

Abstract
In this study we aimed to provide an in-depth proteomic analysis of differentially expressed proteins in the hearts of transgenic mouse models of pathological and physiological cardiac hypertrophy using tandem mass tag labeling and liquid chromatography tandem mass spectrometry. The Δ43 mouse model, expressing the 43-amino-acid N-terminally truncated myosin essential light chain (ELC) served as a tool to study the mechanisms of physiological cardiac remodeling, while the pathological hypertrophy was investigated in A57G (Alanine 57 → Glycine) ELC mice. The results showed that 30 proteins were differentially expressed in Δ43 versus A57G hearts as determined by multiple pair comparisons of the mutant versus wild-type (WT) samples with P < 0.05. The A57G hearts showed differential expression of nine mitochondrial proteins involved in metabolic processes compared to four proteins for Δ43 hearts when both mutants were compared to WT hearts. Comparisons between Δ43 and A57G hearts showed an upregulation of three metabolically important mitochondrial proteins but downregulation of nine proteins in Δ43 hearts. The physiological model of cardiac hypertrophy (Δ43) showed no changes in the levels of Ca2+-binding proteins relative to WT, while the pathologic model (A57G) showed the upregulation of three Ca2+-binding proteins, including sarcalumenin. Unique differences in chaperone and fatty acid metabolism proteins were also observed in Δ43 versus A57G hearts. The proteomics data support the results from functional studies performed previously on both animal models of cardiac hypertrophy and suggest that the A57G- and not Δ43- mediated alterations in fatty acid metabolism and Ca2+ homeostasis may contribute to pathological cardiac remodeling in A57G hearts.
Keywords: Myosin ELC, Transgenic mice, Molecular proteomics, Pathological hypertrophy
Introduction
The heart, in response to various types of stimuli such as mechanical, hemodynamic or genetic, has the ability to adapt to increased workloads through the hypertrophy of muscle cells (Hunter and Chien 1999; Lorell and Carabello 2000). The nature of the workload increase may vary depending on the stimulus. If the heart fully adapts to the new loading condition, reaching new equilibrium, the hypertrophic response is considered physiological. This type of hypertrophy is characterized by a normal organization of cardiac structure, normal or enhanced cardiac function, relatively normal patterns of gene expression, and is also reversible (Bernardo et al. 2010). On the other hand, genetic hypertrophic cardiomyopathy (HCM), which originates from mutations in all major sarcomeric proteins of the heart, can be manifested by pathological cardiac remodeling, left ventricular (LV) enlargement, fibrosis, filament disarray and sudden cardiac death (SCD) (Maron 2002; Moolman et al. 1997; Spirito et al. 2000).
Cardiac muscle contraction consists of the ATP-dependent cyclic attachments and detachments of the myosin cross-bridges to the actin-tropomyosin-troponin filaments (Geeves and Holmes 2005). The myosin cross-bridge is the molecular motor of the heart; it binds ATP and actin and its lever arm region, supported by the regulatory (RLC) and essential light chains (ELC), amplifies small conformational changes generated in the motor domain into large movements needed to produce force and sarcomere shortening (Rayment et al. 1993). Myosin ELC constitutes an essential part of the lever arm structure and plays a role in force development and muscle contraction (Kazmierczak et al. 2009; Miller et al. 2005; Timson 2003; VanBuren et al. 1994). Similar to other sarcomeric proteins, mutations in the ventricular ELC (encoded by MYL3) are implicated in cardiomyopathies. Compared to β-myosin heavy chain (MHC) or myosin binding protein-C, mutations in the ELC are quite rare, but they are also associated with malignant outcomes (Kaski et al. 2009; Lee et al. 2001; Morita et al. 2008; Olson et al. 2002; Poetter et al. 1996; Richard et al. 2003).
This study focuses on two types of hypertrophic remodeling of the heart associated with mutations in the myosin ventricular ELC (UniProt—P08590) (Hernandez et al. 2007). The A57G (Alanine57 → Glycine) variant of ELC was found in patients diagnosed with hypertrophic cardiomyopathy (HCM) with a classic asymmetric septal hypertrophy phenotype and occurrences of SCD (Lee et al. 2001). The A57G mutation in the human ventricular myosin ELC has been expressed in transgenic mouse hearts (Kazmierczak et al. 2013) and investigated for the mechanisms specific to HCM. Another type of hypertrophy associated with the myosin ELC that we explore here is of physiological nature and characterized by a normal cardiac function. In the Δ43 mouse model, the endogenous mouse ELC is partially replaced by the 43-amino-acid N-terminally truncated human ventricular ELC protein (Kazmierczak et al. 2009). Our published data on Δ43 mice show that as the mice grow older, the hearts of Δ43 mice hypertrophy, but the ventricles do not show any pathologic phenotype (Kazmierczak et al. 2009, 2014). Magnetic resonance imaging confirmed normal cardiac function in >7 month-old Δ43 mice compared with age matched controls (Kazmierczak et al. 2009, 2014). These two ELC animal models were investigated for the physiological (Δ43) versus pathological (A57G) manifestations of cardiac remodeling using molecular proteomics approaches, and the results were compared with those obtained for transgenic WT-line 1 expressing full length non-mutated human ventricular ELC (Kazmierczak et al. 2009; Muthu et al. 2011).
Proteomics is a powerful technique (Cui et al. 2011; Dewey et al. 2013) which allows us to compare the classes of biological, cellular and functional processes that are similar and/or different between these two hypertrophic phenotypes. The isobaric chemical labeling, utilizing the tandem mass tag (TMT) labeling method, was used because this technique has been shown to be efficient in the quantification accuracy, precision, and reproducibility of results (Li et al. 2012; Megger et al. 2014). TMT labeling reagents share an identical structure and therefore enable the identification and quantitation of proteins in different samples using tandem mass spectrometry (MS/MS) (Rauniyar and Yates 2014). Briefly, protein extracts from tissues or cells are reduced, alkylated and digested. After proteolysis the samples are labeled separately with different TMT isotopic variants and then combined before sample fractionation (Li et al. 2012; Rauniyar and Yates 2014). The TMT label contain three functional parts: a reporter ion group, a mass normalization group, and an amine-reactive group (Rauniyar and Yates 2014). The amine-reactive group reacts with the N-terminal amine groups and epsilon-amine groups of lysine residues, while MS fragmentation of a susceptible linker between the reporter ion group and the mass normalization group results in reporter ions of different masses being dissociated from isolated peptides. The relative abundance of the peptide labeled with the specific TMT tag variant is determined by the relative intensity of the reporter ions associated with the specific peptide (Rauniyar and Yates 2014).
Materials and methods
All animal studies were conducted in accordance with institutional guidelines. The University of Miami has an Animal Welfare Assurance (A-3224-01, effective November 23, 2011) on file with the Office of Laboratory Animal Welfare (OLAW), National Institutes of Health.
Sample preparation
Three hearts from each group of mice (3.5–4 month-old females) were subjected to the proteomic studies. Transgenic mice expressing mutations in the human ventricular myosin ELC were previously generated and characterized (Kazmierczak et al. 2013, 2009, 2014; Muthu et al. 2011). After euthanasia, the hearts were immediately excised and washed briefly with cold PBS, then weighed and flash frozen. It has been demonstrated previously that hearts prepared in this manner are of excellent quality for subsequent proteomic investigation (Gomes et al. 2009; Gomes et al. 2006).
Protein digestion
Urea (8 M, 300 μL) containing protease and phosphatase inhibitors (Sigma Chemical Company, St. Louis, MO) was added to powdered cardiac muscle (20 mg) from WT, Δ43, and A57G mice. Each tissue was then lightly sonicated and mixed for 1 h followed by centrifugation at 15,800×g for 10 min. Protein concentration was determined using the Bio-Rad RC/DC method. 100 μg of each sample was placed into polypropylene microcentrifuge tubes. 45 μL of 100 mM triethyl ammonium bicarbonate (TEAB) were added to each sample and the final volume adjusted to 100 μL with ultrapure water. 5 μL of 200 mM tris(2-carboxyethyl)phosphine (TCEP) were added to each sample and the samples were incubated at 55 °C for 1 h. 5 μL of 375 mM iodoacetamide were then added to the samples, and left for 30 min in the dark. Six volumes of pre-chilled (−20 °C) acetone were subsequently added to each sample, and they were left overnight at −20 °C. Samples were then centrifuged at 8000×g for 10 min at 4 °C and supernatants removed without disturbing the pellet. The pellets were redissolved in 100 μL of TEAB and 2.5 μg of tryspin (trypsin gold sequencing grade, Promega, Madison, WI) were then added (ratio 2.5 μg of trypsin per 100 μg of protein), and the samples were left to digest overnight at 37 °C.
