trial, evaluating the safety and immunogenicity of candidate tuberculosis vaccines AERAS-402 and MVA85A, administered by prime-boost regime in BCG-vaccinated healthy adults. PLoS One 10: e0141687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameris, M, Hokey, DA, Nduba, V, Sacarlal, J, Laher, F, Kiringa, G et al. (2015). A double-blind, randomised, placebo-controlled, dose-finding trial of the novel tuberculosis vaccine AERAS-402, an adenovirus-vectored fusion protein, in healthy, BCG-vaccinated infants. Vaccine 33: 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, MJ, Björkström, NK, Petrovas, C, Liang, F, Gall, JG, Loré, K et al. (2014). Type I interferon-dependent activation of NK cells by rAd28 or rAd35, but not rAd5, leads to loss of vector-insert expression. Vaccine 32: 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, MJ, Petrovas, C, Yamamoto, T, Lindsay, RW, Loré, K, Gall, JG et al. (2012). Type I IFN induced by adenovirus serotypes 28 and 35 has multiple effects on T cell immunogenicity. J Immunol 188: 6109–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan, M, Damjanovic, D, Shaler, CR, Lai, R, Wortzman, M, Yin, C et al. (2013). Differentially imprinted innate immunity by mucosal boost vaccination determines antituberculosis immune protective outcomes, independent of T-cell immunity. Mucosal Immunol 6: 612–625. [DOI] [PubMed] [Google Scholar]
- McNab, F, Mayer-Barber, K, Sher, A, Wack, A and O’Garra, A (2015). Type I interferons in infectious disease. Nat Rev Immunol 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles, RM, Graeber, TG, Krutzik, SR, Montoya, D, Schenk, M, Lee, DJ et al. (2013). Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339: 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S, Gao, G, Lu, Y, Zhou, X, Lock, M, Calcedo, R et al. (2004). Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Hum Gene Ther 15: 519–530. [DOI] [PubMed] [Google Scholar]
- Stylianou, E, Griffiths, KL, Poyntz, HC, Harrington-Kandt, R, Dicks, MD, Stockdale, L et al. (2015). Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine 33: 6800–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan, M, Thanthrige-Don, N, Afkhami, S, Lai, R, Damjanovic, D, Zganiacz, A et al. (2015). Novel chimpanzee adenovirus-vectored respiratory mucosal tuberculosis vaccine: overcoming local anti-human adenovirus immunity for potent TB protection. Mucosal Immunol 8: 1373–1387. [DOI] [PubMed] [Google Scholar]
- Lambert, LC and Fauci, AS (2010). Influenza vaccines for the future. N Engl J Med 363: 2036–2044. [DOI] [PubMed] [Google Scholar]
- Hoelscher, MA, Garg, S, Bangari, DS, Belser, JA, Lu, X, Stephenson, I et al. (2006). Development of adenoviral-vector-based pandemic influenza vaccine against antigenically distinct human H5N1 strains in mice. Lancet 367: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kampen, KR, Shi, Z, Gao, P, Zhang, J, Foster, KW, Chen, DT et al. (2005). Safety and immunogenicity of adenovirus-vectored nasal and epicutaneous influenza vaccines in humans. Vaccine 23: 1029–1036. [DOI] [PubMed] [Google Scholar]
- Gurwith, M, Lock, M, Taylor, EM, Ishioka, G, Alexander, J, Mayall, T et al. (2013). Safety and immunogenicity of an oral, replicating adenovirus serotype 4 vector vaccine for H5N1 influenza: a randomised, double-blind, placebo-controlled, phase 1 study. Lancet Infect Dis 13: 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S, Kobinger, GP, Lin, J, Figueredo, J, Calcedo, R, Kobasa, D et al. (2007). Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine 25: 6845–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antrobus, L, Berthoud, TK, Dicks, MD, Hill, AVS, Lambe, T, Gilbert, SC et al. (2014). Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved influenza a antigens. Mol Ther 22: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, GE, Soboleski, MR, Lo, CY, Misplon, JA, Quirion, MR, Houser, KV et al. (2010). Single-dose mucosal immunization with a candidate universal influenza vaccine provides rapid protection from virulent H5N1, H3N2 and H1N1 viruses. PLoS One 5: e13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, EH, Park, HJ, Han, GY, Song, MK, Pereboev, A, Hong, JS et al. (2014). Intranasal adenovirus-vectored vaccine for induction of long-lasting humoral immunity-mediated broad protection against influenza in mice. J Virol 88: 9693–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer, F and Palese, P (2014). Universal influenza virus vaccines: need for clinical trials. Nat Immunol 15: 3–5. [DOI] [PubMed] [Google Scholar]
- McMichael, AJ, Borrow, P, Tomaras, GD, Goonetilleke, N and Haynes, BF (2010). The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol 10: 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, EA et al. (2013). Comparison of systemic and mucosal immunization with helper-dependent adenoviruses for vaccination against mucosal challenge with SHIV. PLoS One 8: e67574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath, BM, Schumann, KE and Boyer, JD (2000). The chimpanzee and other non-human-primate models in HIV-1 vaccine research. Trends Microbiol 8: 426–431. [DOI] [PubMed] [Google Scholar]
- Shiver, JW, Fu, TM, Chen, L, Casimiro, DR, Davies, ME, Evans, RK et al. (2002). Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415: 331–335. [DOI] [PubMed] [Google Scholar]
- Priddy, FH, Brown, D, Kublin, J, Monahan, K, Wright, DP, Lalezari, J et al. Merck V520-016 Study Group (2008). Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis 46: 1769–1781. [DOI] [PubMed] [Google Scholar]
- Buchbinder, SP, Mehrotra, DV, Duerr, A, Fitzgerald, DW, Mogg, R, Li, D et al. Step Study Protocol Team (2008). Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372: 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, GE, Allen, M, Moodie, Z, Churchyard, G, Bekker, LG, Nchabeleng, M et al. HVTN 503/Phambili study team. (2011). Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: a double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect Dis 11: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr, A, Huang, Y, Buchbinder, S, Coombs, RW, Sanchez, J, del Rio, C et al. Step/HVTN 504 Study Team. (2012). Extended follow-up confirms early vaccine-enhanced risk of HIV acquisition and demonstrates waning effect over time among participants in a randomized trial of recombinant adenovirus HIV vaccine (Step Study). J Infect Dis 206: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi, H, Genescà, M, Fritts, L, McChesney, MB, Robert-Guroff, M and Miller, CJ (2014). Infection with host-range mutant adenovirus 5 suppresses innate immunity and induces systemic CD4+ T cell activation in rhesus macaques. PLoS One 9: e106004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, SM, Sobieszczyk, ME, Janes, H, Karuna, ST, Mulligan, MJ, Grove, D et al. HVTN 505 Study Team (2013). Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N Engl J Med 369: 2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herath, S, Le Heron, A, Colloca, S, Bergin, P, Patterson, S, Weber, J et al. (2015). Analysis of T cell responses to chimpanzee adenovirus vectors encoding HIV gag-pol-nef antigen. Vaccine 33: 7283–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm, N, DeCamp, AC, Friedrich, DP, Carter, DK, Defawe, OD, Kublin, JG et al. (2012). Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest 122: 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]