

ABSTRACT
Small cell lung cancer (SCLC) is an aggressive tumor type with high mortality. One promising approach for SCLC treatment would be to utilize agents targeting molecular abnormalities regulating resistance to apoptosis. BH3 mimetic antagonists, such as ABT-737 and its orally available derivative ABT-263 (navitoclax) have been developed to block the function of pro-survival BCL-2 family members. The sensitivity of SCLC to these drugs varies over a broad range in vitro and in clinical trials. We have previously shown that the expression of Noxa, a BH3-only pro-apoptotic BCL-2 family protein, is a critical determinant of sensitivity to ABT-737. Thus, pharmacological up-regulation of Noxa could enhance cell death induced by the BH3 mimetics. We find that the combination of ABT-263 and a HDAC inhibitor, vorinostat, efficiently induces apoptosis in a variety of SCLC cell lines, including ABT-263 resistant cell lines. Cell death induced by combined treatment is Noxa- and/or BIM-dependent in some cell lines but in others appears to be mediated by down-regulation of BCL-XL and release of BAK from BCL-XL and MCL-1. These results suggest that combination of HDAC inhibitors and BCL-2 inhibitors could be an alternative and effective regimen for SCLC treatment.
KEYWORDS: ABT-263, apoptosis, BCL-2 family, BCL-XL, Noxa, small cell lung cancer, vorinostat
Abbreviations
- SCLC
small cell lung cancer
- BCL
B-cell lymphoma
- BH
BCL-2 homology
- HDAC
histone deacetylase
- BIM
BCL-2 interacting mediator of cell death
- MCL-1
myeloid cell leukemia sequence 1
- shRNA
short-hairpin RNA.
Introduction
Lung cancer is the most common cause of cancer related mortality worldwide, with small cell lung cancer (SCLC) representing 15% of cases and accounting for 30,000 new cases in the US each year.1 While SCLC is initially a highly chemotherapy-responsive disease, relapse and progressive development of chemotherapy resistance is the rule. SCLC has not seen substantial improvements in therapies over the past 4 decades and no targeted therapies have been approved for SCLC.2,3 One promising approach to overcome the therapeutic resistance would be to utilize agents targeting molecular abnormalities regulating resistance to apoptosis.
Tumor cell death induced by chemotherapy or lack of appropriate cellular survival signals is mediated by the BCL-2 family-dependent mitochondrial apoptotic pathway.4,5 We and others have shown that the pro-survival member BCL-2, as well as BCL-XL and MCL-1, are overexpressed in SCLC.6,7 BH3 mimetic antagonists have been developed to block the function of pro-survival BCL-2 family members. ABT-737 and its orally available derivative ABT-263 bind to and block BCL-2 and BCL-XL, but not MCL-1 function.8-10 Although SCLC is the only non-hematologic malignancy against which ABT-737/ABT-263 are effective as single agents, the sensitivity of SCLC to these drugs varies over a broad range in vitro and in clinical trials.11-13 We have shown in a model SCLC system that expression of Noxa, a BH3-only pro-apoptotic BCL-2 family protein, is a critical determinant of ABT-737 sensitivity, demonstrating that Noxa specifically binds to and recruits MCL-1 from the cytosol to the mitochondria which results in its phosphorylation, ubiquitination and eventual degradation. 14,15
Recent integrative genome analysis in SCLC implicates histone modification as a major feature of SCLC.16 It has also been demonstrated that Noxa is transcriptionally activated by histone deacetylase (HDAC) inhibitors.17 Thus, we hypothesized that alteration of histone modification by a HDAC inhibitor, vorinostat could induce Noxa to enhance cell death by ABT-737/263 treatment. We find that combination of ABT-263 with vorinostat efficiently induces apoptosis in a variety of SCLC cell lines, even in ABT-263 resistant cells. Cell death induced by combined treatment is Noxa- and/or BIM-dependent in some cell lines but in others appears to be mediated by downregulation of BCL-XL and release of BAK from BCL-XL and MCL-1. These results suggest that combination of HDAC inhibitors and BCL-2 inhibitors could be an alternative and effective regimen for SCLC treatment.