TMT labeling and peptide fractionation
After proteolysis, heart samples were labeled separately with different isotopic variants of TMT (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions and then combined. 6-Plex TMT was utilized, allowing the comparison of up to 6 heart samples in a single LC–MS/MS analysis. Each set of TMT labeling was carried out using pooled WT (TMT label 126), WT-line 1 (TMT label 127), Δ43 (TMT label 129), and A57G (TMT label 130) (Kazmierczak et al. 2009; Muthu et al. 2011). Three independent sets of TMT labeling were carried out on each set using lysates from different hearts with the exception of the pooled WT sample. The pooled WT used in these proteomic experiments contained pooled lysates from three different wild-type line 1 mice (Kazmierczak et al. 2009; Muthu et al. 2011). The pooled WT sample was the same for all three independent sets of TMT labeling allowing us to compare the labeling efficiency and reproducibility of mass spectrometry runs because the identical pooled WT sample was labeled independently in each of the three independent TMT experiments.
Briefly, TMT tags were dissolved in anhydrous acetonitrile and added to the digested heart samples and incubated for 1 h at room temperature. Quenching of excess TMT tags was carried out by adding 10 % (w/v) hydroxylamine to a final concentration of 0.5 % and incubating for 15 min. TMT labeled peptides were fractionated using SCX SpinTips (Protea Biosciences, Inc, Morgantown, WV). Stepwise elution of peptides from the SCX columns was carried out using 20, 60, 80, 125, 150, 200, 400, and 500 mM ammonium formate in 10 % acetonitrile at pH 3. Eluted fractions were desalted using SDB columns (GL Sciences, Tokyo, Japan).
LC–MS/MS
Labeled peptides were analyzed by LC–MS/MS on a Thermo Scientific Q ExactivePlus Orbitrap Mass spectrometer with an attached Proxeon nanospray source and a Waters UPLC (Waters Corporation, Milford, MA, USA). Digested peptides were loaded onto a 100 micron × 25 mm Magic C18 100Å 5U reverse phase trap (using material from Bruker, Billerica, MA) where they were desalted online before being separated using a 75 micron × 150 mm Magic C18 200Å 3U reverse phase column (packed using material from Bruker). Elution of peptides occurred over a 60 min gradient with a flow rate of 300 nL/min. MS survey scans were obtained for the m/z range 300–1600, and MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the MS spectra were subjected to HCD (High Energy Collisional Dissociation). The mobile phases were A: water with 0.1 % TFA and B: 100 % acetonitrile. The mobile phases at different gradient time points: 0 min 98 % A/2 % B, 45 min 65 % A/35 % B, 50 min 20 % A/80 % B, 51 min 98 % A/2 % B, 60 min 98 % A/2 % B. An isolation mass window of 2.0 m/z was utilized for the precursor ion selection, and a normalized collision energy of 30 % was used for fragmentation. A 5 s duration was used for the dynamic exclusion.
Protein identification
Protein identification and initial quantification were performed with Proteome Discoverer (ver. 1.4.0.288, Thermo Fisher Scientific). Three different sets of TMT 6-Plex were carried out and analyzed by LC–MS/MS. The same master mix of WT hearts was used as one of the labeled samples in all TMT-6 plex LC–MS/MS runs to provide a reference set to allow all the runs to be compared. The UniProt database used (version 02192014) contained 28266 proteins and 29282 decoy sequences. Parameters used included cleavage semi set as yes, missed cleavage sites set at 2, static modifications: carbamidomethyl (C), TMT 6-Plex (K), and TMT 6-Plex (N-term), variable modifications: oxidation (M), deamidation (N, Q). The fragment mass tolerance was set at 0.1 Da and the precursor mass tolerance set at 20 ppm. Peptide identifications were validated based upon q-value using the percolator function in Proteome Discoverer. Peptides with false discovery rates >1 % (q value >0.01) were discarded.
Quantitative data analysis
Scaffold Q + (version 4.4.1, Proteome Software Inc., Portland, OR) was used to quantitate label based quantitation (TMT) peptide and protein identifications using quantitative files obtained from Proteome Discoverer. Peptide identifications were accepted if they could be established at greater than 99.0 % probability by the Peptide Prophet algorithm (Keller et al. 2002) with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 7.0 % probability to achieve an FDR less than 2.5 % and contained at least 3 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al. 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Channels were corrected by the matrix [0.000,0.000,1.000,0.000,0.000]; [0.000,0.000,1.000,0.000,0.000]; [0.000,0.000,1.000,0.000,0.000]; [0.000,0.000,1.000,0.000,0.000]; [0.000,0.000,1.000,0.000,0.000]; [0.000,0.000,1.000,0.000,0.000] in all samples according to the algorithm described in i-Tracker (Shadforth et al. 2005). Acquired intensities in the experiment were globally normalized across all acquisition runs. Individual quantitative samples were normalized within each acquisition run. Intensities for each peptide identification were normalized within the assigned protein. The reference channels were normalized to produce a 1:1 fold change. All normalization calculations were performed using medians to multiplicatively normalize data. Differentially expressed proteins were determined using Mann–Whitney Test analysis which was Bonferroni Corrected for multiple sample analysis. The coefficient of variance (CV) for each protein identified was calculated using the Scaffold program. Independently, the pooled WT sample data from the three runs were normalized to total intensity, averaged, and accuracy estimated by calculating the CV (S.D./mean × 100) of the measurements.
Western blotting
Independent validation of some of the MS data was done by Western blotting using protein specific antibodies. Changes in total protein were determined with 20 μg (for vinculin blot) or 16 μg (all other blots) of each muscle lysate. Heart homogenates were separated on a 12 % polyacrylamide gel (for vinculin blot, Bio-Rad, Hercules, CA, USA) or 4–20 % Stain-Free Criterion Gels (all other blots, Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad). Stain-Free gels were activated by UV transillumination for 1 min prior to transferring. The membranes were then probed with the respective antibodies. All Western blotting procedures were carried out at room temperature with agitation except when stated otherwise. Blocking for vinculin was done in 1:1 (v/v) of TBS (5 mM NaCl, 50 mM Tris, pH 7.5) and blocking buffer (Rockland, Limerick, PA, USA blocking buffer for fluorescent Western blotting; cat# MB-070-010) for 1 h at room temperature. The membranes were then incubated in primary antibody in TBST (5 mM NaCl, 50 mM Tris, pH 7.5, 0.05 % (v/v) Tween-20) : Blocking buffer (1:1 v/v) overnight at 4 °C, washed and incubated in secondary antibody in TBST: Blocking buffer (1:1 v/v) for 1 h at room temperature and washed. For all other blots, the membranes were blocked in 3 % non-fat milk (w/v) in TBST for 1 h, then incubated in primary antibody in TBST (no blocking reagent) for 2 h, washed, incubated in secondary antibody in TBST (no blocking reagent) for 1 h, and washed. Immunoblotting for vinculin was done using rabbit monoclonal antibody specific for vinculin (E1E9 V, Cat. #13901, Cell Signaling, Danvers, MA, USA; dilution 1:1000) with GAPDH (Sigma; Cat# G8795, dilution 1:10,000) used as a loading control. A goat anti-rabbit antibody conjugated with the fluorescent dye, IR red 800 (Rockland, Cat #611-132-002, dilution 1:4000) and goat anti-mouse antibody conjugated with fluorescent Cy5 (Rockland, Cat. #611-610-110-121, dilution 1:4000) were used as secondary antibodies. Immunoblotting for other proteins of interest was carried out using the following antibodies at the dilutions indicated: PSMA6 1:10,000 (Cat. #3782-1, Epitomics, now Abcam, Cambridge, MA, USA), PSMA3 1:10,000 (Cat. #376601, Epitomics), Rpt4 1:2000 (Cat. #PW8830, Biomol), Ezrin 1:1000 (4A5, Cat. #sc-32759, Santa Cruz, Dallas, TX, USA), HSP-90β 1:1000 (D-19, Cat #sc-1057, Santa Cruz). Peroxidase-conjugated rabbit anti-mouse (Cat. #A9044, Sigma) and goat anti-rabbit (Cat. #A0545, Sigma) secondary antibodies were used at a dilution of 1:40,000. These blots were normalized to total protein (determined by quantification of the total protein on membranes transferred from Stain-Free gels). The vinculin blot was scanned using the Odyssey Infrared Imaging System (LI-COR Inc., Lincoln, NE, USA), and the band intensities were quantified using Image J software (imageJ.en.softonic.com) as described earlier (Yuan et al. 2015). All other blots were incubated with Westar Supernova ECL Substrate (Cod. XLS3, 0200, Cyanagen, Bologna, Italy) and imaged using the Chemidoc MP (Bio-Rad). The band intensities were quantified using ImageLab Version 5 (Bio-Rad).
