

Abstract
MK-1775 is the first-in-class selective Wee1 inhibitor which has been demonstrated to synergize with CHK1 inhibitors in various malignancies. In this study, we report that the pan-histone deacetylase inhibitor (HDACI) panobinostat synergizes with MK-1775 in acute myeloid leukemia (AML), a malignancy which remains a clinical challenge and requires more effective therapies. Using both AML cell line models and primary patient samples, we demonstrated that panobinostat and MK-1775 synergistically induced proliferation arrest and cell death. We also demonstrated that panobinostat had equal anti-leukemic activities against primary AML blasts derived from patients either at initial diagnosis or at relapse. Interestingly, treatment with panobinostat alone or in combination with MK-1775 resulted in decreased Wee1 protein levels as well as downregulation of the CHK1 pathway. shRNA knockdown of CHK1 significantly sensitized AML cells to MK-1775 treatment, while knockdown of Wee1 significantly enhanced both MK-1775- and panobinostat-induced cell death. Our results demonstrate that panobinostat synergizes with MK-1775 in AML cells, at least in part through downregulation of CHK1 and/or Wee1, providing compelling evidence for the clinical development of the combination treatment in AML.
Keywords: Acute myeloid leukemia, CHK1, MK-1775, Panobinostat, Wee1
Introduction
Acute myeloid leukemia (AML) has a guarded prognosis, with overall survival rates of 25% and 65% in the adult and pediatric populations, respectively.1,2 Standard treatment for AML consists primarily of a combination of cytarabine and an anthracycline.3 These drugs are thought to act by targeting DNA in dividing cells, leading to DNA damage.4,5 The DNA damage triggers activation of the cell cycle checkpoints, leading to cell cycle arrest and DNA damage repair. Therefore, activation of the cell cycle checkpoints remains a possible mechanism of drug resistance leading to treatment failure for AML.
Checkpoint kinase 1 (CHK1) plays an important role in replication initiation and fork stability, homologous recombination repair, progression of the cell cycle, and the S and G2/M cell cycle checkpoints.6-9 In response to DNA damage or replication stress, CHK1 phosphorylates CDC25 phosphatases, inhibiting activation of CDK1/CDK2 and arresting cell cycle progression allowing for DNA repair and cell survival.6,10-12 Another important checkpoint kinase is Wee1, whose primary function is inhibitory phosphorylation of CDK1 and CDK2 on Tyr-15 (Y15), which prevents cell cycle progression.13,14 Thus, targeting these checkpoint kinases would interfere with the DNA damage response, allowing for accumulation of irreparable DNA damage and eventually leading to cell death. Therefore, inhibition of checkpoint kinases in combination with DNA damaging agents may become a therapeutic strategy for the treatment of various malignancies.15-18
Histone deacetylase (HDAC) inhibitors (HDACIs) have demonstrated anticancer activity in numerous malignancies including AML.19-26 They have been demonstrated to induce cell cycle arrest, differentiation, and apoptosis in cancer cells, but less so in normal cells.19-26 Though their single-agent efficacy in the clinic has been modest,27-31 there are many clinical trials investigating combination therapies (NCT01242774, NCT01742793, NCT02061449, and NCT02145715, clinicaltrials.gov). Previously, we demonstrated that panobinostat, a pan-HDACI which was recently approved by the US FDA for the treatment of multiple myeloma, downregulates the CHK1 pathway in AML cells.32,33 Also, we demonstrated that MK-1775, a potent and selective Wee1 inhibitor, synergizes with LY2603618, a CHK1 selective inhibitor, in AML cells.34 Therefore, it is conceivable that the combination of panobinostat and MK-1775 would result in synergistic anti-leukemic activity in AML cells.
In this study, we demonstrate that panobinostat synergizes with MK-1775 to induce proliferation arrest and cell death in AML cell lines and primary patient samples. The combined panobinostat and MK-1775 treatment results in downregulation of Wee1 and CHK1. We also demonstrate that panobinostat treatment alone downregulates CHK1 and Wee1. Furthermore, shRNA knockdown of CHK1 enhances MK-1775-induced cell death, while shRNA knockdown of Wee1 enhances single drug panobinostat- and MK-1775-induced cell death. The results presented here support the clinical development of panobinostat in combination with MK-1775 for the treatment of AML.
Results
Panobinostat cooperates with MK-1775 in AML cells
To determine if panobinostat synergizes with MK-1775 in AML cells, we first assessed the effects of the 2 agents, alone or combined, on cell proliferation in U937 cells by MTT assays. The combined treatment resulted in synergistic anti-leukemic interactions, as determined by standard isobologram analyses, regardless of drug administration schedule (Fig. 1A-C). For ease of experimental setup, simultaneous treatment was used from here on out. We then confirmed the additive-to-synergistic anti-leukemic activities of the 2 agents in CTS, MOLM-13, and OCI-AML3 AML cell lines (Fig. 1D-F).
Figure 1.

Panobinostat synergizes with MK-1775 to induce proliferation arrest in AML cells. Panels A-C: U937 cells were treated for 72 h simultaneously with MK-1775 -> Panobinostat (Pan), pretreated with MK-1775 for 24 h then simultaneous for 48 h (MK-1775 Panobinostat), or pretreated with panobinostat for 24 h then simultaneous for 48 h (Panobinostat -> MK-1775). Panels D-F: CTS (Panel D), MOLM-13 (Panel E), and OCI-AML3 (Panel F) cells were treated simultaneously with the 2 agents for 72 h. Viable cells post drug treatments were detected using MTT reagent. Anti-leukemic interactions between MK-1775 and panobinostat were determined by standard isobologram analyses. The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism whereas those above the line indicate antagonism. MTT assays were repeated at least 3 times and the data are presented as mean values ± s.e.m.