Results
Combined treatment with ABT-263 and vorinostat efficiently induces cell death in a variety of SCLC cell lines
We have shown that the ectopic expression of Noxa or BIM in H209 cells, in which the levels of Noxa and BIM are originally undetectable, restores sensitivity to ABT-737.15 It has also been demonstrated that Noxa and/or Bim can be transcriptionally activated by HDAC inhibitors.17 Thus, we hypothesized that alteration of histone modification by the HDAC inhibitor vorinostat could induce Noxa and/or BIM to enhance cell death by ABT-737/263 treatment. In this study, we used ABT-263, an orally available derivative of ABT-737 that is in clinical trials. We first determined the IC50 for ABT-263 or vorinostat alone in a representative panel of SCLC cell lines along with the basal expression of anti- and pro-apoptotic BCL-2 family proteins (Fig. 1). The IC50 for vorinostat was in a tight distribution from 0.5 to 2 µM without any obvious correlation to Bcl-2 family protein expression. In contrast, the IC50 for ABT-263 varied over 2 logs from 0.05 to >3 µM. The level of Noxa expression correlated with ABT-263 sensitivity: higher in H2171, H2141, and H69 that are relatively sensitive to ABT-263, and lower in H526, H82, and H209 that are resistant. The level of BIM was variable from undetectable in H209 to high expression in H2141 and H526. The levels of anti-apoptotic MCL-1 and BCL-XL were similar across the SCLC cell lines that we examined. The level of BCL-2 varied from undetectable in H82 to high expression in H2171, H2141, and H526, without correlation to ABT-263 sensitivity. Based on the results of IC50 testing, we designated H2171, H69, and H2141 cells as sensitive, and H526, H82, and H209 cells as resistant to ABT-263.
Figure 1.
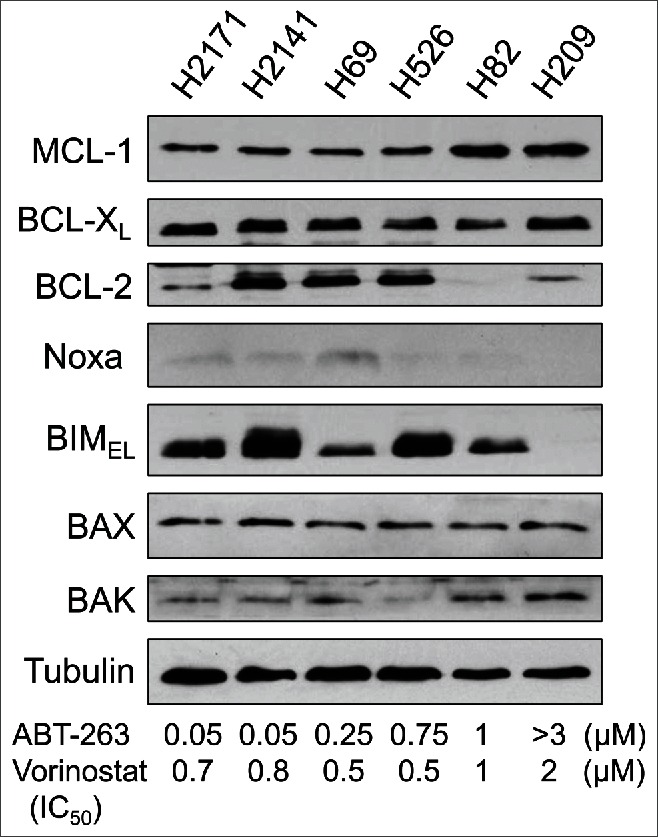
The expression levels of Noxa in SCLC cells correlate with sensitivity to ABT-263. Expression of the BCL-2 family proteins in SCLC cell lines was determined by immunoblot analysis. The IC50 of ABT-263 and vorinostat in each cell line was determined by WST-1 assay at 72 hours after treatment.