Statistics
Proteomic results
Differentially expressed proteins had a P value <0.05; at least two labeled unique peptides from the same protein were detected in all three independent experiments. P values were calculated using Bonferroni Corrected Mann–Whitney tests (Scaffold 4.0), which takes into account a multiple sample analysis.
Western blotting
Mean ± standard deviation (SD) are shown (n = 4 hearts per experimental group). Comparisons were performed by one-way ANOVA. P < 0.05 was considered statistically significant.
Results
In this study, we performed an in-depth proteomic analysis to identify the major signaling pathways involved in cardiac remodeling in the hearts of 3.5–4 month-old of pathologic (A57G) versus physiologic (Δ43) mouse models of hypertrophic cardiomyopathy. The results collected using transgenic (Tg) mice expressing the 43 amino acid N-terminal truncated ELC (Δ43) and A57G mutated ELC were compared to those of age and gender matched transgenic WT-ELC mice. TMT labeling and liquid chromatography tandem mass spectrometry (LC–MS/MS) were used to pinpoint the differences in the signaling cascades involved in these two different models of cardiac growth. Although a label-free approach has been found to outperform a TMT approach for proteome coverage (Megger et al. 2014), we used the TMT approach because the label-free method was observed to be less accurate than the TMT approach (Megger et al. 2014). Isotopic variants of TMT contain an amine-reactive group which reacts with the N-terminal amine groups and epsilon-amine groups of the lysine residues of peptides. The TMT labels contain the mass normalization groups which allow the different isotopic variants of TMT tag on the peptide to have the same mass.
Using a TMT quantitative proteomic approach we identified the proteins differentially expressed in the hearts of Δ43 versus A57G mice and differentially expressed in each hypertrophy model versus WT-line 1 controls. The identified proteins and the group comparisons are presented in Tables 1, S1, S2 and S3. Figure 1 presents a bar chart comparing the number of the up- and down-regulated proteins in all heart models of cardiac hypertrophy. The analysis of the results shows that 30 proteins were different in Δ43 versus A57G (Table 1). Examples include mitochondrial NAD(P) transhydrogenase and aldehyde dehydrogenase, which were both 1.46-fold downregulated in Δ43 relative to A57G hearts (Table 1). There were also differences between the two mouse models of hypertrophy and WT hearts with 35 proteins differentially expressed in Δ43 versus WT (Table S1), and 51 that were differently expressed in A57G versus WT (Table S2; Fig. 1).
Table 1.
Proteins differentially expressed in the ELC transgenic mouse models of physiological (Δ43) versus pathological (A57G) cardiac hypertrophy
| Protein | Accession number | Molecular weight (kDa) | Mann–Whitney test (Bonferroni corrected) P value |
Δ43 versus A57G* (FC) |
|---|---|---|---|---|
| Elongation factor 2 | EF2_MOUSE | 95 | <0.0001 | −1.16 |
| Complement C3 | CO3_MOUSE | 186 | <0.0001 | −1.15 |
| Aldehyde dehydrogenase, mitochondrial | ALDH2_MOUSE | 57 | <0.0001 | −1.46 |
| Heat shock protein HSP 90-beta | HS90B_MOUSE | 83 | <0.0001 | −1.14 |
| Leucine-rich PPR motif-containing protein, mitochondrial | LPPRC_MOUSE | 157 | <0.0001 | −1.17 |
| NAD(P) transhydrogenase, mitochondrial | NNTM_MOUSE | 114 | <0.0001 | −1.46 |
| Delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial | AL4A1_MOUSE | 62 | <0.0001 | −1.25 |
| Stress-induced-phosphoprotein 1 | STIP1_MOUSE | 63 | <0.0001 | −1.15 |
| Cluster of Troponin T, cardiac muscle | K3W4R6_MOUSE | 35 | 0.00018 | 1.14 |
| Mitochondrial 2-oxoglutarate/malate carrier protein | M2OM_MOUSE | 34 | 0.0017 | −1.29 |
| Alpha-1B-glycoprotein | A1BG_MOUSE | 57 | 0.0018 | 1.39 |
| Elongation factor 1-gamma | EF1G_MOUSE | 50 | 0.0025 | −1.14 |
| Cluster of Myosin-6 | MYH6_MOUSE | 224 | 0.0039 | 1.11 |
| Succinyl-CoA ligase [GDP-forming] subunit beta, mitochondrial | SUCB2_MOUSE | 47 | 0.0068 | −1.14 |
| Cluster of Inorganic pyrophosphatase 2, mitochondrial | D3Z636_MOUSE | 38 | 0.0069 | −1.25 |
| Coiled-coil-helix-coiled-coil-helix domain-containing protein 3, mitochondrial | CHCH3_MOUSE | 26 | 0.0083 | 1.18 |
| Cluster of Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | MLRV_MOUSE | 19 | 0.011 | 1.08 |
| Histidine rich calcium binding protein, isoform CRA_a | G5E8J6_MOUSE | 85 | 0.017 | −1.10 |
| Clathrin heavy chain 1 | CLH1_MOUSE | 192 | 0.017 | 1.25 |
| Sarcalumenin | SRCA_MOUSE | 99 | 0.02 | −1.12 |
| Cytochrome c oxidase subunit 5B, mitochondrial | COX5B_MOUSE | 14 | 0.02 | 1.12 |
| Serpin H1 | SERPH_MOUSE | 47 | 0.021 | −1.15 |
| Calcium-binding mitochondrial carrier protein Aralar1 | CMC1_MOUSE | 75 | 0.024 | −1.12 |
| Short-chain specific acyl-CoA dehydrogenase, mitochondrial | ACADS_MOUSE | 45 | 0.031 | −1.15 |
| Heterogeneous nuclear ribonucleoprotein U | G3XA10_MOUSE | 87 | 0.038 | −1.19 |
| Cluster of Long-chain-fatty-acid–CoA ligase 1 | ACSL1_MOUSE | 78 | 0.039 | −1.05 |
| Citrate lyase subunit beta-like protein, mitochondrial | CLYBL_MOUSE | 38 | 0.042 | −1.12 |
| Cytochrome b-c1 complex subunit 7 | Q9CQB4_MOUSE | 14 | 0.046 | 1.15 |
| Isovaleryl-CoA dehydrogenase, mitochondrial | IVD_MOUSE | 46 | 0.048 | −1.11 |
| NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial | E9QPX3_MOUSE | 20 | 0.048 | 1.19 |
Negative fold change (FC) values indicate proteins that were significantly downregulated in Δ43 versus A57G hearts, while positive values indicate proteins that were significantly upregulated in Δ43 versus A57G hearts with significance (*) defined as P < 0.05
Fig. 1.