Next, we investigated panobinostat treatment in primary patient samples. Panobinostat IC50s, as determined by MTT assays, for diagnostic AML blast samples obtained from patients at initial diagnosis (n = 32) or at relapse (n = 7) were similar (median IC50 15.9 and 24.8 nM, respectively, p = 0.129, Fig. 2A). Then we determined Wee1 transcript levels by real-time RT-PCR in the primary patient samples. Interestingly, panobinostat IC50s positively correlated with Wee1 transcript levels (Fig. 2B, r = 0.37, p = 0.025), suggesting that Wee1 may play an important role in panobinostat sensitivity in AML. We then tested the combined drug treatment in primary patient samples (n = 11), which had sufficient number of cells, by MTT assays. Consistent with the results obtained in the AML cell lines, MTT assays and CompuSyn software analyses revealed that the combination of panobinostat and MK-1775 resulted in additive-to-synergistic anti-leukemic interactions in all of the primary patient samples tested (Table 1). MK-1775 has been reported to reach maximal plasma concentrations of 400-500 nM when administered orally twice daily for 2.5 days35-37 and the steady-state plasma concentrations of panobinostat range from 15-22 nM over 48 h (Novartis Investigator's brochure). Our MTT data shows ex vivo panobinostat IC50s were clinically achievable or slightly higher than clinically achievable concentrations, while MK-1775 IC50s varied widely, ranging from 233 nM to almost 6 μM and were at or significantly higher than maximal plasma concentrations. However, when clinically achievable concentrations of panobinostat were combined with MK-1775, the IC50s for MK-1775 for all but one of the patient samples was at or below maximal MK-1775 plasma concentrations. Together, our results demonstrate global additive-to-synergistic anti-leukemic interactions between MK-1775 and panobinostat in AML.
Figure 2.
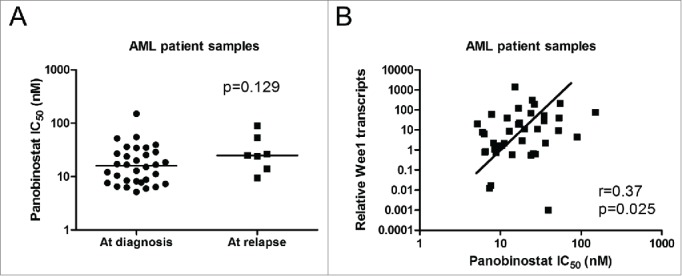
Wee1 transcript levels positively correlate with panobinostat IC50s in ex vivo primary AML patient samples. Panel A: Ex vivo panobinostat sensitivity was determined by MTT assays in diagnostic AML blast samples. The horizontal lines indicate median panobinostat IC50 in each group of AML patient samples. Statistical significance between panobinostat IC50 for cases at initial diagnosis (n = 32) and cases at relapse (n = 7) was calculated using Mann-Whitney 2-sample U test. Panel B: Total RNAs were isolated from primary patient samples and Wee1 transcript levels were quantified by Real-time RT-PCR. The relative Wee1 transcript levels (normalized to GAPDH) were calculated using the comparative Ct method, and graphed versus the panobinostat IC50s. The relationship between Wee1 transcript levels and panobinostat IC50s was determined by the nonparametric Spearman rank correlation coefficient.
Table 1.
Effects of panobinostat (Pan) on MK-1775 anti-leukemic sensitivity in diagnostic AML blasts
| MK-1775 IC50 (nM) in the absence or presence of panobinostat |
|||||
|---|---|---|---|---|---|
| AML patient samples | Panobinostat IC50 (nM) | 0 nM Pan | 5 nM Pan | 10 nM Pan | 20 nM Pan |
| AML#0 | 27.0 | 5996.5 | 2506.0 (0.61) | 1408 (0.54) | 157.1 (0.77) |
| AML#1 | 16.7 | 2448.5 | 684.7 (0.58) | 200.7 (0.68) | ND |
| AML#2 | 34.9 | 3204.0 | 1682.0 (0.66) | 1156.0 (0.65) | 925.0 (0.86) |
| AML#3 | 55.2 | 461.0 | 383.6 (0.92) | 307.4 (0.85) | 236.4 (0.87) |
| AML#4 | 53.3 | 844.0 | 591.3 (0.79) | 734.0 (0.70) | 262.2 (0.69) |
| AML#5 | 14.0 | 948.3 | 449.2 (0.83) | 130.1 (0.85) | ND |
| AML#6 | 34.6 | 233.7 | 168.3 (0.86) | 139.8 (0.88) | 113 (1.05) |
| AML#7 | 26.3 | 325.0 | 244.5 (0.94) | 186.6 (0.95) | 67.6 (0.97) |
| AML#8 | 36.1 | 887.9 | 651.6 (0.87) | 528.3 (0.86) | 302.7 (0.89) |
| AML#10 | 25.4 | 439.7 | 309.6 (0.90) | 210.8 (0.87) | 91.9 (1.00) |
| AML#13 | 17.0 | 3579.2 | 657.2 (0.47) | 396.8 (0.70) | ND |
Note: IC50s are presented as mean of duplicates from one experiment. Numbers in the parentheses represent the combination index values. CI<0.9 indicates synergistic, 0.9<CI<1.1 indicates additive, and CI>1.1 indicates antagonistic anti-leukemic interactions. ND - not determined. AML#4, AML#5, and AML#7 were derived from patients at relapse. The rest of the primary AML patient samples were derived from patients at first diagnosis.
Panobinostat synergistically enhances MK-1775-induced cell death in AML cells
We next assessed the effects of clinically achievable concentrations of panobinostat and MK-1775 treatments on cell death in 2 representative AML cell lines, U937 and CTS. The cells were treated with panobinostat and MK-1775, alone or in combination, for 24 h and then subjected to annexin V/propidium iodide (PI) staining and flow cytometry analyses. Panobinostat treatments significantly enhanced MK-1775-induced cell death in both U937 and CTS cells (Fig. 3A-D). The combination index (CI) values (determined using CompuSyn software) for the U937 cells treated with 10 nM panobinostat in combination with 250 nM or 500 nM MK-1775 were 0.95 and 0.73, while cells treated with 20 nM panobinostat in combination with 250 nM or 500 nM MK-1775 were 0.39 and 0.40, respectively. These results revealed that the drug combination did indeed result in additive to synergistic induction of cell death in U937 cells. Similar results were obtained in the CTS, though the CI values (10 nM panobinostat in combination with 125 nM or 250 nM MK-1775 were 0.6 and 0.6, while 20 nM panobinostat in combination with 125 nM or 250 nM MK-1775 were 0.5 and 0.5) indicated that all of the combinations tested were synergistic. Similar results were also obtained in 3 primary patient samples, though 2 of the data points for one patient sample (CI values of 1.0 and 0.9) indicated additivity, the majority (ranging from 0.4 to 0.8) indicated synergy (Fig. 3E and F, these samples were chosen based solely on the availability of sufficient number of cells for analysis). These results demonstrate that the combination of MK-1775 and panobinostat causes cell death in AML cells rather than merely causing proliferation arrest.