We then examined the effect of combining ABT-263 and vorinostat. Based on the respective IC50, we treated each cell line with combinations of suboptimal therapeutically achievable concentrations of ABT-263 and vorinostat which did not as single agents induce cell death efficiently. All cell lines, including ABT-263 resistant cell lines, showed enhanced cell death with combined treatment of ABT-263 and vorinostat (Fig. 2). Of note, cell death was efficiently induced with vorinostat alone in H82 cells (which express no detectable BCL-2), with relatively minimal enhancement on addition of ABT-263 compared to other cell lines. We confirmed that apoptosis was efficiently induced by combined treatment as judged by PARP cleavage on Western blot analyses (Fig. 3). Consistently, Noxa was induced by the combined treatment except in H209 cells, which still possessed undetectable levels of Noxa and BIM protein, although both mRNAs were slightly induced by vorinostat (Fig. 3 and data not shown).
Figure 2.

Co-treatment with ABT-263 and vorinostat efficiently induces cell death in SCLC cells. Each SCLC cell line was treated with ABT-263 and/or vorinostat at the concentrations (µM) indicated in each graph. Cell viability was determined by WST-1 assay at 72 hours after treatment. Average values from triplicate samples are shown as representative of 2 independent experiments. Con, vehicle control; V (µM), vorinostat; ABT (µM), ABT-263. *P < 0.01 compared with control.
Figure 3.
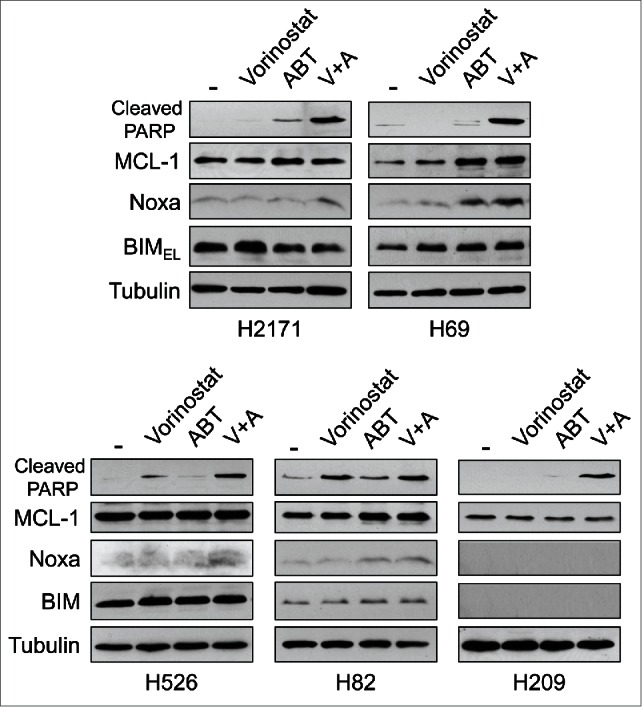
Co-treatment with ABT-263 and vorinostat induces Noxa followed by apoptosis in SCLC cells.SCLC cells were treated with ABT-263 and/or vorinostat for 24 hours at concentrations indicated in Figure 2. Equal amounts of total extracts were subjected to immunoblot analysis using the indicated antibodies.
Noxa and/or BIM contribute to apoptosis induced by ABT-263 and vorinostat co-treatment
Since Noxa induction was detected with ABT-263 and vorinostat co-treatment even in the ABT-263-resistant H82 and H526 cells (Fig. 3), we examined the role of Noxa in apoptosis induced by combined treatment. In H82 cells, in which the basal expression of BCL-2 was low, treatment with vorinostat alone induced mRNA of Noxa, but not Bim (Fig. 4A). When Noxa was down-regulated by shRNA in H82 cells, vorinistat-induced apoptosis was significantly reduced, indicating the contribution of Noxa (Fig. 4B and C). Interestingly, down-regulation of BIM by shRNA also significantly reduced apoptosis induced by vorinostat, suggesting a contribution by BIM as well.