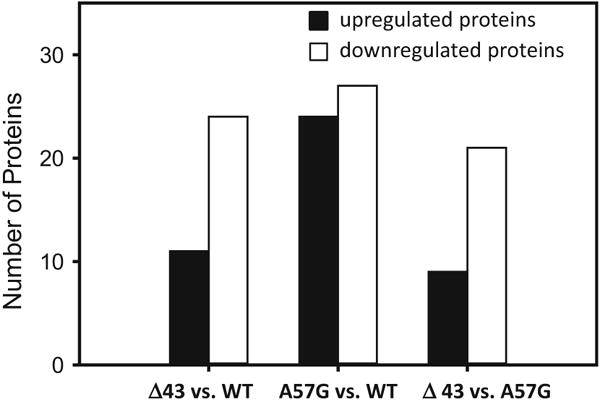
Summary of proteomic analysis showing the number of statistically significantly upregulated and downregulated proteins in the hearts of physiological (Δ43) and pathological (A57G) mouse models of hypertrophy and in the hearts of transgenic WT-ELC mice
To quantify the reproducibility of the experiments, CV of the intensity values across all runs were determined using the scaffold program (Table S3). 86 % of the quantified peptides showed a CV equal to or below 25 %, and the average coefficient of variance of all quantified peptides was 22.5 %. For the 3 replicates of pooled WT samples (used in runs 1–3), CVs of less than 4 % were obtained. Sample wide fold changes between different samples show that very little changes occurred between the pooled WT runs (Fig. S1). Significant differences in sample wide fold changes were observed when pooled WT or individual WT samples were compared to Δ43 or A57G hearts (Fig. S1). Figure S2 shows a Venn diagram of the number of proteins identified with two TMT labeled unique peptides in the WT pooled sample in the three different runs (Fig. S2). Altogether 26 % of the proteins were detected in only one run. For the proteins that were found to be differentially expressed in Δ43 relative to A57G hearts the number of unique peptides that were quantified was ≥7 (Table S4). A sample file of cytochrome b-c1 complex peptides detected and the relative intensity of the different peptides in the different heart samples is shown in Table S5.
Comparison between physiological and pathological hypertrophy
Tables 1 and S1, and Fig. 2 present differentially expressed proteins in the hearts of Δ43 versus A57G mice. Signaling pathways different between these two animal models included proteins involved in metabolic processes. Classification of differentially expressed proteins into pathways using the Panther program, which utilizes the Gene Ontology (GO) classification scheme, revealed 19 proteins involved in metabolic processes that were significantly different in Δ43 versus A57G hearts. There were also significant differences between heart models in biogenesis, cellular organization, and biological regulation (Fig. 2a; Tables 1, S1). In addition to biological processes (Fig. 2a), many significant differences between physiological (Δ43) and pathological (A57G) hypertrophy heart models were noted in pathways involved in cellular processes (Fig. 2b), and in molecular functions (Fig. 2c). An example of a protein downregulated in Δ43 relative to A57G is Succinyl-CoA ligase [GDP-forming] subunit beta (mitochondrial) (Table 1). This protein is involved in the tricarboxylic acid cycle and catalyzes the GTP-dependent ligation of succinate and CoA to form succinyl-CoA.
Fig. 2.
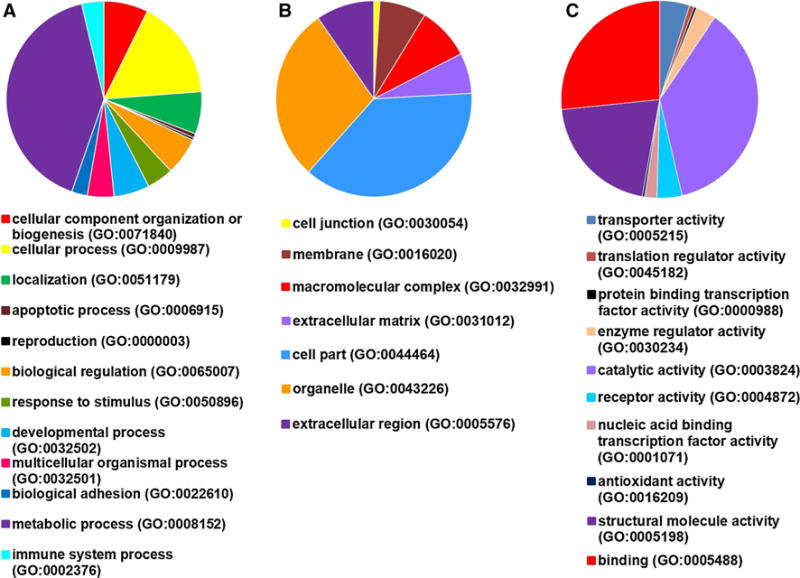
Processes affected in physiological (Δ43) versus pathological (A57G) cardiac hypertrophy. a Biological processes; b cellular processes; c molecular function
Metabolic changes with respect to the actomyosin ATPase activity in skinned papillary muscle fibers from Δ43 hearts suggested that pathways involved in ATP production might be affected in this model of cardiac hypertrophy (Kazmierczak et al. 2009). Indeed, aldehyde dehydrogenase was downregulated while trifunctional enzyme subunit alpha and 3-ketoacyl-CoA thiolase expression levels were higher only in Δ43 hearts when compared to control hearts (Table 2). These enzymes play important roles in metabolism of corticosteroids and lipid peroxidation (aldehyde dehydrogenase), fatty acid metabolism (trifunctional enzyme and 3-ketoacyl-CoA thiolase), and in other processes such as growth control and may regulate the energetic and metabolic pathways specific for the physiological type of hypertrophy observed in transgenic Δ43 hearts.
Table 2.
Mitochondrial proteins involved in metabolic processes that show altered expression levels in transgenic ELC mice
| Protein name | UniProt | Gene | PANTHER protein class | A57G versus WT (FC) | Δ43 versus WT (FC) | Δ43 versus A57G (FC) |
|---|---|---|---|---|---|---|
| Aldehyde dehydrogenase, mitochondrial | P47738 | ALDH2_MOUSE | Dehydrogenase | 1.20* | −1.21* | −1.46* |
| Atpase inhibitor, mitochondrial | O35143 | E9PV44_MOUSE | – | −1.58* | −1.20 | 1.32 |
| Citrate lyase subunit beta-like protein, mitochondrial | Q8R4N0 | CLYBL_MOUSE | – | 1.07 | −1.03 | −1.11* |
| Cytochrome C oxidase subunit 5B, mitochondrial | P19536 | COX5B_MOUSE | Oxidase | −1.12 | 1.04 | 1.12* |
| Cytochrome B-C1 complex subunit 7 | Q9CQB4 | Q9CQB4_MOUSE | – | −1.07 | 1.07 | 1.15* |
| Delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial | Q8CHT0 | AL4A1_MOUSE | Dehydrogenase | −1.14* | −1.42* | −1.25* |
| Isovaleryl-COA dehydrogenase, mitochondrial | Q9JHI5 | IVD_MOUSE | Dehydrogenase | 1.10 | 1.00 | −1.11* |
| Cluster of inorganic pyrophosphatase 2, mitochondrial | D3Z636 | D3Z636_MOUSE | – | 1.19* | −1.03 | −1.25* |
| FUMARATE hydratase, mitochondrial | P97807 | FUMH_MOUSE | Lyase | 1.14* | 1.03 | −1.09 |
| 3-Ketoacyl-COA thiolase, mitochondrial | Q8BWT1 | THIM_MOUSE | Acetyltransferase | 1.14 | 1.16* | −1.03 |
| Mitochondrial 2-oxoglutarate/malate carrier protein | Q9CR62 | M2OM_MOUSE | Calmodulin | 1.28* | 1.00 | −1.29* |
| NAD(P) Transhydrogenase, mitochondrial | Q61941 | NNTM_MOUSE | Dehydrogenase | 1.13* | −1.29 | −1.46* |
| NADH dehydrogenase [ubiquinone] 1 alpha Subcomplex subunit 9, mitochondrial | Q9DC69 | NDUA9_MOUSE | Dehydrogenase | 1.13* | 1.00 | −1.13 |
| NADH dehydrogenase [ubiquinone] iron-sulphur protein 4, mitochondrial | E9QPX3 | E9QPX3_MOUSE | – | −1.07 | 1.13 | 1.19* |
| succinyl-COA ligase [GDP-forming] subunit beta, mitochondrial | Q9Z2I8 | SUCB2_MOUSE | Ligase | 1.20* | 1.07 | −1.14* |
| Short-chain specific acyl-COA dehydrogenase, mitochondrial | Q07417 | ACADS_MOUSE | Dehydrogenase | 1.13 | 1.00 | −1.15* |
| Trifunctional enzyme subunit alpha, mitochondrial | Q8BMS1 | ECHA_MOUSE | Dehydrogenase, hydratase, epimerase | 1.10 | 1.13* | 1.03 |
P < 0.05. FC fold change. Negative FC values indicate proteins that were downregulated, while positive FC values those which were upregulated