Figure 3.
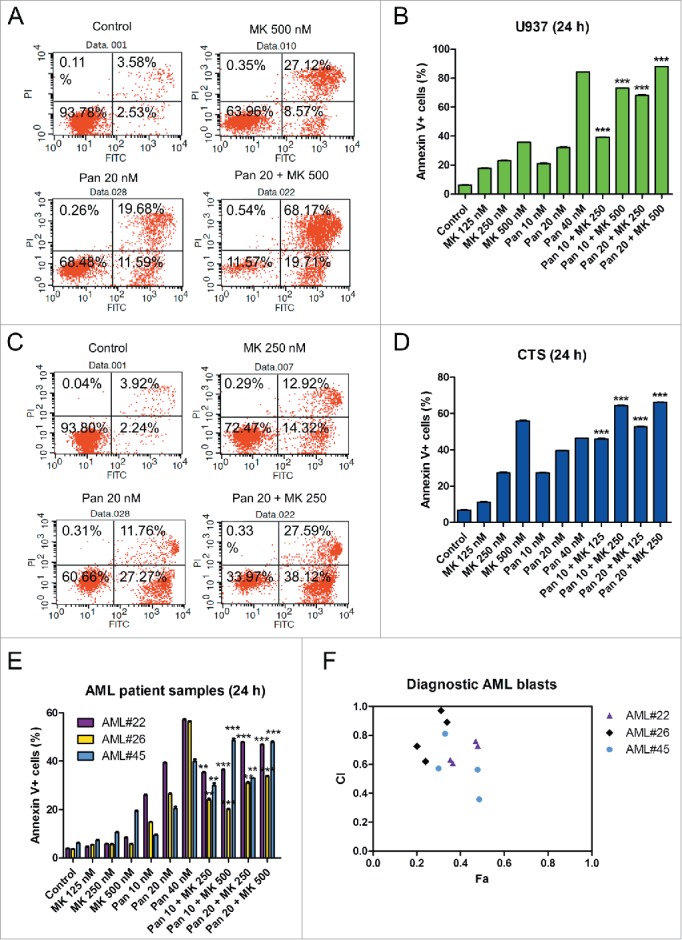
Panobinostat synergizes with MK-1775 to induce cell death in AML cells. Panels A-D: U937 (Panels A and B) and CTS (Panels C and D) cells were treated with the indicated drugs for 24 h. Cell death was determined by annexin V/PI staining and flow cytometry analyses. The experiment was performed at least 3 independent times. Representative annexinV/PI dot plots are shown in Panels A and C. The data are presented as means ± s.e.m of triplicates from one representative experiment (Panels B and D). Statistical significance was calculated using the pair-wise 2-sample t-test. ***indicates p<0.0005. Panel E: Primary patient samples were treated with the indicated drugs for 24 h. Cell death was determined by annexin V/PI staining and flow cytometry analyses. The data are presented as means of triplicates ± s.e.m. from one experiment due to limited sample. Panel F: CI vs. Fa plot (combination index vs. fraction affected) for the data presented in panel E. The CI values were calculated using CompuSyn software.
Panobinostat enhances MK-1775-induced cell death at least partially through downregulation of CHK1 and Wee1 in AML cells
Then we investigated the effects of panobinostat and MK-1775 on cell cycle progression in the CTS cell line. Panobinostat treatment resulted in S and G2\M arrest, while MK-1775 treatment resulted in abrogation of the G2\M checkpoint (as indicated by the significant decrease of the percentage of G2\M phase cells, Fig. 4). The combined treatment resulted in abrogation of the G2\M cell cycle checkpoint and significantly increased cell death (indicated by the percent of cells with sub-G1 DNA content, Fig. 4). In the U937 cells, panobinostat treatment resulted in abrogation of the S and G2\M checkpoint, while MK-1775 resulted in decreased G0\G1. Similar to the CTS cells, U937 cells treated with the drug combination resulted in significant decrease of cells in G2\M.
Figure 4.
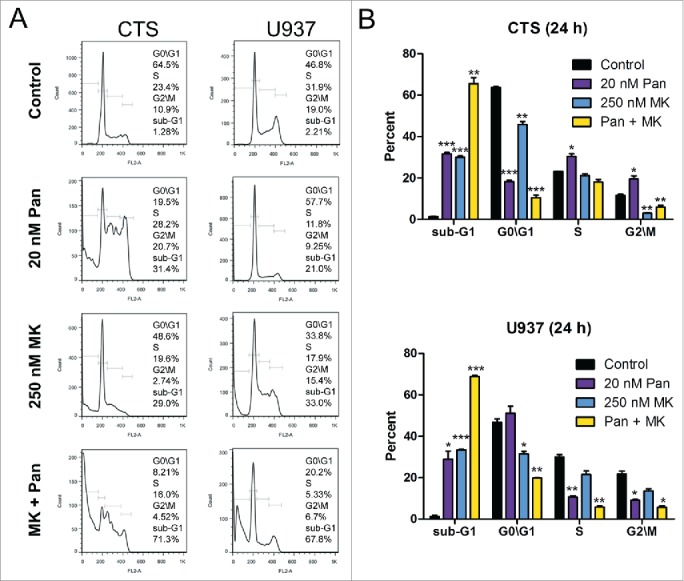
Effects of panobinostat and MK-1775, alone or in combination, on cell cycle progression in CTS and U937 cells. Panel A: CTS and U937 cells were treated with 20 nM panobinostat, 250 nM MK-1775, or in combination for 24 h. Then the cells were fixed with ethanol, stained with PI, and subjected to flow cytometry analyses. Panel B: The percentage of cells in each cell cycle phase after drug treatment are presented as means of triplicates ± s.e.m from one representative experiment. *indicates p<0.05, **indicates p<0.005, and ***indicates p<0.0005.
