

ABSTRACT
Members of krüppel-like factor family have been shown to play critical roles in the development, metabolism and tumorigenesis. Our previous study demonstrated that up-regulationupregulation of KLF2 in livers from obese mice could promote hepatosteatosis. However, little is known about its effects in hepatocellular carcinogenesis. In the present study, we found that mRNA and protein expression of KLF2 was increased in primary HCC, when compared with that in adjacent normal liver. In vitro studies further showed that enforced overexpression of KLF2 expression enhanced, while its knockdown inhibited cell proliferation and invasion. At the molecular level, c-myc was a direct transcriptional target of KLF2 and a KLF2-binding site in the c-myc promoter bound specifically to KLF2 protein. Indeed, ablation of c-myc largely attenuated the proliferative roles of KLF2 in HCC cells. Therefore, our data highlight an important role for KLF2/c-myc pathway in HCC development and progression.
KEYWORDS: Cell proliferation, c-myc, hepatocellular carcinoma, KLF2, transcription
Introduction
Hepatocellular carcinoma (HCC), a primary malignancy of the liver, has become one of the most lethal human cancers in China.1,2 Although great efforts have been made in the understanding the genetic alterations in its development,3 key molecular profiles involvement in HCC pathogenesis remain incompletely understood.
Recent studies demonstrate that members of the krüppel-like factor (KLF) transcription factor family play critical roles in the initiation and (or) progression of several human cancers.4,5 For instance, KLF4 expression was decreased or lost in HCC patients and, in particular, with lymph node metastases.6,7 KLF4 restoration could inhibit cell migration, invasion and growth both in vitro and in vivo through induction of vitamin D receptor (VDR).6 Besides, KLF6 overexpression was shown to protect HCC-derived cells from apoptosis by regulation of p53 and Bcl-xL.8 Therefore, investigation of this protein family might be helpful for designing novel preventive and therapeutic strategies to control HCC development.