Figure 4.

Noxa and BIM contribute to apoptosis induced by vorinostat in H82 cells.(A) The levels of Noxa and Bim mRNA in H82 cells treated with vorinostat (1 µM) for 8 hours were determined by qPCR. Average values from triplicate samples are shown. NT, control. (A) and (C) H82 cells were infected with lentivirus-encoding shRNA for non-targeting control, Noxa, or Bim. Forty-eight hours after the infection, cells were then treated with vorinostat (1 µM) for 24 hours and equal amounts of total extracts were subjected to immunoblot analysis using the indicated antibodies. Cell viability was determined by WST-1 assay at 72 hours after treatment. *P < 0.02 compared to sh-Control with co-treatment.
In H526 cells, both Noxa and Bim mRNAs were induced by vorinostat alone and in combination with ABT-263 (Fig. 5A). However, downregulation of Noxa in H526 cells did not affect apoptosis induced by co-treatment. In contrast, down-regulation of BIM partially inhibited apoptosis, suggesting the contribution of BIM (Fig. 5B and C). These results may be in part reflective of the basal levels of BIM and Noxa proteins, which are relatively high and low, respectively, in H526 cells compared to those in other cell lines (Fig. 1; see also Discussion).
Figure 5.

BIM contributes to apoptosis induced by vorinostat and ABT-263 in H526 Cells.(A) The levels of Noxa and Bim mRNA in H526 cells treated with vorinostat (0.5 µM) for 8 hours were determined by qPCR. Average values from triplicate samples are shown. NT, control. (B) and (C) H526 cells were infected with lentivirus-encoding shRNA for non-targeting control, Noxa, or Bim. Cells were then treated with vorinostat (0.5 µM) and/or ABT-263 (0.1 µM) for 24 hours and equal amounts of total extracts were subjected to immunoblot analysis using the indicated antibodies. Cell viability was determined by WST-1 assay at 72 hours after treatment. *P < 0.02 compared to sh-Control with co-treatment.
The reduction of BCL-XL by ABT-263 and vorinostat co-treatment contributes to apoptosis in H209 cells
In contrast to the other SCLC cells, H209 cells express very low (undetectable by immunoblot analyses) basal levels of Noxa and BIM (Fig. 1). When H209 cells were treated with vorinostat and ABT-263 in combination, apoptosis was efficiently induced (Figs. 2 and 3). However, we still could not detect either protein, although both mRNAs were slightly induced (data not shown). We therefore investigated the expression of other BCL-2 family proteins. Surprisingly, the protein expression of BCL-XL was dramatically reduced only by the treatment with ABT-263 and vorinostat in combination, although the expression of BCL-2 and MCL-1 remained at similar levels under all conditions (Fig. 6A). Similar phenomena were also observed in H526 cells (Fig. S1A). The decrease of BCL-XL protein could not be explained by the mRNA levels, since treatment with vorinostat or ABT-263 alone also reduced the BCL-XL mRNA expression but not the protein (Fig. 6B). Furthermore, the decrease of BCL-XL protein was not altered by a pan-caspase inhibitor, Q-VD-OPh, but was reversed by a proteasome inhibitor, MG-132 (Fig. S2A). These data suggest that the reduction of BCL-XL protein by the combination treatment is caused by proteasome-dependent, but caspase-independent post-translational mechanisms. We also profiled the levels of BH3-only proteins (Fig. S2B). None of them (Noxa, BIM, and BIK were undetectable) were increased by co-treatment with ABT-263 and vorinostat. However, BAK was released from BCL-XL and MCL-1 and became an active form (Fig. 6C). These results suggest that the drug combination results in the activation of BAK by means other than increased expression of BH3-only proteins, which, along with BCL-XL downregulation, may be sufficient to induce apoptosis by ABT-263 and vorinostat co-treatment.