As expected, increased levels of acetylated histone H4 were detected after panobinostat treatment of the CTS cells (Fig. 5A). In addition, treatment with MK-1775 or panobinostat alone resulted in significantly decreased levels of p-CDK1 and p-CDK2 compared to the no drug treatment control, which was not due to decrease of total CDK1 and CDK2 levels as the total levels remained largely unchanged or slightly increased (p<0.05, Fig. 5A and B). Compared to individual drug treatment, the combined drug treatment did result in further decrease of both p-CDK1 and p-CDK2 (p<0.05, Fig. 5A and B). Significantly, increased γH2AX was detected after individual drug treatments and was further increased after combined drug treatment (p<0.05, Fig. 5A and B). Consistent with our previous studies,38 CHK1 levels were decreased following panobinostat treatment and were accompanied by decreased levels of p-CDC25C. Surprisingly, MK-1775 treatment alone also caused significant decrease of CHK1, though it had no effect on CDC25C phosphorylation (Fig. 5A and B). CHK1 levels were further decreased in the combined drug treatment compared to individual drug treatment and p-CDC25C levels were decreased compared to MK-1775 treatment, but not to panobinostat treatment. Interestingly, panobinostat treatment caused downregulation of Wee1, which was further decreased in the combined drug treatment. In contrast, MK-1775 treatment had no effect on Wee1 level. Similar results were obtained in U937 cells, except panobinostat or MK-1775 treatment alone resulted in decreased CHK1, yet neither had an effect on CDC25C phosphorylation (Fig. 5C and D). These results provide evidence that cooperative downregulation of CHK1 and Wee1 by panobinostat and MK-1775 may represent a critical molecular mechanism underlying the synergistic anti-leukemic interactions between panobinostat and MK-1775 in AML cells.
Figure 5.
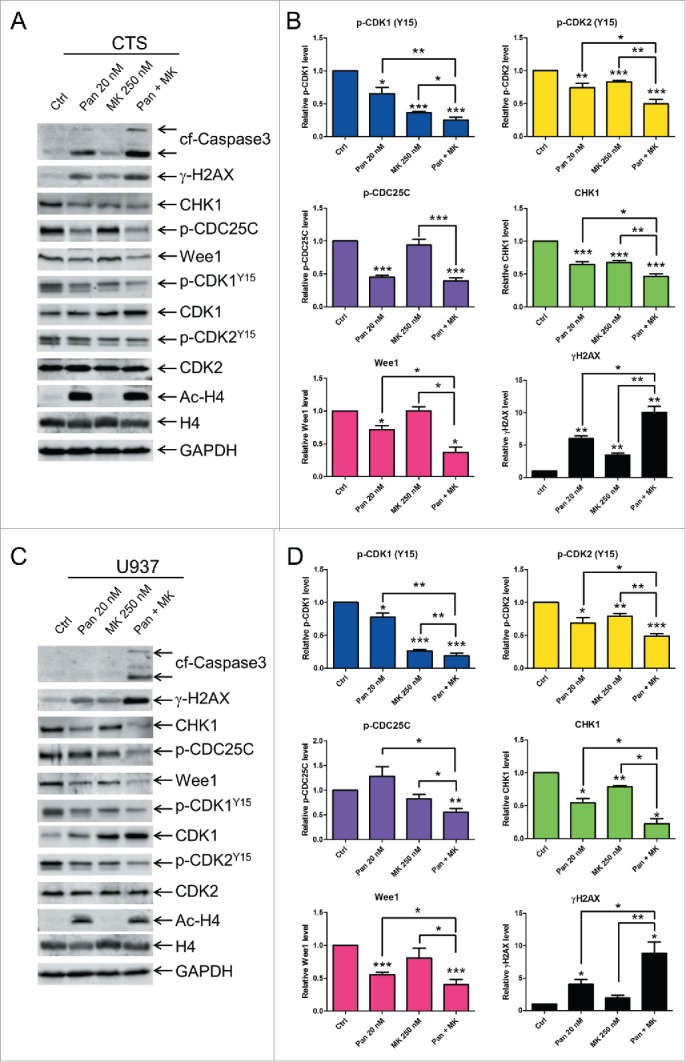
Cooperative downregulation of CHK1 and Wee1 by panobinostat and MK-1775 is common in different subtypes of AML. Panels A and C: CTS (Panel A) and U937 (Panel C) cells were treated with panobinostat, MK-1775, or in combination. Whole cell lysates were subjected to Western blotting and probed with anti-cleaved caspase-3 (cf-cleaved fragment), -p-CDK1, -CDK1, p-CDK2, -CDK2, -γH2AX, -CHK1, -p-CDC25c, -Wee1, -ac-H4, -H4, or -GAPDH antibody. Representative Western blots are shown. Panels B and D: Densitometry for protein expression, corresponding to experiments presented in Panels A and C, were measured using Oddyssey V3.0, normalized to GAPDH, and graphed as mean fold change (relative to no drug control) ± s.e.m from at least 3 independent experiments. For p-CDK1 and p-CDK2 the densitometry measurements were first normalized to the total CDK1 and CDK2 levels, respectively, then to GAPDH. *indicates p<0.05, **indicates p<0.005, and ***indicates p<0.0005.
To provide evidence that CHK1 and Wee1 play critical roles in the anti-leukemic activities of MK-1775 and panobinostat, we used lentivirus shRNA to knockdown CHK1 and Wee1 in U937 cells (Fig. 6). Knockdown of CHK1 and Wee1 resulted in significantly increased MK-1775-induced cell death. Knockdown of Wee1 also significantly enhanced panobinostat-induced cell death, whereas CHK1 knockdown did not appear to have an effect on panobinostat-induced cell death (Fig. 6B and D).
Figure 6.