In our previous study, we found that mRNA and protein levels of KLF2 were significantly elevated in livers from obese mice.9 Adenoviruses-mediated overexpression of KLF2 promoted, while its silencing ameliorated triglycerides accumulation in mice.9 Moreover, our results established CD36, a fatty acid transporter, as a direct transcriptional target of KLF2.9 In the present study, we show that KLF2 acts as an oncogene in HCC. Ectopic KLF2 expression in HCC cells enhanced cell proliferation and invasion, whereas KLF2 knockdown suppresses the phenotypes. We further identify c-myc as a transcription target of KLF2, and find that c-myc deficiency partially rescues the oncogenic roles of KLF2.
Results
Up-regulation of KLF2 in HCC tissues
To investigate the roles of KLF2 in HCC pathogenesis, we first analyzed its mRNA expression in 60 primary HCC and adjacent normal specimens by quantitative real-time PCR. We observed a drastic up-regulationupregulation of KLF2 expression levels in HCC tissues (Fig. 1A), which was further confirmed by western blot (Fig. 1B and Supplementary Fig. 1).
Figure 1.
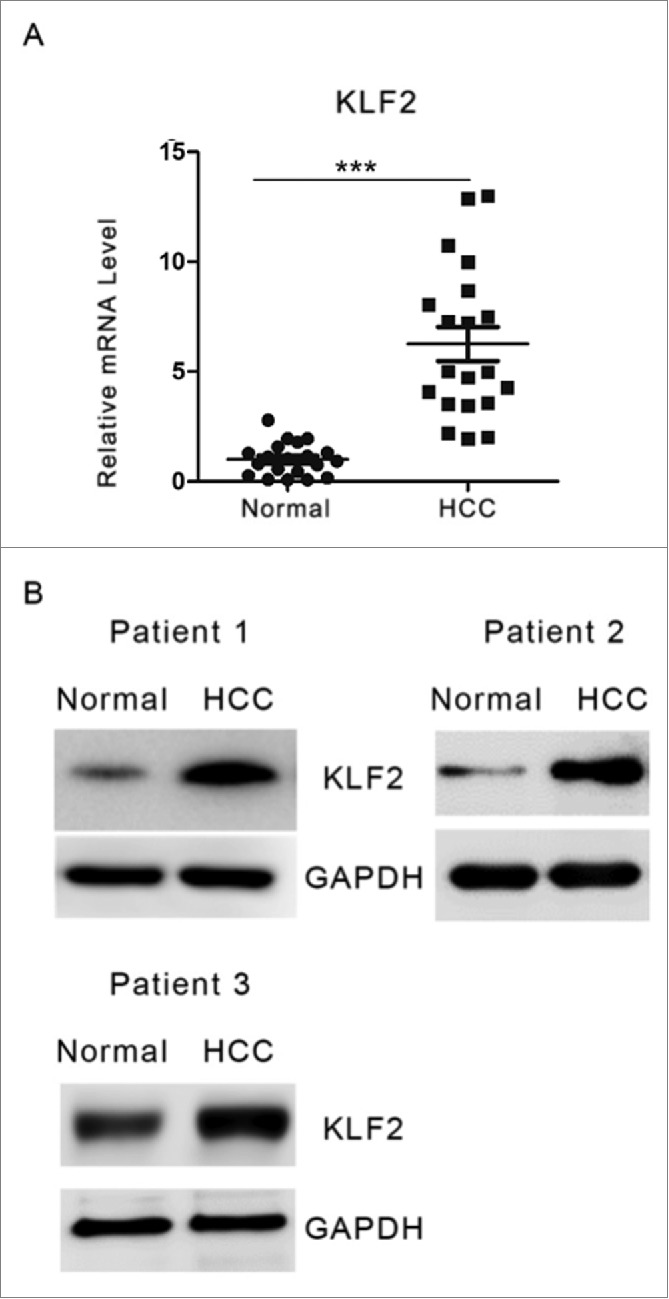
KLF2 expression in HCC and normal tissues. (A) Relative mRNA levels of KLF2 were determined by quantitative real-time PCR analysis in 60 primary HCC and adjacent normal liver tissues. (B) Representative protein levels of KLF2 in HCC and normal liver tissues from three 3 patients.
KLF2 promotes cell proliferation
We then examined the impact of ectopic KLF2 expression on cell proliferation associated phenotypes in HuH7 cells (Fig. 2A). We found that KLF2 overexperssion significantly enhanced cell proliferation, cell-cycle progression and invasion abilities (Fig. 2B–D). Cell colony formation in soft agar was also increased by KLF2 (Fig. 2E). Similar results were obtained in another human HCC cell line HepG2 (Supplementary Fig. 2A–C). Conversely, knockdown of KLF2 by small interfering RNA (siRNA) oligos in HCC cells suppressed proliferation, invasion abilities and colony formation (Fig. 3A–E, Supplementary Fig. 3A–C). Therefore, our results indicate that KLF2 acts as a positive regulator in HCC cell proliferation.
Figure 2.
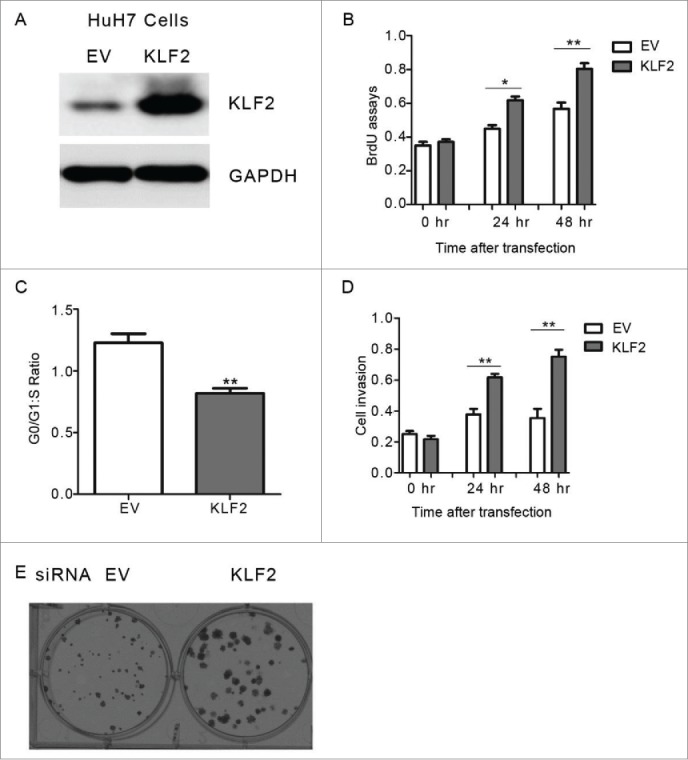
The effect of KLF2 overexpression on HCC growth in vitro (A) Representative protein levels of KLF2 in HuH7 cells transfected with KLF2 expression plasmids or empty vector (EV) for 24 hr. 25 μg protein lysis were used to perform western blot analysis. (B-D) Cell proliferation (B), cell-cycle progression (C) and invasion (D) assays in HuH7 cells expressing KLF2 or empty vector (EV). (E) Representative anchorage-independent growth activity for HuH7 cells with ectopic KLF2 expression and its corresponding control (EV). The colonies were observed at lower magnification (40X).