Figure 6.

Survival of H209 cells is dependent on BCL-XL and MCL-1 expression. (A) H209 cells were treated with vorinostat (1 µM) and/or ABT-263 (1 µM) for 24 hours and equal amounts of total extracts were subjected to immunoblot analysis using the indicated antibodies. (B) The levels of Bcl-XL and Mcl-1mRNA treated with vorinostat (1 µM) and/or ABT-263 (1 µM) for 8 hours were determined by qPCR. Average values from triplicate samples are shown. (C) H209 cells were treated with vorinostat (V; 1 µM) and ABT-263 (A; 1 µM) for 24 hours. Total cell extracts were immunoprecipitated with anti-BAK (06–536) (upper panel) or conformation-specific anti-BAK (Ab-1) (lower panel) antibodies. Immunoblot analyses were carried out on precipitated samples with the indicated antibodies. Ig-H, immunoglobulin heavy-chain; Ig-L, immunoglobulin light-chain. (D) and (E) H209 cells were infected with lentivirus-encoding shRNA for non-targeting control or Bcl-XL. Cells were then treated with vorinostat (1 µM) and/or ABT-263 (1 µM) for 24 hours and equal amounts of total extracts were subjected to immunoblot analysis using the indicated antibodies. Cell viability was determined by WST-1 assay at 72 hours after treatment. *P < 0.01 compared to sh-Control with vehicle control. (F) H209 cells were infected with lentivirus-encoding shRNA for non-targeting control, Bcl-XL, Bcl-2, or Mcl-1 for 72 hours. Equal amounts of total extracts were then subjected to immunoblot analysis using the indicated antibodies. Different target sequences for BCL-XL shRNAs were used in (D) and (F).
To confirm the significance of BCL-XL protein expression, we downregulated the expression of BCL-XL by shRNA. Efficient downregulation of BCL-XL alone strongly induced apoptosis in H209 cells, and apoptosis was slightly augmented by co-treatment (Fig. 6D and 6E). We further addressed whether reduction of each anti-apoptotic BCL-2 family protein could induce apoptosis in SCLC cells. We introduced shRNAs that were specific for BCL-XL, BCL-2 or MCL-1 in H209 cells. Down-regulation of each protein induced apoptosis, but the effect was much stronger when BCL-XL or MCL-1 was reduced by shRNA (Fig. 6F). Furthermore, compared with the control, we could not obtain stable cell clones with BCL-XL or MCL-1 shRNA and obtained a lesser number of clones with BCL-2 shRNA (encoded by a vector containing a puromycin resistance gene) after selection with puromycin (data not shown). Similar data were obtained with H526 cells (Fig. S1B). These results suggest that these SCLC cell lines preferentially depend on BCL-XL and MCL-1 for survival.