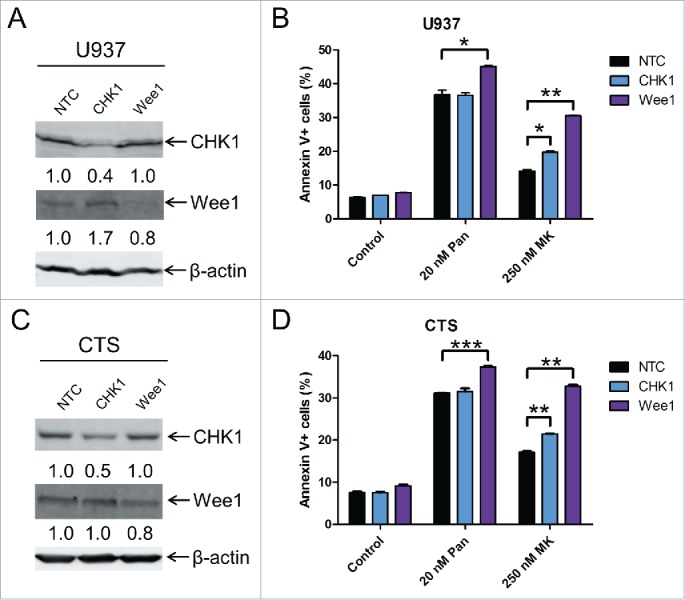
CHK1 and Wee1 play important roles in the anti-leukemic activity of MK-1775. Panel A: U937 cells were infected with CHK1, Wee1 or non-target control (NTC) shRNA lentivirus overnight, washed, and then incubated for 48 h. Whole cell lysates were subjected to Western blotting and probed with anti-CHK1 (Santa Cruz Biotechnology), -Wee1 (Cell Signaling Technology), or -β-actin antibody. Panel B: U937 cells were infected with CHK1, Wee1 or non-target control (NTC) shRNA lentivirus overnight, washed, incubated for 24 h. Then the cells were treated with panobinostat or MK-1775 for 24 h. Cell death was determined by annexin V/PI staining and flow cytometry analyses. Panel C: CTS cells were infected with CHK1, Wee1 or NTC shRNA lentivirus overnight, washed, and then incubated for 48 h. Whole cell lysates were subjected to Western blotting and probed with anti-CHK1, -Wee1, or -β-actin antibody. Panel D: CTS cells were infected with CHK1, Wee1 or NTC shRNA lentivirus overnight, washed, incubated for 24 h, and then the cells were treated with no drug control, panobinostat or MK-1775 for 24 h. Cell death was determined by annexin V/PI staining and flow cytometry analyses.
Discussion
MK-1775 has demonstrated promising preclinical results in combination with DNA damaging agents.16-18,39,40 Recent studies have investigated the combination of Wee1 and CHK1 inhibitors in preclinical models of different malignancies and have also found promising activity.39,41,42 In addition, we have previously demonstrated that activation of CHK1 was a potential mechanism of resistance to MK-1775 which could be overcome by the addition of a CHK1 selective inhibitor.34 HDACIs have shown promising results in combination with other chemotherapy agents and have been demonstrated to downregulate CHK1 in various malignancies, including AML;38,43-45 therefore they could be used as a means to downregulate the CHK1 pathway. Many HDACIs are currently under clinical development for treating cancer and several of them have been FDA approved for the treatment of a number of malignancies,46,47 which makes them attractive options for the clinical development of combination therapies with MK-1775. Of note, pracinostat has been granted Orphan Drug status by the FDA for the treatment of AML48 and panobinostat has recently received FDA approval after being granted “priority review” status for the treatment of multiple myeloma.32,33
In this study, we demonstrated that combined panobinostat and MK-1775 treatment resulted in synergistic anti-leukemic activity in AML cell lines and ex vivo primary patient samples. In addition, we provide evidence to suggest that cooperative downregulation of CHK1 and Wee1 plays an important role in the synergistic anti-leukemic activity. While we were performing our studies, Zhou et al. published their study investigating the combination of Vorinostat, a pan-HDACI, with MK-1775 in AML.45 In their study, the authors found diminished p-CDK1 following Vorinostat or MK-1775 treatment and further decrease after combined drug treatment,45 which are similar to our results with panobinostat and MK-1775 treatment (Fig. 5). In contrast to Zhou et al., we found that panobinostat treatment alone resulted in decreased Wee1 protein levels. These differences may be due to the nature of the different HDACIs. Additionally, the HDACIs Vorinostat and valproic acid have been demonstrated to downregulate Wee1 expression in glioma cells49 and panobinostat has been demonstrated to downregulate Wee1 transcript levels in T-cell leukemia.50 Our Wee1 knockdown demonstrated that Wee1 is important for panobinostat- and MK-1775-induced cell death. We also found that Wee1 transcript levels positively correlated with panobinostat IC50s in primary patient samples (Fig. 2B). Others have demonstrated that inhibition of CHK1 enhances MK-1775 activity39,41,42 and we have previously demonstrated that CHK1 plays a role in MK-1775 resistance.34 In line with those studies, our CHK1 knockdown further confirms that CHK1 plays a role in MK-1775-induced cell death. Based on all of these findings, the synergistic anti-leukemic interaction of panobinostat and MK-1775 is likely due to the cooperative downregulation of Wee1 and CHK1 in combination with inhibition of Wee1.
In addition to HDACIs, MK-1775 has been combined with other agents, such as cytarabine, in preclinical AML models.36,40,51,52 The combined cytarabine and MK-1775 treatments resulted in synergistic inhibition of proliferation, regardless of p53 status.40 It has been demonstrated that cytarabine-induced S-phase cell cycle arrest was overcome by the addition of MK-1775.40,51,52 Tibes et al. used an RNAi screening approach to identify kinases involved in cytarabine sensitivity and found ATR, PKMYT1, and CHK1, among others, as kinases involved in cytarabine sensitivity.36 In addition, they demonstrated synergistic anti-leukemic activity for combined MK-1775 and cytarabine treatment in cell line models. In contrast to MK-1775 combined with cytarabine, Van Linden et al. found that doxorubicin in combination with MK-1775 was largely antagonistic.40
In conclusion, the present findings indicate that panobinostat potentiates the anti-leukemic activity of MK-1775 in AML cell lines and primary patient samples at clinically achievable concentrations. Panobinostat treatment downregulates the CHK1 pathway and/or Wee1 expression levels in AML cells. As demonstrated by our shRNA knockdown studies in the U937 cells, the synergy between panobinostat and MK-1775 may be more likely due to panobinostat's ability to downregulate Wee1 than CHK1. However, panobinostat has been shown to regulate expression of many genes and acetylation of various proteins, which leads to cell death and tumor growth inhibition in various malignancies.44,53-55 Therefore, we cannot rule out the possible involvement of other proteins in addition to CHK1 and Wee1.38,53,56 The present findings provide support for the clinical development of panobinostat in combination with MK-1775 for the treatment of AML.
Methods
Drugs
MK-1775 (MK) and panobinostat (Pan) were purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
The U937 cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). The OCI-AML3 cell line was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). MOLM-13 cells were purchased from AddexBio (San Diego, CA, USA). The CTS cell line was a gift from Dr. A Fuse from the National Institute of Infectious Diseases, Tokyo, Japan. The cell lines were cultured in RPMI 1640 (except OCI-AML3, which was cultured in alpha-MEM) with 10-15% fetal bovine serum (Hyclone, Logan, UT, USA), 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. All cells were cultured in a 37°C humidified atmosphere containing 5% CO2/95% air.