Figure 3.
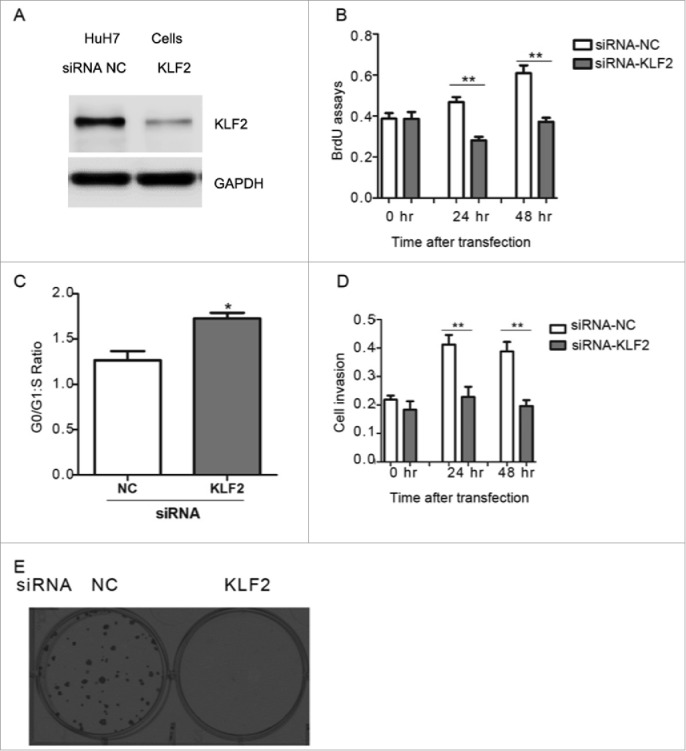
Knockdown of KLF2 inhibits HCC cell proliferation (A) Representative protein levels of KLF2 in HuH7 cells transfected with siRNA oligos targeting KLF2 or negative control (NC) for 24 hr. 40 μg protein lysis were used to perform western blot analysis. (B-C) Cell proliferation (B), cell-cycle progression (C) and invasion (D) assays in HuH7 cells with KLF2 knockdown. (E) Representative anchorage-independent growth activity for HuH7 cells with KLF2 deficiency and its corresponding control (NC). The colonies were observed at lower magnification (40X).
KLF2 up-regulated c-myc expression in HCC cells
To understand the molecular mechanisms underlying KLF2-mediated cell proliferation, real-time PCR analysis of proliferation-related genes was performed. As a result, KLF2 overexpression resulted in an increased expression of c-myc, but not other biomarkers including p53, E2F1 and β-catenin (Fig. 4A and Supplementary Fig. 4A). The up-regulation of c-myc protein levels was also confirmed by western blot (Fig. 4B and Supplementary Fig. 4B). In contrast, knockdown of KLF2 did the opposite (Fig. 4C–D, Supplementary Fig. 4C–D). Moreover, expression levels of cell-cycle regulators, Cyclin A and Cyclin B1, which are down-stream target genes of c-myc, were also modulated by KLF2 (Fig. 4E–F). Importantly, we observed a significant positive correlation between KLF2 and c-myc expression in HCC and normal liver specimens (Fig. 4G), further suggesting that c-myc might be a direct transcription target of KLF2 in HCC.
Figure 4.