Several calcium-binding proteins were altered in the hypertrophied transgenic hearts (Table 3). Interestingly, the myosin light polypeptide 6 (gene MYL6) was down-regulated in the pathological hypertrophy heart model compared with WT (Tables 3, S2). MYL6 encodes for the smooth muscle myosin ELC, which is essential for myosin’s contractile function. In addition, MYL2, depicting the mouse ventricular myosin regulatory light chain (UniProt # P51667, MLRV_MOUSE) was significantly downregulated in A57G versus Δ43 hearts (Table 3). As a part of the myosin lever arm, the light chain structurally supports myosin during the power stroke and force generation, but also was shown to play a role in the regulation of gene expression (Li and Sarna 2009). In addition it was shown to function as a transcription factor to promote NOX2 (NADPH oxidase 2) expression following myocardial ischemia/reperfusion injury in a phosphorylation-dependent manner (Zhang et al. 2015a, 2015b). Other altered calcium-binding proteins in A57G mice included sarcalumenin, calcium-binding mitochondrial carrier protein ARALAR1 and mitochondrial 2-oxoglutarate/malate carrier protein, which were all higher in A57G hearts when compared to WT hearts (Tables 3, S1). Upregulation of ARALAR1 may have important physiological implications, as this calcium-binding protein localizes to mitochondria and is involved in catalysis of the calcium-dependent exchange of cytoplasmic glutamate with mitochondrial aspartate across the mitochondrial inner membrane, and thus functions as a calcium-regulated metabolite carrier (Del Arco et al. 2000). Likewise, the mitochondrial 2-oxoglutarate/malate carrier protein, which transports 2-oxoglutarate across the inner mitochondrial membrane, was upregulated in the A57G model (Tables 3, S1). The transport of 2-oxoglutarate across the inner mitochondrial membrane is important in several metabolic processes, including the malate-aspartate shuttle, the oxoglutarate/isocitrate shuttle, gluconeogenesis from lactate, and in nitrogen metabolism. The Δ43 hearts showed no statistically significant changes in the levels of calcium-binding proteins relative to WT hearts (Table 3). When compared with pathologic model of cardiac hypertrophy, sarcalumenin (gene SRCA), ARALAR1 (gene SMC1), sarcoplasmic reticulum histidine-rich calcium-binding protein, isoform CRA_a (gene G5E8J6), and mitochondrial 2-oxoglutarate/malate carrier protein (gene M2OM) were all lower in Δ43 compared with A57G hearts (Tables 3, S2). The histidine-rich calcium-binding protein, which exhibits high-capacity calcium-binding properties, has been suggested to be a potential regulator of both SERCA2a and RyR2 functions (Bers and Guo 2005). Overexpression of histidine-rich calcium-binding protein in neonatal or adult rat cardiomyocytes increased SR calcium storage capacity and decreased sarcoplasmic reticulum calcium-induced calcium-release (Fan et al. 2004).
Table 3.
Calcium-binding proteins with altered expression levels in transgenic ELC mice
| Protein name | UniProt | Gene | PANTHER protein class | A57G versus WT (FC) | A57G Δ43 versus WT (FC) | A57G Δ43 versus A57G (FC) | A57G versus Δ43 (FC) |
|---|---|---|---|---|---|---|---|
| Sarcalumenin | Q7TQ48 | SRCA_MOUSE | Calcium-binding protein | 1.12* | 1.03 | −1.12* | 1.12* |
| Mitochondrial 2-oxoglutarate/malate carrier protein | Q9CR62 | M2OM_MOUSE | Calmodulin | 1.28* | 1.00 | −1.29* | 1.29* |
| Calcium-binding mitochondrial carrier protein Aralar1 | Q8BH59 | CMC1_MOUSE | Calmodulin | 1.17* | 1.07 | −1.12* | 1.12* |
| Sarcoplasmic reticulum histidine-rich calcium-binding protein, isoform CRA_A | G5E8J6 | G5E8J6_MOUSE | – | −1.11 | 1.13 | −1.10* | 1.10* |
| Myosin regulatory light chain 2, ventricular/cardiac muscle form | P51667 | MLRV_MOUSE | Calmodulin | −1.15* | −1.08 | 1.08* | −1.08* |
| Myosin light polypeptide 6 | Q60605 | MYL6_MOUSE | Calmodulin | −1.44* | −1.22 | 1.17 | −1.17 |
P < 0.05; FC fold change, negative FC values indicate proteins that were downregulated, while positive FC values those which were upregulated
Among chaperones that constitute another important group of proteins, many were differentially altered in both models of hypertrophy when compared with WT hearts (Table 4). Several heat shock proteins were downregulated in both A57G and Δ43 mice relative to WT hearts. Heat shock protein (HSP)-90β and stress-induced phosphoprotein 1 were both found to be downregulated in Δ43 hearts relative to A57G hearts (Tables 1, 4). In an autoimmune myocarditis-induced chronic heart failure rat model HSP-90β was suggested to play a protective role (Sanzen et al. 2010). Increased levels of stress-induced phosphoprotein-1 have been associated with cancer (Cho et al. 2014).
Table 4.
Chaperone proteins with altered expression levels in transgenic ELC mice
| Protein name | UniProt | Gene | PANTHER protein class | A57G versus WT (FC) | Δ43 versus WT (FC) | Δ43 versus A57G (FC) |
|---|---|---|---|---|---|---|
| Alpha-crystallin B chain | P23927 | CRYAB_MOUSE | Chaperone | −1.21* | −1.21 | 1.00 |
| Stress-70 protein, mitochondrial | P38647 | GRP75_MOUSE | Hsp70 family chaperone | −1.18* | −1.13* | 1.04 |
| Heat shock protein beta-1 | P14602 | HSPB1_MOUSE | Chaperone | −1.34 | −1.43* | 1.00 |
| Endoplasmin-like protein-related | P08113 | ENPL_MOUSE | Hsp90 family chaperone | −1.19* | 1.07 | 1.07 |
| BAG family molecular chaperone regulator 3 | Q9JLV1 | BAG3_MOUSE | Chaperone | −1.24* | −1.12 | 1.13 |
| 10 KDA heat shock protein, mitochondrial | Q64433 | CH10_MOUSE | Chaperonin | −1.25* | −1.24* | 1.00 |
| Heat shock protein HSP 90-alpha | P07901 | HS90A_MOUSE | Hsp90 family chaperone | −1.22 | −1.36* | −1.17 |
| Heat shock protein HSP 90-beta | P11499 | HS90B_MOUSE | Hsp90 family chaperone | 1.03 | −1.10* | −1.14* |
| Heat shock 70 KDA protein 4 | Q3U2G2 | Q3U2G2_MOUSE | Hsp70 family chaperone | −1.07 | −1.16* | −1.08 |
| Stress-induced-phosphoprotein 1 | Q60864 | STIP1_MOUSE | Chaperone | 1.00 | −1.13* | −1.15* |
| Heat shock cognate 71 KDA protein | P63017 | HSP7C_MOUSE | Hsp70 family chaperone | −1.03 | −1.17* | −1.12 |
P < 0.05; FC fold change, negative FC values indicate proteins that were downregulated, while positive FC values those which were upregulated
Table 5 presents cytoskeletal proteins that were differentially altered in both models of cardiac hypertrophy. Changes in the levels of important cytoskeletal proteins included cofilin-2, meosin, and cytoplasmic dynein 1 heavy chain 1 (Table 5). Moesin is an important cytoskeletal protein which has been shown to interact with ezrin and radixin (Amsellem et al. 2014). Interestingly, meosin and stress-induced phosphoprotein-1, which were both downregulated in Δ43 hearts and not in A57G hearts, have been recently suggested to be possible sero-diagnostic markers of psoriasis (Maejima et al. 2014). Among cytoskeletal proteins altered in pathologic (A57G) versus physiologic (Δ43) hypertrophy was myosin heavy chain 6 (α-MHC) downregulated in A57G hearts (Table 5). The upregulation of the β-MHC in mouse myocardium and the switch from the α-MHC to β-MHC has been previously reported in older A57G animals (Kazmierczak et al. 2013). Upregulation of β-MHC was also seen in other mouse models of heart disease (Barefield et al. 2014) and occurred in response to a wide variety of pathological insults (reviewed in (Harvey and Leinwand 2011)).