Discussion
SCLC is the only solid tumor against which ABT-263 is effective as a single agent, albeit with a broad range of sensitivities. While the drug could eventually result in a significant advance in the treatment of SCLC, considerable work must be done to optimize the likelihood of therapeutic success. Next-generation sequencing analyses to date have yet to identify clear SCLC-mutated drug targets. Under these circumstances, Peifer et al.16 using an integrative genome analysis in SCLC has suggested histone modification as a major feature of SCLC. We thus examined the effect of a combination of ABT-263 and vorinostat, a HDAC inhibitor that has been approved for treatment of cutaneous T-cell lymphoma treatment. We found that treatment with vorinostat overcomes ABT-263 resistance of SCLC cell lines such as H526, H82, and H209. Among them, vorinostat treatment alone induced significant cell death in H82, likely because expression of BCL-2 and BCL-XL, which are the targets of ABT-263, are very low in this cell line to begin with. Combination with ABT-263/-737 and vorinostat also induces synergistic cell death in mantle cell lymphoma,18 squamous cell carcinoma (SCC),19 diffuse large B-cell lymphoma,20 and the Eµ-myc mouse lymphoma model.21 In SCC, vorinostat induces Noxa and BIM while suppressing MCL-1, thereby shuttling BIM from MCL-1 to BCL-2/BCL-XL, which primes cells for sensitivity to ABT-737 and results in synergy for combination therapy, suggesting that MCL-1 is a dominant survival factor in SCC. As observed in other tumors, Noxa and/or BIM were also induced by vorinostat treatment in all SCLC cell lines except H209 cells (Fig. 3–5). Downregulation of Noxa or BIM by shRNA confirmed the contribution of these proteins to the efficacy of ABT-263 and vorinostat co-treatment (Figs. 4 and 5). However, depending on the expression of other BCL-2 family proteins in addition to Noxa and BIM, the contribution of these proteins may differ between cell lines. For example, both Noxa and BIM play a role in cell death with the combined treatment in H82 cells. Since both BCL-2 and BCL-XL expression are low, survival of this cell line could largely depend on MCL-1. Noxa and BIM have a strong affinity for MCL-1, thus the induction of Noxa or BIM by vorinostat is sufficient to induce apoptosis. In contrast, BIM, but not Noxa, contributes to apoptosis induced by co-treatment of H526 cells. Since survival of this cell line is dependent more on BCL-XL and MCL-1 than BCL-2 (Fig. S1) and the basal expression of BIM is high (Fig. 1), the vorinostat-induced Noxa expression appears to be relatively dispensable.
Although apoptosis was efficiently induced by co-treatment in H209 cells, the expression of Noxa and BIM remained undetectable (Fig. 3), suggesting an alternative mechanism of sensitization by vorinostat. It does not appear that increases in BH3-only proteins are responsible for this phenomenon since their levels were either decreased or unchanged by co-treatment (Fig. S2B). We then turned our attention to the expression of anti-apoptotic BCL-2 family proteins and found that BCL-XL expression was specifically reduced by co-treatment. In this situation, BAK was released from BCL-XL and MCL-1 and became an active form (Fig. 6C). We speculate that BAK/MCL-1 interaction may be disrupted by unknown mechanisms, which could be regulated by vorinostat. Using shRNAs to down-regulate BCL-XL expression, we confirmed the survival dependency of BCL-XL in H209 cells (Fig. 6). In this regard, ABT-263/-737 alone could not effectively induce apoptosis in H209 cells, probably because BCL-2 is a better target of these compounds than BCL-XL22,23 and MCL-1 provides a potent survival stimulus in the absence of BIM and Noxa expression. It has been recently demonstrated that a BCL-XL specific inhibitor, A-1155463 has a capability to induce cell death in H146 SCLC cells in which survival is dependent on BCL-XL.24 On the other hand, ABT-199, a BCL-2 specific inhibitor is effective in chronic lymphocytic leukemia treatment in clinical trials, probably because the survival of CLL cells is highly dependent on BCL-2 expression.25,26 Thus, different tumor types may depend on different anti-apoptotic BCL-2 family members for survival and even within tumor types dependency may vary based on the relative expression of anti- and pro-apoptotic BCL-2 family members.
We have shown that in SCLC vorinostat alters BCL-2 family expression by enhancing Noxa and BIM and decreasing BCL-XL expression, resulting in marked activity for the combination with ABT-263. The combination of vorinostat with ABT-263 at therapeutically achievable concentrations resulted in ≥90% loss of viability in 5/6 cell lines, suggesting that it may have clinical applicability. One concern in this regard, however, is that both drugs produce thrombocytopenia as a major adverse reaction. It has been clearly documented that ABT-263 produces thrombocytopenia by inactivating platelet BCL-XL.27,28 The mechanism of vorinostat-induced thrombocytopenia is unclear, but based on our data it is interesting to speculate that it may also involve down-regulation of BCL-XL, which is required for viability of circulating platelets. However, HDAC inhibitors globally alter gene transcription, thus other targets in addition to the BCL-2 family proteins may also contribute to both activity and toxicity. Our results (Fig. S2A) suggest that the reduction of BCL-XL is regulated by caspase-independent, proteasome-dependent post-translational mechanisms, so it is possible that gene products affected by vorinostat could modify the protein stability of BCL-XL and other proteins as well.