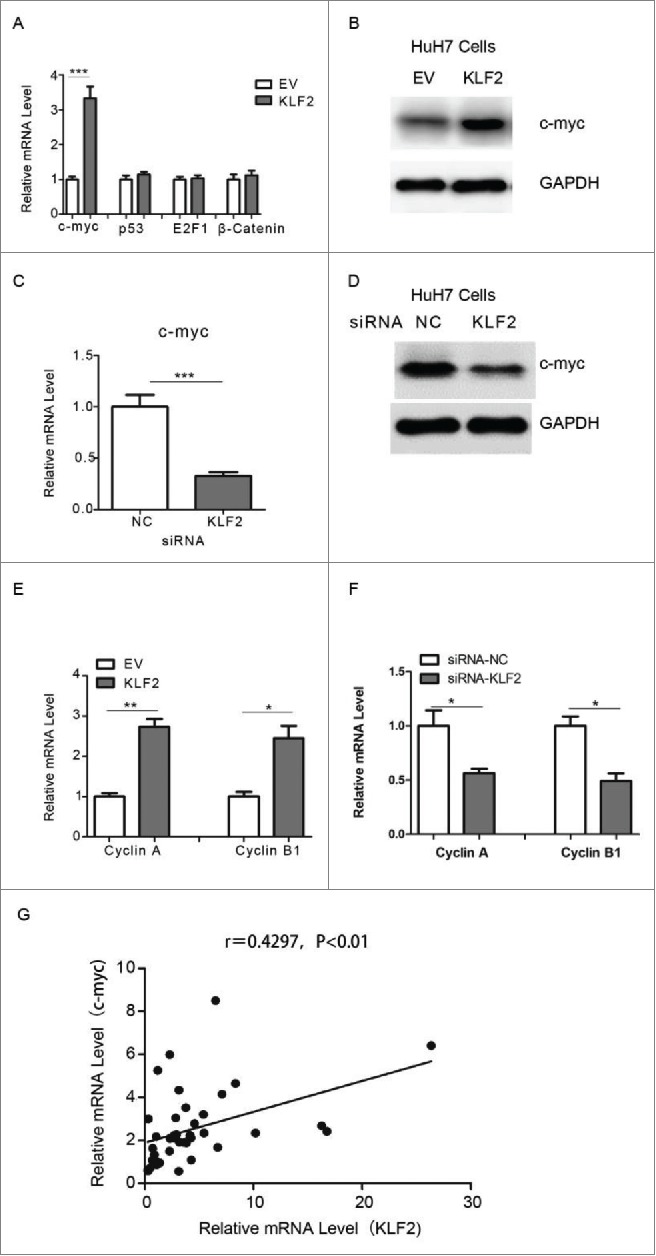
KLF2 regulates c-myc expression in HCC cells (A) Relative mRNA levels of c-myc, p53, E2F1 and β-catenin in HuH7 cells overexpressing KLF2 or empty vector (EV). (B) Representative protein levels of c-myc in HuH7 cells. (C-D) Relative mRNA (C) and protein (D) levels of c-myc in HuH7 cells transfected with siRNA oligos targeting KLF2 or negative control (NC). (E-F) Relative mRNA levels of Cyclin A and Cyclin B1 in HuH7 cells with KLF2 overexpression (E) or knockdown (F). (G) A positive correlation between KLF2 and c-myc expression in 60 paired of HCC and normal liver tissues.
Positive regulation of c-myc promoter by KLF2
To further explore the molecular mechanisms of up-regulationupregulation of c-myc expression by KLF2, the promoter region of human c-myc gene was cloned into a luciferase reporter vector. Luciferase reporter activity increased in a dose-dependent manner in HuH7 cells transfected with KLF2 expression compared with empty vector (Fig. 5A). A potential regulatory site containing KLF protein binding site (CACCC) was also identified, which located at −546 bp (Fig. 5B). Indeed, mutations of the c-myc promoter reporter in this site significantly abolished the roles of KLF2 overexpression (Fig. 5B). To be noted that, although the binding site is conserved between KLF proteins, we did not observe the up-regulation of c-myc promoter activity by other KLFs, such as KLF4 and KLF9 (Supplementary Fig. 5).
Figure 5.
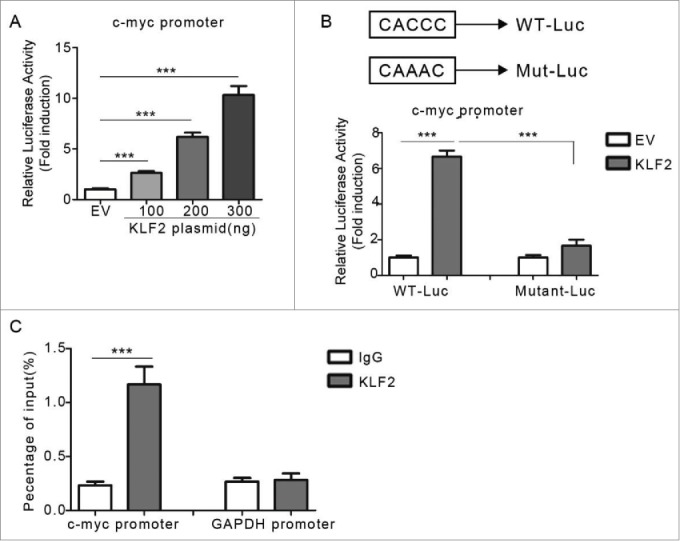
Direct regulation of c-myc promoter by KLF2 (A) The transcriptional activity of the c-myc promoter. HuH7 cells were co-transfected with c-myc promoter reporter and KLF2 expression plasmids as indicated. (B) The c-myc promoter constructs containing a potential KLF2 motif (−546 to −542 bp). Point mutations were induced in the KLF2 motif (CACCC to CAAAC). (C) ChIP assays by using KLF2 antibody or control IgG showing the direct binding of KLF2 to the c-myc promoter. GAPDH promoter region was used as a negative control.