Table 5.
Cytoskeletal proteins with altered expression levels in transgenic ELC mice
| Protein name | UniProt | Gene | PANTHER protein class | A57G versus WT (FC) | Δ43 versus WT (FC) | Δ43 versus A57G (FC) |
|---|---|---|---|---|---|---|
| Moesin | P26041 | MOES_MOUSE | Actin family cytoskeletal protein | −1.15 | −1.16* | −1.04 |
| Cofilin-2 | P45591 | COF2_MOUSE | Non-motor actin binding protein | −1.29* | −1.22* | 1.04 |
| Cytoplasmic dynein 1 heavy chain 1 | Q9JHU4 | DYHC1_MOUSE | Microtubule binding motor protein | −1.03 | −1.08* | −1.07 |
| Glutathione S-transferase Omega-1 | O09131 | GSTO1_MOUSE | Cytoskeletal protein | −1.43* | −1.31* | 1.10 |
| Myosin-6 | Q02566 | MYH6_MOUSE | Actin binding motor protein | −1.07* | 1.00 | 1.11* |
| Myosin light polypeptide 6 | Q60605 | MYL6_MOUSE | Actin family cytoskeletal protein, calmodulin | −1.43* | −1.22 | 1.17 |
| Elongation factor 1-gamma | Q9D8N0 | EF1G_MOUSE | Cytoskeletal protein, translation elongation factor | 1.10 | −1.07 | −1.14* |
P < 0.05; FC fold change, negative FC values indicate proteins that were downregulated, while positive FC values those which were upregulated
Independent validation of HSP-90β by Western blotting (Fig. 3a) showed that this protein was downregulated in Δ43 hearts relative to both WT and A57G hearts. This result is consistent with the mass spectrometry data which also showed downregulation of HSP-90β in Δ43 hearts relative to both WT and A57G hearts (Tables 1, 4). Collectively, these results suggest that altered heat shock protein expression occurs differently in the two transgenic heart models. Investigation of cytoskeletal and proteasome proteins that were not differentially expressed in transgenic hearts by Western blotting showed that vinculin (Fig. 3b), two 20S proteasome subunits, PSMA3 (Fig. 3c), PSMA6 (Fig. 3d) and one 19S proteasome subunit, Rpt4 (Fig. 3e) were all unchanged similar to what was detected by the proteomic results. The proteasome is a complex unit made up of over 32 subunits (Cui et al. 2014), and changes in expression have often been observed in cardiac diseases (Powell et al. 2012). However, no changes in expression of proteasome subunits were observed in our study in either A57G or Δ43 heart models. Another cytoskeletal protein, ezrin, which was not differentially expressed in the proteomic studies was found to be greater in the A57G hearts when compared to Δ43 hearts (P < 0.05) (Fig. 3f), while it was similar among Δ43 and WT hearts. The proteomic results for ezrin, although not statistically significant, suggested a trend towards increased expression levels of ezrin in A57G hearts relative to Δ43 hearts (Table S3). Bar graphs in Fig. 3 represent the data (average ± SD) from Western blots performed on four hearts per group of mice.
Fig. 3.
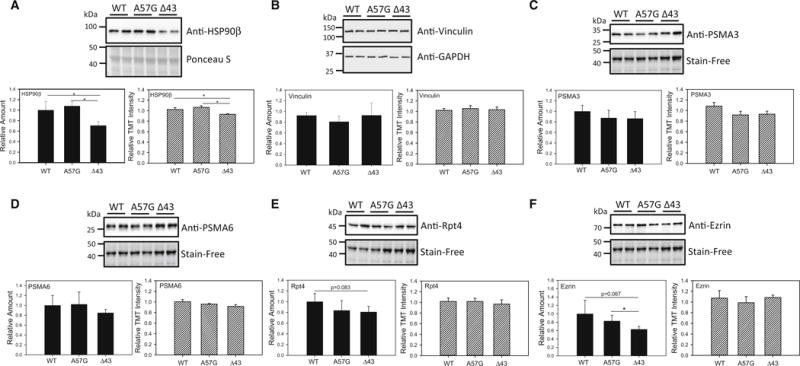
Validation of proteomic analyses by Western blotting in WT, Δ43 and A57G hearts. a Expression of HSP-90β. b Expression of Vinculin; c expression of 20S proteasome subunit, PSMA3. d Expression of 20S proteasome subunit, PSMA6. e Expression of 19S proteasome subunit, Rpt4. f Expression of Ezrin. Bar charts containing black bars indicate the protein expression determined by Western blotting, while striped white bars represent the protein expression as determined by TMT labeling of heart lysates and quantification by mass spectrometry. Note no changes in expression of any proteasome subunits between A57G and Δ43 hearts, while differences between pathological (A57G) and physiological (Δ43) hypertrophy were noted in expression of HSP-90β and Ezrin
Discussion
Hypertrophic cardiac remodeling is a highly important phenomenon; however, the molecular mechanisms and signaling pathways underlying this process are not fully understood. Recognition of functional, structural, metabolic, and molecular differences between non-toxic and disease-causing mutations is critical in the effort to develop potential target-specific therapeutic approaches. Disease severity depends on various factors, e.g. disease-modifying genes, cardiac load, and non-genetic modifiers including epigenetic modifications, post-translational modifications of proteins, and environmental disease triggers (van der Velden et al. 2015). In addition to distinguishable morphological and functional phenotypes between HCM and physiological cardiac hypertrophy, the signaling cascades were shown to be distinctive (Chung and Leinwand 2014; McMullen et al. 2007). The physiologic and pathological growth of the myocardium is typically initiated by membrane-bound receptors that promote intracellular signaling through multiple GTPase proteins, kinases, and phosphatases (Chang et al. 2015; Chen et al. 2015a; Heineke and Molkentin 2006). The MAPK (mitogen-activated protein kinase) signaling pathways consist of a sequence of successively functioning kinases that ultimately result in the dual phosphorylation and activation of p38, c-Jun N-terminal kinases (JNKs) and extracellular signal-regulated kinases (ERKs) (Garrington and Johnson 1999; Yujiri et al. 1999).
In this report, we investigated the molecular pathways leading to HCM versus physiologic hypertrophy using a quantitative proteomic approach applied to transgenic mouse hearts carrying mutations in the human cardiac myosin ELC (Kazmierczak et al. 2014). Gene-manipulated mouse models that closely recapitulate the clinical phenotypes of human inherited cardiomyopathies are invaluable in demonstrating the true cause of the disease and are useful in exploring pathogenic mechanisms. Our previous investigations showed that even though transgenic Δ43 mice develop substantial cardiac hypertrophy, there were no signs of histopathology or fibrosis and no altered cardiac function (Kazmierczak et al. 2009; Muthu et al. 2011). Therefore, this mouse model served as an excellent tool to study the signaling pathways that might underlie the physiological type of cardiac hypertrophy. On the other hand, the A57G mice showed clear evidence of pathological morphology and function (Kazmierczak et al. 2013; Muthu et al. 2011), and our mechanics studies suggested that the A57G allele may cause HCM by means of a discrete modulation of myofilament function, reduced maximal force, and increased myofilament Ca2+ sensitivity (Kazmierczak et al. 2013). Therefore, the A57G mouse model was ideally suited to investigate the pathways specific to pathological ventricular remodeling.
The proteomic data suggest that multiple signaling pathways might be affected in the hearts of Δ43 and A57G mice. The A57G hearts showed more proteins that were altered in this mouse model compared with Δ43 when both heart models were compared to WT hearts. Importantly, while the Δ43 hearts showed no changes in the levels of calcium-binding proteins relative to WT hearts, the A57G hearts demonstrated an upregulation or downregulation of various calcium-binding proteins localized in different cellular compartments, when compared with WT or Δ43 hearts. Alterations in calcium-binding proteins in A57G mice observed in our proteomic experiments are in accordance with functional studies on papillary muscle fibers from Tg-A57G mice showing significant changes in the Ca2+-mediated regulation of muscle contraction in the A57G versus WT myocardium (Kazmierczak et al. 2013; Kazmierczak et al. 2014). In contrast, no disruption of Ca2+-dependent force development was observed in transgenic Δ43 versus WT hearts (Kazmierczak et al. 2009, 2014).