Diagnostic AML blast samples derived from patients either at initial diagnosis or at relapse were purified by standard Ficoll-Hypaque density centrifugation, then cultured in RPMI 1640 with 20% fetal bovine serum supplemented with ITS solution (Sigma-Aldrich, St. Louis, MO, USA) and 20% supernatant of the 5637 bladder cancer cell line (as a source of granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, interleukin-1 beta, macrophage colony-stimulating factor, and stem cell factor).57-59
Clinical samples
Diagnostic AML blast samples were obtained from the First Hospital of Jilin University. Written informed consent was provided according to the Declaration of Helsinki. This study was approved and carried out in accordance with the guidelines set forth by the Human Ethics Committee of the First Hospital of Jilin University. Clinical samples were screened for FLT3-ITD, NPM1, C-kit, CEBPA, IDH1, IDH2 and DNMT3A gene mutations. The samples were also screened for the following fusion genes by real-time RT-PCR: PML-RARα, BCR-ABL, AML1-MDS1, MLL-AF10, MLL-AF4, MLL-ELL, SET-CAN, TLS-ERG, NPM-RARα, E2A-PBX1, AML1-EAP, MLL-AF17, MLL-AF6, MLL-ENL, SIL-TAL1, HOX11, PLZF-RARα, TEL-AML1, DEK-CAN, MLL-AF1p, MLL-AF9, NPM-ALK, TEL-ABL, EIP1L1-PDGFRA, AML1-ETO, CBFB-MYH11, E2A-HLF, MLL-AF1q, MLL-AFX, NPM-MLF1, dupMLL, and TEL-PDGFB. Patient characteristics are presented in Table S1.
In vitro cytotoxicity assays
In vitro cytotoxicities of the AML cells were measured by using MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazoliumbromide, Sigma-Aldrich), as previously described.60,61 Briefly, the cells were treated with variable concentrations of MK-1775, panobinostat, or in combination for 72 hours. MTT was added to a final concentration of 1 mM and cells were incubated for 4 hours at 37°C. The cells were lysed overnight using 10% SDS in 10 mM HCL and plates were read at 590 nm using a microplate reader. IC50 values were calculated as drug concentrations necessary to inhibit 50% growth compared to vehicle control treated cells. The IC50 values for the cell lines are presented as mean values ± standard errors from at least 3 independent experiments. The IC50 values for the patient samples are means of duplicates from one experiment, due to limited sample. Patient samples for the combined drug treatments were chosen based on sample availability. The combination index (CI) values were determined using CompuSyn software. CI<0.9 indicates synergistic, 0.9<CI<1.1 indicates additive, and CI>1.1 indicates antagonistic anti-leukemic interactions.62
Quantification of gene expression by real-time RT-PCR
Total RNA was extracted using TRIzol (Life Technologies, Carlsbad, CA, USA) and cDNAs were prepared from 2 µg total RNA using random hexamer primers and a RT-PCR kit (Life Technologies), and purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA) as previously described.58,61,63 Wee1 (Hs01119384_g1) transcripts were quantitated using TaqMan probes (Life Technologies) and a LightCycler® 480 real-time PCR machine (Roche Diagnostics, Indianapolis, IN, USA), based on the manufacturer's instructions. Real-time PCR data are presented as means of duplicates from one experiment, due to limited sample, and results were normalized to GAPDH (4333764) transcripts. Fold changes were calculated using the comparative Ct method.64
Western blot analysis
Cells were lysed in the presence of protease and phosphatase inhibitors (Roche Diagnostics). Whole cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, USA) and immunoblotted with anti-p-CDK2 (Y15) (ab76146) (Abcam, Hong Kong, China), p-CDK1 (Y15) (5757-1), -CDK1 (1484-1), -Wee1 (S2798), -p-CDC25C (S216) (1190-1) -CDK2 (1134-1) (Epitomics, Burlingame, CA, USA), -H4 (07-108), -ac-H4 (06-598) (Millipore, Billerica, MA, USA), -CHK1 (10362-1-AP), (Proteintech, Chicago, IL, USA), -cleaved caspase-3 (9661), -γH2AX (2577), -Wee1 (4936), -GAPDH (2118) (Cell Signaling Technology, Danvers, MA, USA), -CHK1 (sc-8408, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or -β-actin antibody (Sigma-Aldrich), as previously described.65,66 Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA), as described by the manufacturer. Western blots were repeated at least 3 times and one representative blot is shown. Densitometry measurements were made using Odyssey software V3.0 (Li-Cor), normalized to GAPDH and graphed as fold change relative to the no drug control.
Annexin V/PI staining
AML cells were treated with MK-1775, panobinostat, or in combination and subjected to flow cytometry analysis to determine drug-induced cell death using an annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis Kit (Beckman Coulter; Brea, CA, USA), as previously described,60,63 and a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Experiments with AML cell lines were performed 3 independent times in triplicates and data presented are from one representative experiment, while patient sample experiments were performed once in triplicates. Based on Galluzi et al.'s review, annexin V+ cells are referred to as dead cells.67 Data are presented as mean percent of annexin V+ cells ± standard errors from one representative experiment. Due to limited sample, only 3 patient samples were evaluated for cell death induced by MK-1775 or panobinostat, alone or in combination, by flow cytometry.
Cell cycle progression
Cells were treated with the indicated drugs for 24 h. The cells were harvested and fixed with ice-cold 80% (v/v) ethanol for 24 h. The cells were pelleted, washed with PBS, and resuspended in PBS containing 50 μg/mL PI, 0.1% Triton X-100 (v/v), and 1 μg/mL DNase-free RNase. DNA content was determined by flow cytometry analysis using a FACS Calibur flow cytometer (Becton Dickinson), as previously described.68 Cell cycle analysis was performed using ModFit LT 3.0 (Becton Dickinson). Histograms were created using FlowJo v7.6.5 (Tree Star, Ashland, OR, USA).