To determine if KLF2 interacted with the endogenous c-myc promoter, we performed chromatin immunoprecipitation (ChIP) assays using anti-KLF2 antibodies and control IgG. As shown in the Fig. 5C, KLF2, but not IgG, could bind to the promoter region of human c-myc gene.
Knockdown of c-myc attenuated the roles of KLF2
Finally, to confirm the relationship between KLF2 and c-myc, HuH7 cells were administrated with siRNA oligos targeting c-myc or negative control, after transfection of KLF2 or empty vector (Fig. 6A). For this experiments, CD36, which has been shown to be regulated by KLF2 in hepatocytes by our previous study,9 was employed as a control (Fig. 6A). As expected, ablation of c-myc largely block the proliferative roles of KLF2 overexpression (Fig. 6B–C), suggesting the oncogenic effects of KLF2, at least in part, depend on its up-regulationupregulation of c-myc expression.
Figure 6.
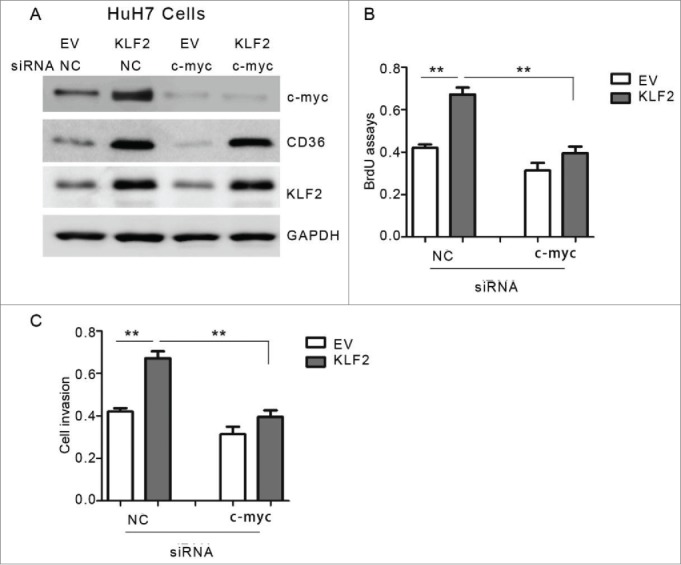
Ablation of c-myc attenuates the effects of KLF2 overexpression (A) Representative protein levels of c-myc, CD36 and KLF2 in HuH7 cells. Cells were transfected with KLF2 overexpression plasmids or empty vector (EV) for 24 hr, and then transfected with siRNA oligos targeting c-myc or negative control (NC) for another 24 hr. (B-C) Cell viability (B) and proliferation (C) assays in HuH7 cells with KLF2 overexpression and (or) c-myc knockdown as indicated.
Discussion
In the present study, our data strongly support the notion that KLF2 acts as an oncogene in HCC progression. Firstly, KLF2 expression was increased in HCC tissues, compared with adjacent normal liver tissues. Secondly, ectopic KLF2 expression promoted, whereas KLF2 silencing inhibited cell proliferation in HCC cells. However, in vivo studies using cancer models or KLF2 knockout mice are still needed to further establish the precise roles of KLF2 in HCC development. Moreover, recent studies demonstrated that promoter hypermethylation contributed to the dys-regulated expression of KLF family proteins in several cancer types.6,10-12 Whether epigenetic changes in the KLF2 promoter occurred in HCC tissues remain to be determined in the future studies.
At the molecular level, our results suggested that KLF2 is a positive regulator of c-myc. It has been shown that KLF proteins could also indirectly regulate the promoter through interactions with other transcription factors.13,14 Here, using both luciferase reporter and ChIP assays, we found that endogenous KLF2-containing transcription complex binds to and activates the c-myc promoter, indicating that c-myc might be a direct target of KLF2.