The expression of other important calcium-binding proteins, e.g. sarcalumenin and the mitochondrial 2-oxoglutarate/malate carrier protein, as well as several chaperone proteins including the heat shock protein beta-1 and 10 kDa heat shock protein, was downregulated in Δ43 versus A57G hearts. Expression of HSP-90β and stress-induced phosphoprotein 1 was also downregulated in Δ43 relative to A57G and WT hearts. The decreased levels of HSP-90β in Δ43 hearts were verified by Western blotting.
Since several heat shock proteins were observed to be involved in cardiovascular diseases, it is conceivable that their levels of expression are differentially altered in the pathologic versus physiologic models of hypertrophy (Chen et al. 2015b). On the other hand, the mitochondrial proteins NADH dehydrogenase (ubiquinone) iron-sulphur protein 4, cytochrome c oxidase subunit 5B and cytochrome b-c1 complex subunit 7 were all upregulated in Δ43 versus A57G hearts. Ubiquinone, cytochrome c oxidase and the cytochrome b-c1 complex are all critical for ATP production in the mitochondria. The group of cytoskeletal proteins that were upregulated in Δ43 versus A57G hearts included the myosin heavy chain 6 (α-MHC) and the myosin light polypeptide 6 (smooth muscle ELC). Interestingly, the switch from the α-MHC to β-MHC has been previously reported in older A57G animals (Kazmierczak et al. 2013), and was also seen in other mouse models of HCM (Barefield et al. 2014; Harvey and Leinwand 2011). The alterations in the smooth muscle ELC expression in Δ43 versus WT hearts (downregulation) or its upregulation in Δ43 versus A57G hearts, could be due to the inability of the Δ43 myocardium to produce the full length ELC protein with the intact N-terminal extension shown to be needed to maintain the proper actomyosin molecular contacts during force generation and muscle contraction (Muthu et al. 2011).
Pathologic cardiac hypertrophy manifests itself through the reappearance of fetal metabolic patterns with decreased fatty acid metabolism (reduced oxidative metabolism) and increased glucose utilization (Allard et al. 1994; Barger and Kelly 1999; Doenst et al. 2010; Razeghi et al. 2001; Uehara et al. 1998). Changes in these two metabolic profiles, specifically metabolic intermediates affecting cardiomyocyte function, might be contributing factors to the development of pathological heart remodeling. It has been reported that increased preference for glucose utilization in overload-induced hypertrophy increases flux through the hexoamine biosynthesis pathway (Young et al. 2007). Other studies reported that upregulation of O-linked β-N-acetylglucosamine (O-GlcNAc) signaling is required for activation of the transcriptional reprogramming that occurs at the onset and progression of cardiac hypertrophy, while cardiac-specific genetic ablation of O-linked β-N-acetylglucosamine transferase, which catalyzes O-GlcNAc synthesis, prevents transverse aortic constriction (TAC)-induced hypertrophy (Facundo et al. 2012; Watson et al. 2010). Furthermore, production and efflux of lactate from the myocardium are increased in hypertrophied hearts (Smith et al. 1990). Two related proteins involved in fatty acid metabolism, trifunctional enzyme subunit alpha, and 3-ketoacyl-COA thiolase were observed to be upregulated in Δ43 hearts relative to WT hearts. Although A57G hearts also show a trend towards increased levels of these two proteins relative to WT hearts, these increases were not statistically significant. 3-ketoacyl-COA thiolase is also called trifunctional enzyme subunit beta, and the mitochondrial trifunctional protein is associated with the inner mitochondrial membrane and involved in the final step of beta-oxidation utilizing long chain fatty acids as substrate. Mice lacking the mitochondrial trifunctional enzyme alpha subunit resulted in fetal growth retardation, neonatal hypoglycemia and sudden death (Ibdah et al. 2001). Lack of the trifunctional protein has also been linked to severe fatty acid oxidation disorder with cardiac and neurologic involvement (den Boer et al. 2003). In addition, reduced rates of fatty acid oxidation are observed in pathologic cardiac hypertrophy, which in turn may lead to accumulation of free fatty acids and their derivatives, and play a role in the development of pathological versus physiological hypertrophy (Stanley et al. 1997).
Changes in the activity of the ubiquitin proteasome system have been reported to play important roles in hypertrophic hearts, and increased protein expression levels together with changes in proteasome activity were shown to be associated with cardiac dysfunction (Depre et al. 2006). Mutants of myosin binding protein C, the thick filament-containing protein, have also been shown to impair the ubiquitin proteasome system (Sarikas et al. 2005). However, no differences were detected in the levels of these proteasome subunits suggesting that the expression levels of the proteasome complex in these heart models were similar.
Conclusions
Collectively, our data suggest that the A57G mediated alterations in Ca2+ homeostasis and fatty acid metabolism may be important in the pathological cardiac remodeling observed in transgenic A57G hearts. The physiological hypertrophy associated with the truncation mutation in myosin ELC does not seem to be driven by changes in Ca2+ homeostasis and may be due to differential regulation of chaperones, metabolic proteins, and cytoskeletal proteins. The proteomic data for the A57G hearts support our functional studies on A57G hearts suggesting that alterations in Ca2+ homeostasis and the α- to β-MHC switch may contribute to the pathological cardiac remodeling and be responsible for the development of HCM. Our findings on Δ43 hearts suggest that increases in the key mitochondrial and cytoskeletal proteins might be to compensate for hypertrophic growth and to prevent the Δ43 hearts from transitioning from physiological to pathological cardiac remodeling and heart failure.
Supplementary Material
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) Grants HL108343 and HL123255 (DSC); HL096819 (AVG), and the American Heart Association Grants 15PRE23020006 (CCY) and 15POST25080302 (ZZ).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10974-015-9434-0) contains supplementary material, which is available to authorized users.
Contributor Information
Aldrin V. Gomes, Email: avgomes@ucdavis.edu.
Danuta Szczesna-Cordary, Email: dszczesna@med.miami.edu.