Lentivirus production and shRNA knockdown of CHK1 and Wee1
The pMD-VSV-G and delta 8.2 plasmids were gifts from Dr. Dong at Tulane University. CHK1, Wee1, and non-target control shRNA lentiviral vectors were purchased from Sigma-Aldrich. Lentivirus production and transduction of U937 cells were carried out as previously described.38
Statistical analysis
Differences between treated (individually or combined) and untreated annexin V + cells were compared using the pair-wise 2-sample t-test. The p value for the differences between panobinostat IC50s for the groups of patient samples was calculated using the Mann-Whitney 2-sample U test. The relationship between Wee1 transcript levels and panobinostat IC50s was determined by the nonparametric Spearman rank correlation coefficient. Statistical analyses were performed with GraphPad Prism 5.0. Error bars represent ± s.e.m. The level of significance was set at p<0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Funding
This study was supported by a Start-up Fund from Jilin University, Changchun, China, and grants from the National Natural Science Foundation of China, NSFC 31271477 and NSFC 31471295, and the Natural Science Fund, Department of Science and Technology, Jilin Province, China. The funders had no role in study design, data collection, analysis and interpretation of data, decision to publish, or preparation of the manuscript.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11-30; PMID:23335087; http://dx.doi.org/ 10.3322/caac.21166 [DOI] [PubMed] [Google Scholar]
- 2.Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, Pounds S, Razzouk BI, Lacayo NJ, Cao X, et al.. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 2010; 11:543-52; PMID:20451454; http://dx.doi.org/ 10.1016/S1470-2045(10)70090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol 2011; 29:487-94; PMID:21220605; http://dx.doi.org/ 10.1200/JCO.2010.30.1820 [DOI] [PubMed] [Google Scholar]
- 4.Grant S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res 1998; 72:197-233; PMID:9338077; http://dx.doi.org/ 10.1016/S0065-230X(08)60703-4 [DOI] [PubMed] [Google Scholar]
- 5.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57:727-41; PMID:10075079; http://dx.doi.org/ 10.1016/S0006-2952(98)00307-4 [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129-39; PMID:11390642; http://dx.doi.org/ 10.1128/MCB.21.13.4129-4139.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 2010; 16:376-83; PMID:20068082; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739-45; PMID:18082599; http://dx.doi.org/ 10.1016/j.molcel.2007.11.015 [DOI] [PubMed] [Google Scholar]
- 10.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997; 277:1501-5; PMID:9278512; http://dx.doi.org/ 10.1126/science.277.5331.1501 [DOI] [PubMed] [Google Scholar]
- 11.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277:1497-501; PMID:9278511; http://dx.doi.org/ 10.1126/science.277.5331.1497 [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al.. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448-59; PMID:10859164; http://dx.doi.org/ 10.1101/gad.840500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992; 257:1955-7; PMID:1384126; http://dx.doi.org/ 10.1126/science.1384126 [DOI] [PubMed] [Google Scholar]
- 14.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J 1995; 14:1878-91; PMID:7743995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, Raghavan M, Letson D, Gillies RJ, Altiok S. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS One 2013; 8:e57523; PMID:23520471; http://dx.doi.org/ 10.1371/journal.pone.0057523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res 2011; 17:5638-48; PMID:21799033; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et al.. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 2010; 9:514-22; PMID:20107315; http://dx.doi.org/ 10.4161/cbt.9.7.11115 [DOI] [PubMed] [Google Scholar]
- 18.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al.. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009; 8:2992-3000; PMID:19887545; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0463 [DOI] [PubMed] [Google Scholar]
- 19.Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011; 25:226-35; PMID:21116282; http://dx.doi.org/ 10.1038/leu.2010.276 [DOI] [PubMed] [Google Scholar]
- 20.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med 2005; 11:71-6; PMID:15619634; http://dx.doi.org/ 10.1038/nm1160 [DOI] [PubMed] [Google Scholar]
- 21.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol 2005; 23:3971-93; PMID:15897549; http://dx.doi.org/ 10.1200/JCO.2005.16.600 [DOI] [PubMed] [Google Scholar]
- 22.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5:769-84; PMID:16955068; http://dx.doi.org/ 10.1038/nrd2133 [DOI] [PubMed] [Google Scholar]
- 23.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 2010; 19:1049-66; PMID:20687783; http://dx.doi.org/ 10.1517/13543784.2010.510514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:38-51; PMID:16397526; http://dx.doi.org/ 10.1038/nrc1779 [DOI] [PubMed] [Google Scholar]
- 25.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 2009; 15:3958-69; PMID:19509172; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2785 [DOI] [PubMed] [Google Scholar]
- 26.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 2010; 1:117-36; PMID:21258646; http://dx.doi.org/ 10.1007/s13148-010-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, et al.. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 2005; 105:959-67; PMID:15466934; http://dx.doi.org/ 10.1182/blood-2004-05-1693 [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, et al.. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008; 111:1060-6; PMID:17962510; http://dx.doi.org/ 10.1182/blood-2007-06-098061 [DOI] [PubMed] [Google Scholar]
- 29.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, et al.. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 2006; 12:4628-35; PMID:16899611; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0511 [DOI] [PubMed] [Google Scholar]
- 30.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol 2008; 81:170-6; PMID:18510700; http://dx.doi.org/ 10.1111/j.1600-0609.2008.01102.x [DOI] [PubMed] [Google Scholar]
- 31.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, et al.. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2007; 109:2781-90; PMID:17179232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garnock-Jones KP. Panobinostat: first global approval. Drugs 2015; 75:695-704; PMID:25837990; http://dx.doi.org/ 10.1007/s40265-015-0388-8 [DOI] [PubMed] [Google Scholar]
- 33.Torimoto Y, Shindo M, Ikuta K, Kohgo Y. Current therapeutic strategies for multiple myeloma. Int J Clin Oncol 2015; 20(3):423-30; PMID:25855312 [DOI] [PubMed] [Google Scholar]
- 34.Qi W, Xie C, Li C, Caldwell JT, Edwards H, Taub JW, Wang Y, Lin H, Ge Y. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J Hematol Oncol 2014; 7:53; PMID:25084614; http://dx.doi.org/ 10.1186/s13045-014-0053-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leijen S, Schellens JH, Shapiro G, Pavlick AC, Tibes R, Demuth T, Viscusi J, Cheng JD, Xu Y, Oza AM. A phase I pharmacological and pharmacodynamic study of MK-1775, a Weel tyrosine kinase inhibitor, in monotherapy and combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol 2010; 28:3067. [Google Scholar]