The c-myc oncogene has been implicated in the pathogenesis and progression of multiple human neoplastic diseases, including HCC.15,16 Besides, several studies have shown that c-myc is required for HCC progression induced by other molecules. For instance, Cheng Q et al. demonstrated that cellular senescence-inhibited gene (CSIG) promotes hepatocellular carcinoma proliferation by activating c-myc expression.17 Yang T et al. showed that serine/threonine kinase 33 (STK33) promotes hepatocellular carcinoma through binding directly to c-myc, by which increases the transcriptional activity of c-myc.18 Additionally, c-myc-mediated epigenetic silencing of certain MicroRNAs contributes to dysregulation of multiple pathways in HCC and is closely associated with poorer prognosis of HCC patients.19 Therefore, these results suggest that c-myc is a critical regulator in the HCC progression and may provide a promising therapeutic candidate for cancer therapy. However, the molecular determinants responsible for c-myc up-regulation in malignances are currently unclear. A recent study showed that JNK1 could control the proliferation of human HCC cells by affecting c-myc expression.20 Besides, phosphorylation, activation, and Tumorigenic potential of c-myc in HCC was shown to be regulated by HMG-CoA Reductase.21 Therefore, our data also provide an alternative transcriptional route to modulate c-myc expression in HCC. In addition, knockdown of c-myc largely attenuated the proliferative roles of KLF2 in HCC cells, suggesting that the oncogenic role of KLF2 in HCC, at least in part, is dependent on its up-regulationupregulation of c-myc. However, KLF2 might regulate HCC progression through other alternative mechanisms. Further studies are still needed to clarify this issue.
In summary, this study provided an important insight into the function of KLF2/c-myc signaling pathway in HCC progression. Given suppression of c-myc might become a novel therapy for HCC,22,23 our results may bring some advances in better drug design, pharmacokinetics and pharmacogenetics in the future.
Materials and methods
Human tissue samples
60 paired of HCC tissues and adjacent non-tumor normal specimens were collected from routine therapeutic surgery at our department. Informed consent was obtained from each patient before surgery. The study protocol was reviewed and approved by the Institutional Review Board of our Hospital Shanghai East Hospital, Tongji University School of Medicine.
Cell culture, transfection and luciferase assays
HCC cell lines (HuH-7 and HepG2 cells) were obtained from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences (CAS, Shanghai). Cells were grown in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Invitrogen) and maintained at 37°C in a humidified atmosphere with 5% CO2. RNAi-mediated depletion of KLF2 or c-myc was achieved by infecting cells with small interfering RNA (siRNA) oligos targeting KLF2 or negative control (GE Dharmacon, Pittsburgh PA, United States). The siRNA sequence for KLF2 is 5′-GCATCAGTGTGCTCGAAATGA-3′, for c-myc is 5′- TCCCTCCACTCGGAAGGAC-3′ and for negative control is 5′-CACAGTGTTCGAGATATTG-3′ . Human c-myc promoter from −1200 bp to −50 bp was amplified by PCR and inserted into the KpnI and XhoI sites of PGL3 empty vector (Promega, Madison, WI, USA). Mutant KLF2 binding site was generated using a PCR mutagenesis kit (Toyobo) with primer 5′-AGCTAAGCCTTGCGAAGACCCATTAGCAGC-3′ and a reverse complement primer (mutation sites underlined). All the transfection experiments were performed when cells were grown to 70% to 80% confluence, using Lipofectamine 2000 reagents (Invitrogen) according to the manufacturer's instructions. For luciferase assays, cells were seeded in 24-well plates and transfection efficiency was normalized by co-transfecting Simian virus 40 (SV40) plasmids (Promega). Luciferase values were measured using the Dual-Luciferase Reporter Assay System (Promega).
BrdU incorporation assays
A cell proliferation enzyme-linked immunosorbent assay (BrdU kit; Beyotime) was used to analyze the incorporation of BrdU during DNA synthesis following the manufacturer's protocols. Absorbance was measured at 450 nm in the Spectra Max 190 ELISA reader (Molecular Devices, Sunnyvale, CA).
Flow cytometry analysis
Cells were trypsinized and fixed in 70% ethanol at −20°C overnight. Cells were then resuspended in PBS containing 40 μg/ml propidium iodide and 100 μg/ml RNase A. After incubation for 1 hr at 37°C, cells were characterized and cell cycle distribution was determined by fluorescence activated cell sorting (FACS). For each sample, 2.0 * 104 cells were analyzed. Data was acquired using a BD LSRII apparatus and analyzed using the FlowJo software.
Cell invasion assays
Cell invasion abilities were determined with extracellular matrix-coated invasion chambers (Millipore) according to the manufacture's instructions. In short, HCC cells were harvested and resuspended in serum-free medium after pre-transfection for 24 hr. Then 2.0 × 105 cells were plated into the top well of an extracellular matrix-coated invasion chamber (for invasion assay). The bottom well of the chamber contained 500 μL Dulbecco's Modified Eagle's Medium supplemented with 10% FBS (for invasion assay). After 24 hours incubation, the noninvading cells were removed with a cotton swab, and the invading cells on the underside of the membrane were stained with cell stain solution (Millipore) for 10 minutes. After washed 3 times with water, the stain of each membrane was removed with 100 μL extraction buffer (Millipore) and quantitated with a colorimetric microplate reader at 570 nm.