References
- Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- Amsellem V, et al. ICAM-2 regulates vascular permeability and N-cadherin localization through ezrin-radixin-moesin (ERM) proteins and Rac-1 signalling. Cell Commun Signal. 2014;12:12. doi: 10.1186/1478-811X-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield D, Kumar M, de Tombe PP, Sadayappan S. Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2014;306:H807–815. doi: 10.1152/ajpheart.00913.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. 1999;318:36–42. doi: 10.1097/00000441-199907000-00006. [DOI] [PubMed] [Google Scholar]
- Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci. 2005;1047:86–98. doi: 10.1196/annals.1341.008. [DOI] [PubMed] [Google Scholar]
- Chang AN, et al. Constitutive phosphorylation of cardiac myosin regulatory light chain in vivo. J Biol Chem. 2015 doi: 10.1074/jbc.M115.642165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, et al. In vivo roles for myosin phosphatase targeting subunit-1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J Physiol. 2015a;593:681–700. doi: 10.1113/jphysiol.2014.283853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TH, et al. Neonatal death and heart failure in mouse with transgenic HSP60 expression. Biomed Res Int. 2015b;2015:539805. doi: 10.1155/2015/539805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, et al. Expression of stress-induced phosphoprotein1 (STIP1) is associated with tumor progression and poor prognosis in epithelial ovarian cancer. Genes Chromosom Cancer. 2014;53:277–288. doi: 10.1002/gcc.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Leinwand LA. Pregnancy as a cardiac stress model. Cardiovasc Res. 2014;101:561–570. doi: 10.1093/cvr/cvu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Dewey S, Gomes AV. Cardioproteomics: advancing the discovery of signaling mechanisms involved in cardiovascular diseases. Am J Cardiovasc Dis. 2011;1:274–292. [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Scruggs SB, Gilda JE, Ping P, Gomes AV. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol. 2014;71:32–42. doi: 10.1016/j.yjmcc.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Agudo M, Satrústegui J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem J. 2000;345:725–732. doi: 10.1042/0264-6021:3450725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer ME, Dionisi-Vici C, Chakrapani A, van Thuijl AO, Wanders RJ, Wijburg FA. Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142:684–689. doi: 10.1067/mpd.2003.231. [DOI] [PubMed] [Google Scholar]
- Depre C, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- Dewey S, Lai X, Witzmann FA, Sohal M, Gomes AV. Proteomic analysis of hearts from Akita mice suggests that increases in soluble epoxide hydrolase and antioxidative programming are key changes in early stages of diabetic cardiomyopathy. J Proteome Res. 2013;12:3920–3933. doi: 10.1021/pr4004739. [DOI] [PubMed] [Google Scholar]
- Doenst T, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res. 2010;86:461–470. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol. 2012;302:H2122–H2130. doi: 10.1152/ajpheart.00775.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GC, Gregory KN, Zhao W, Park WJ, Kranias EG. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am J Physiol Heart Circ Physiol. 2004;287:H1705–H1711. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Geeves MA, Holmes KC. The molecular mechanism of muscle contraction. Adv Protein Chem. 2005;71:161–193. doi: 10.1016/S0065-3233(04)71005-0. [DOI] [PubMed] [Google Scholar]
- Gomes AV, et al. Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol Cell Proteomics. 2009;8:302–315. doi: 10.1074/mcp.M800058-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AV, et al. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006;99:362–371. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- Harvey PA, Leinwand LA. Cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Hernandez OM, Jones M, Guzman G, Szczesna-Cordary D. Myosin essential light chain in health and disease. Am J Physiol Heart Circ Physiol. 2007;292:H1643–H1654. doi: 10.1152/ajpheart.00931.2006. [DOI] [PubMed] [Google Scholar]
- Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med. 1999;341:1276–1283. doi: 10.1056/NEJM199910213411706. [DOI] [PubMed] [Google Scholar]
- Ibdah JA, et al. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest. 2001;107:1403–1409. doi: 10.1172/JCI12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaski JP, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- Kazmierczak K, et al. Discrete effects of A57G-myosin essential light chain mutation associated with familial hypertrophic cardiomyopathy American journal of physiology. Heart Circ Physiol. 2013;305:H575–H589. doi: 10.1152/ajpheart.00107.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak K, Xu Y, Jones M, Guzman G, Hernandez OM, Kerrick WGL, Szczesna-Cordary D. The role of the N-terminus of the myosin essential light chain in cardiac muscle contraction. J Mol Biol. 2009;387:706–725. doi: 10.1016/j.jmb.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak K, Yuan C-C, Liang J, Huang W, Rojas AI, Szczesna-Cordary D. Remodeling of the heart in hypertrophy in animal models with myosin essential light chain mutations. Front Physiology. 2014;5:353. doi: 10.3389/fphys.2014.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Lee W, et al. Different expressivity of a ventricular essential myosin light chain gene Ala57Gly mutation in familial hypertrophic cardiomyopathy. Am Heart J. 2001;141:184–189. doi: 10.1067/mhj.2001.112487. [DOI] [PubMed] [Google Scholar]
- Li Q, Sarna SK. Nuclear myosin II regulates the assembly of preinitiation complex for ICAM-1 gene transcription. Gastroenterology. 2009;137(1051–1060):e1051–e1053. doi: 10.1053/j.gastro.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Adams RM, Chourey K, Hurst GB, Hettich RL, Pan C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res. 2012;11:1582–1590. doi: 10.1021/pr200748h. [DOI] [PubMed] [Google Scholar]
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. doi: 10.1161/01.CIR.102.4.470. [DOI] [PubMed] [Google Scholar]
- Maejima H, Nagashio R, Yanagita K, Hamada Y, Amoh Y, Sato Y, Katsuoka K. Moesin and stress-induced phosphoprotein-1 are possible sero-diagnostic markers of psoriasis. PLoS One. 2014;9:e101773. doi: 10.1371/journal.pone.0101773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ. The young competitive athlete with cardiovascular abnormalities: causes of sudden death, detection by preparticipation screening, and standards for disqualification. Card Electrophysiol Rev. 2002;6:100–103. doi: 10.1023/A:1017903709361. [DOI] [PubMed] [Google Scholar]
- McMullen JR, et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megger DA, et al. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim Biophys Acta. 2014;1844:967–976. doi: 10.1016/j.bbapap.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Miller MS, et al. The essential light chain N-terminal extension alters force and fiber kinetics in mouse cardiac muscle. J Biol Chem. 2005;280:34427–34434. doi: 10.1074/jbc.M508430200. [DOI] [PubMed] [Google Scholar]
- Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29:549–555. doi: 10.1016/s0735-1097(96)00530-x. [DOI] [PubMed] [Google Scholar]
- Morita H, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthu P, et al. Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J. 2011;25:4394–4405. doi: 10.1096/fj.11-191973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Olson TM, Karst ML, Whitby FG, Driscoll DJ. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation. 2002;105:2337–2340. doi: 10.1161/01.cir.0000018444.47798.94. [DOI] [PubMed] [Google Scholar]
- Poetter K, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- Powell SR, Herrmann J, Lerman A, Patterson C, Wang X. The ubiquitin-proteasome system and cardiovascular disease. Prog Mol Biol Trans Sci. 2012;109:295–346. doi: 10.1016/B978-0-12-397863-9.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13:5293–5309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- Richard P, et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. and erratum (2004) 2109(2225):3258. [DOI] [PubMed] [Google Scholar]
- Sanzen Y, et al. Functional proteomic analysis of experimental autoimmune myocarditis-induced chronic heart failure in the rat. Biol Pharm Bull. 2010;33:477–486. doi: 10.1248/bpb.33.477. [DOI] [PubMed] [Google Scholar]
- Sarikas A, et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Shadforth IP, Dunkley TP, Lilley KS, Bessant C. i-Tracker: for quantitative proteomics using iTRAQ. BMC Genom. 2005;6:145. doi: 10.1186/1471-2164-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SH, Kramer MF, Reis I, Bishop SP, Ingwall JS. Regional changes in creatine kinase and myocyte size in hypertensive and nonhypertensive cardiac hypertrophy. Circ Res. 1990;67:1334–1344. doi: 10.1161/01.res.67.6.1334. [DOI] [PubMed] [Google Scholar]
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- Timson DJ. Fine tuning the myosin motor: the role of the essential light chain in striated muscle myosin. Biochimie. 2003;85:639–645. doi: 10.1016/s0300-9084(03)00131-7. [DOI] [PubMed] [Google Scholar]
- Uehara T, et al. Myocardial glucose metabolism in patients with hypertrophic cardiomyopathy: assessment by F-18-FDG PET study. Ann Nucl Med. 1998;12:95–103. doi: 10.1007/BF03164836. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Ho CY, Tardiff JC, Olivotto I, Knollmann BC, Carrier L. Research priorities in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:449–456. doi: 10.1093/cvr/cvv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBuren P, Waller GS, Harris DE, Trybus KM, Warshaw DM, Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc Natl Acad Sci USA. 1994;91:12403–12407. doi: 10.1073/pnas.91.26.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LJ, et al. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci USA. 2010;107:17797–17802. doi: 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ME, et al. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul Syst Biol. 2007;1:251–262. doi: 10.4137/grsb.s222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CC, et al. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2015 doi: 10.1073/pnas.1505819112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yujiri T, Fanger GR, Garrington TP, Schlesinger TK, Gibson S, Johnson GL. MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J Biol Chem. 1999;274:12605–12610. doi: 10.1074/jbc.274.18.12605. [DOI] [PubMed] [Google Scholar]
- Zhang YS, et al. Nuclear cardiac myosin light chain 2 modulates NADPH oxidase 2 expression in myocardium: a novel function beyond muscle contraction. Basic Res Cardiol. 2015a;110:38. doi: 10.1007/s00395-015-0494-5. [DOI] [PubMed] [Google Scholar]
- Zhang YS, et al. A novel function of nuclear nonmuscle myosin regulatory light chain in promotion of xanthine oxidase transcription after myocardial ischemia/reperfusion. Free Radic Biol Med. 2015b;83:115–128. doi: 10.1016/j.freeradbiomed.2015.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.