- 36.Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, Gonzales IM, Demuth T, Slack J, Mesa RA, et al.. RNAi screening of the kinome with cytarabine in leukemias. Blood 2012; 119:2863-72; PMID:22267604; http://dx.doi.org/ 10.1182/blood-2011-07-367557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol 2015; 33:3409-15; PMID:25964244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie C, Drenberg C, Edwards H, Caldwell JT, Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD, et al.. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS One 2013; 8:e79106; PMID:24244429; http://dx.doi.org/ 10.1371/journal.pone.0079106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, McNally A, Gojo I, Karp JE, Mesa RA, et al.. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014; 99:688-96; PMID:24179152; http://dx.doi.org/ 10.3324/haematol.2013.093187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P, Porter CC. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther 2013; 12:2675-84; PMID:24121103; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrassa L, Chila R, Lupi M, Ricci F, Celenza C, Mazzoletti M, Broggini M, Damia G. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012; 11:2507-17; PMID:22713237; http://dx.doi.org/ 10.4161/cc.20899 [DOI] [PubMed] [Google Scholar]
- 42.Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, Wallace E, Woessner RD, Gross S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther 2011; 12:788-96; PMID:21892012; http://dx.doi.org/ 10.4161/cbt.12.9.17673 [DOI] [PubMed] [Google Scholar]
- 43.Wang G, Edwards H, Caldwell JT, Buck SA, Qing WY, Taub JW, Ge Y, Wang Z. Panobinostat synergistically enhances the cytotoxic effects of cisplatin, doxorubicin or etoposide on high-risk neuroblastoma cells. PLoS One 2013; 8:e76662; PMID:24098799; http://dx.doi.org/ 10.1371/journal.pone.0076662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brazelle W, Kreahling JM, Gemmer J, Ma Y, Cress WD, Haura E, Altiok S. Histone deacetylase inhibitors downregulate checkpoint kinase 1 expression to induce cell death in non-small cell lung cancer cells. PLoS One 2010; 5:e14335; PMID:21179472; http://dx.doi.org/ 10.1371/journal.pone.0014335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou L, Zhang Y, Chen S, Kmieciak M, Leng Y, Lin H, Rizzo KA, Dumur CI, Ferreira-Gonzalez A, Dai Y, et al.. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015; 29:807-18; PMID:25283841; http://dx.doi.org/ 10.1038/leu.2014.296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014; 124:30-9; PMID:24382387; http://dx.doi.org/ 10.1172/JCI69738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poole RM. Belinostat: first global approval. Drugs 2014; 74:1543-54; PMID:25134672; http://dx.doi.org/ 10.1007/s40265-014-0275-8 [DOI] [PubMed] [Google Scholar]
- 48.Bose P, Grant S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leuk Res 2014; 38:862-5; PMID:24996975; http://dx.doi.org/ 10.1016/j.leukres.2014.06.007 [DOI] [PubMed] [Google Scholar]
- 49.Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, Comella JX, Sanchis D, Llovera M. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis 2014; 5:e1435; PMID:25275596; http://dx.doi.org/ 10.1038/cddis.2014.412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hasegawa H, Yamada Y, Tsukasaki K, Mori N, Tsuruda K, Sasaki D, Usui T, Osaka A, Atogami S, Ishikawa C, et al.. LBH589, a deacetylase inhibitor, induces apoptosis in adult T-cell leukemia/lymphoma cells via activation of a novel RAIDD-caspase-2 pathway. Leukemia 2011; 25:575-87; PMID:21242994; http://dx.doi.org/ 10.1038/leu.2010.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, Zaberezhnyy V, Patel PR, Gao D, Tan AC, et al.. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia 2012; 26:1266-76; PMID:22289989; http://dx.doi.org/ 10.1038/leu.2011.392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caldwell JT, Edwards H, Buck SA, Ge Y, Taub JW. Targeting the wee1 kinase for treatment of pediatric Down syndrome acute myeloid leukemia. Pediatr Blood Cancer 2014; 61:1767-73; PMID:24962331; http://dx.doi.org/ 10.1002/pbc.25081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 2009; 280:233-41; PMID:19344997; http://dx.doi.org/ 10.1016/j.canlet.2009.02.019 [DOI] [PubMed] [Google Scholar]
- 54.Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P, Pullarkat V, Bhatia R, Forman S, Yen Y, et al.. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood 2008; 111:5093-100; PMID:18349321; http://dx.doi.org/ 10.1182/blood-2007-10-117762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther 2011; 10:2034-42; PMID:22072815; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541-52; PMID:17694093; http://dx.doi.org/ 10.1038/sj.onc.1210620 [DOI] [PubMed] [Google Scholar]
- 57.Taub JW, Matherly LH, Stout ML, Buck SA, Gurney JG, Ravindranath Y. Enhanced metabolism of 1-β-D-arabinofuranosylcytosine in Down syndrome cells: a contributing factor to the superior event free survival of Down syndrome children with acute myeloid leukemia. Blood 1996; 87:3395-403; PMID:8605357 [PubMed] [Google Scholar]
- 58.Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, Taub JW, Li C, Lin H, Ge Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014; 28:1557-60; PMID:24531733; http://dx.doi.org/ 10.1038/leu.2014.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quentmeier H, Zaborski M, Drexler HG. The human bladder carcinoma cell line 5637 constitutively secretes functional cytokines. Leuk Res 1997; 21:343-50; PMID:9150352; http://dx.doi.org/ 10.1016/S0145-2126(96)00132-4 [DOI] [PubMed] [Google Scholar]
- 60.Xie C, Edwards H, Xu X, Zhou H, Buck SA, Stout ML, Yu Q, Rubnitz JE, Matherly LH, Taub JW, et al.. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res 2010; 16:5499-510; PMID:20889917; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PLoS One 2011; 6:e17138; PMID:21359182; http://dx.doi.org/ 10.1371/journal.pone.0017138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621-81; PMID:16968952; http://dx.doi.org/ 10.1124/pr.58.3.10 [DOI] [PubMed] [Google Scholar]
- 63.Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, Taub JW, Matherly LH, Ge Y. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood 2009; 114:2744-52; PMID:19638627; http://dx.doi.org/ 10.1182/blood-2008-09-179812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 65.Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, et al.. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood 2006; 107:1570-81; PMID:16249385; http://dx.doi.org/ 10.1182/blood-2005-06-2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, Ravindranath Y, Matherly LH, Taub JW. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 2005; 97:226-31; PMID:15687366; http://dx.doi.org/ 10.1093/jnci/dji026 [DOI] [PubMed] [Google Scholar]
- 67.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, et al.. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16:1093-107; PMID:19373242; http://dx.doi.org/ 10.1038/cdd.2009.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, Azmi A, Mohammad RM, Dong Y, Kong W, et al.. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS One 2012; 7:e52095; PMID:23251689; http://dx.doi.org/ 10.1371/journal.pone.0052095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.