Colony formation assays
HCC cells were seeded in a 6-well plate 48 hours post-transfection and cultured for 8 to 10 days d at 37°C in 5% CO2. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), washed twice with PBS, and stained with a crystal violet solution (1% crystal violet, 10% ethanol in water). Stained cells were washed thrice with water and counted by under an optical microscope.
Real-time PCR analysis
Total RNA from tissues and cells was extracted using the TRIzol Kit (Invitrogen) according to the manufacturer's instructions. cDNA was transcribed from 1 μg of total RNA following the manufacturer's instructions (Promega). Quantitative real-time PCR was performed using SYBR Premix Ex Taq reagents (Takara, Shiga, Japan). Relative transcript quantities were calculated using the 2−ΔΔCt method with 36B4 as the endogenous reference gene. The primer sequences were listed as below: KLF2 (Forward:5′-CTACACCAAGAGTTCGCATCTG-3′,Rev-erse:5′-CCGTGTGCTTTCGGTAGTG-3′); c-myc(Forward: 5′-GTCAAGAGGCGAACACACAAC-3′, Reverse: 5′-TTGGACGGACAGGATGTATGC-3′);CyclinA(Forward:5′-TGGAAAGCAAACAGTAAACAGCC-3′, Reverse: 5′-GGGCATCTTCACGCTCTATTT-3′); Cyclin B1 (Forward:5′-GTCAAGAGGCGAACACACAAC-3′,Reverse:5′-TTGGACGGACAGGATGTATGC-3′); 36B4 (Forward:5′-GCAGCATCTACAACCCTGAAG-3′, Reverse: 5′-CACTGGCAACATTGCGGAC-3′).
Western blot
Cells and tissues were harvested and lysed with ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM 2-Mercaptoethanol, 2% w/v SDS, 10% glycerol). After centrifugation at 10,000× g for 10 min at 4 °C, proteins in the supernatants were quantified, separated by 10% SDS PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 10% nonfat milk and then incubated with different primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The following primary antibodies were used: anti-KLF2 (Abcam, ab17008, 1:2000); c-myc (Santa Cruz, sc-40, 1:1000); CD36 (Abcam, ab78054, 1:500). Protein levels were normalized to total GAPDH (Santa Cruz, sc-365062, 1:5000). The proteins were visualized by an ECL chemiluminescence detection kit (Amersham Biosciences, Buckinghamshire, UK).
Statistical analysis
Data were expressed as meanm±estandardan±standard error of the mean (SE). Analysis was conducted with GraphPad Prism version 5.01 (GraphPad Software). Significance between two 2g groups was analyzed using the unpaired 2-tailed t test (*p < 0.05, **p < 0.01, ***p < 0.001).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
This study is supported by the National High Technology Research (863) Project of China (2012AA020204).
References
- 1.Villanueva A, Minguez B, Forner A, Reig M, Llovet JM. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med 2010; 61: 317-28; PMID:20059340; http://dx.doi.org/ 10.1146/annurev.med.080608.100623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanaka M, Katayama F, Kato H, Tanaka H, Wang J, Qiao YL, Inoue M. Hepatitis B and C virus infection and hepatocellular carcinoma in China: a review of epidemiology and control measures. J Epidemiol 2011; 21: 401-16; PMID:22041528; http://dx.doi.org/ 10.2188/jea.JE20100190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010; 29: 4989-5005; PMID:20639898; http://dx.doi.org/ 10.1038/onc.2010.236 [DOI] [PubMed] [Google Scholar]
- 4.Jiang W, Cui J, Xie D, Wang L. Sp/KLF family and tumor angiogenesis in pancreatic cancer. Curr Pharm Des 2012; 18: 2420-31; PMID:22372503; http://dx.doi.org/ 10.2174/13816128112092420 [DOI] [PubMed] [Google Scholar]
- 5.McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev 2010; 90: 1337-81; PMID:20959618; http://dx.doi.org/ 10.1152/physrev.00058.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin ZS, Chu HC, Yen YC, Lewis BC, Chen YW. Kruppel-like factor 4, a tumor suppressor in hepatocellular carcinoma cells reverts epithelial mesenchymal transition by suppressing slug expression. PLoS One 2012; 7: e43593; PMID:22937066; http://dx.doi.org/ 10.1371/journal.pone.0043593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q, Gao Y, Jia Z, Mishra L, Guo K, Li Z, Le X, Wei D, Huang S, Xie K. Dysregulated Kruppel-like factor 4 and vitamin D receptor signaling contribute to progression of hepatocellular carcinoma. Gastroenterology 2012; 143: 799-810.e1-2; PMID:22677193; http://dx.doi.org/ 10.1053/j.gastro.2012.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirach E, Bureau C, Peron JM, Pradayrol L, Vinel JP, Buscail L, Cordelier P. KLF6 transcription factor protects hepatocellular carcinoma-derived cells from apoptosis. Cell Death Differ 2007; 14: 1202-10; PMID:17347668; http://dx.doi.org/ 10.1038/sj.cdd.4402114 [DOI] [PubMed] [Google Scholar]
- 9.Chen JL, Lu XJ, Zou KL, Ye K. Kruppel-like factor 2 promotes liver steatosis through upregulation of CD36. J Lipid Res 2014; 55: 32-40; PMID:23861552; http://dx.doi.org/ 10.1194/jlr.M039453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, Wu TT, Huang S, Xie K. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res 2005; 65: 2746-54; PMID:15805274; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3619 [DOI] [PubMed] [Google Scholar]
- 11.Zammarchi F, Morelli M, Menicagli M, Di Cristofano C, Zavaglia K, Paolucci A, Campani D, Aretini P, Boggi U, Mosca F, et al.. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. Am J Pathol 2011; 178: 361-72; PMID:21224073; http://dx.doi.org/ 10.1016/j.ajpath.2010.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakahara Y, Northcott PA, Li M, Kongkham PN, Smith C, Yan H, Croul S, Ra YS, Eberhart C, Huang A, et al.. Genetic and epigenetic inactivation of Kruppel-like factor 4 in medulloblastoma. Neoplasia 2010; 12: 20-7; PMID:20072650; http://dx.doi.org/ 10.1593/neo.91122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ai W, Liu Y, Langlois M, Wang TC. Kruppel-like factor 4 (KLF4) represses histidine decarboxylase gene expression through an upstream Sp1 site and downstream gastrin responsive elements. J Biol Chem 2004; 279: 8684-93; PMID:14670968; http://dx.doi.org/ 10.1074/jbc.M308278200 [DOI] [PubMed] [Google Scholar]
- 14.Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, Lu W. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell 2013; 13: 36-47; PMID:23747203; http://dx.doi.org/ 10.1016/j.stem.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 15.Abou-Elella A, Gramlich T, Fritsch C, Gansler T. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod Pathol 1996; 9: 95-8; PMID:8657726 [PubMed] [Google Scholar]
- 16.Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer 2004; 4: 562-8; PMID:15229481; http://dx.doi.org/ 10.1038/nrc1393 [DOI] [PubMed] [Google Scholar]
- 17.Cheng Q, Yuan F, Lu F, Zhang B, Chen T, Chen X, Cheng Y, Li N, Ma L, Tong T. CSIG promotes hepatocellular carcinoma proliferation by activating c-MYC expression. Oncotarget 2015; 6:4733-44; PMID:25749381; http://dx.doi.org/ 10.18632/oncotarget.2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang T, Song B, Zhang J, Yang GS, Zhang H, Yu WF, Wu MC, Lu JH, Shen F. STK33 promotes hepatocellular carcinoma through binding to c-Myc. Gut 2014 Nov 14. pii: gutjnl-2014-307545.; PMID:25398772; http://dx.doi.org/24002871 10.1136/gutjnl-2014-307545 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Zhang X, Jia LT, Hu SJ, Zhao J, Yang JD, Wen WH, Wang Z, Wang T, Zhao J, et al.. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology 2014; 59:1850-63; PMID:24002871; http://dx.doi.org/ 10.1002/hep.26720 [DOI] [PubMed] [Google Scholar]
- 20.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest 2008; 118: 3943-53; PMID:19033664; http://dx.doi.org/ 10.1172/JCI37156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS, Felsher DW. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011; 71: 2286-97; PMID:21262914; http://dx.doi.org/17159602 10.1158/0008-5472.CAN-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin CP, Liu JD, Chow JM, Liu CR, Liu HE. Small-molecule c-Myc inhibitor, 10058-F4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 2007; 18: 161-70; PMID:17159602; http://dx.doi.org/ 10.1097/CAD.0b013e3280109424 [DOI] [PubMed] [Google Scholar]
- 23.Lin CP, Liu CR, Lee CN, Chan TS, Liu HE. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol 2010; 2: 16-20; PMID:21160952; dx.doi.org/ 10.4254/wjh.v2.i1.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.