Materials and methods
Cell lines and cell culture
Human small cell lung cancer (SCLC) cell lines and 293T cells were purchased from the American Type Culture Collection (Manassas, VA). H69, H526, H82, and H209 were cultured in RPMI 1640 (Life Technologies, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 100 μg/ml penicillin G/streptomycin at 37°C in a humidified, 5% CO2 incubator. H2171 and H2141 were cultured in DMEM/F12 (Life Technologies) supplemented with 5% FBS, extra 2 mM L-glutamine, insulin-transferrin-sodium selenite supplement (Roche, Indianapolis, IN) and 100 μg/ml penicillin G/streptomycin. 293T cells were cultured in DMEM supplemented with 10% FBS and 100 μg/ml penicillin G/streptomycin.
Plasmid construction and virus infection
The lentiviral short-hairpin RNA (shRNA)-expressing constructs were purchased from Open Biosystems (Huntsville, AL) or Sigma-Aldrich (St. Louis, MO). The constructs were transfected into 293T packaging cells along with the packaging plasmids (Addgene; Cambridge, MA) and the lentivirus-containing supernatants were used to transduce SCLC cells.
Chemicals and antibodies
ABT-263 and vorinostat were purchased from Selleckchem (Houston, TX) and Cayman Chemical (Ann Arbor, MI), respectively. Antibodies were purchased as follows: BIM (C34C5), BCL-XL (54H6), and Cleaved PARP (D64E10) from Cell Signaling Technology (Danvers, MA); Noxa (114C307.1) from Thermo Fisher Scientific (Waltham, MA); MCL-1 from Enzo Life Sciences (Farmingdale, NY); BAX (N-20) and α-Tubulin (sc-8035) from Santa Cruz Biotechnology (Santa Cruz, CA); BAK (06–536) and BAK (Ab-1) from Millipore (Billerica, MA).
Immunoprecipitation and immunoblot analyses
Whole cell lysates were prepared with CHAPS lysis buffer [20 mM Tris (pH 7.4), 137 mM NaCl, 1 mM dithiothreitol (DTT), 1% CHAPS (3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate), a protease inhibitor cocktail, and phosphatase inhibitor cocktails (Sigma)]. For immunoprecipitation, equal amounts of protein were pre-cleared with protein A/G beads (Pierce, Rockford, IL), and incubated with the appropriate antibodies on ice for 2 hours. Then the antibody complexes were captured with protein A/G beads at 4°C for 1 hour. After washing 3 times with the same lysis buffer, the beads were re-suspended in the sample buffer and separated by SDS-PAGE. For Western blot analyses, equal amounts of proteins were loaded on a SDS polyacrylamide gel, transferred to a nitrocellulose membrane and analyzed by immunoblotting.
Cell viability assay
SCLC cells were seeded in triplicate in microtiter plates (96 wells) with a concentration of 4 × 104 cells per well in 100 μl medium and were treated with 0–3 μM ABT-263 or 0–10 μM vorinostat for 72 hours. Cell survival was measured by the WST-1 assay, which is based on the tetrazolium salt WST-1 (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulphonate), according to the manufacturer's instruction (Roche; Mannheim, Germany).
Quantitative RT-PCR
Total RNA was extracted with the Quick-RNA MiniPrep (Zymo Research, Irvine, CA) from SCLC cell lines. One-microgram of RNA was reverse-transcribed by High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. Using Taqman Gene Expression Assay probe/primer [Hs00197982 for Bim, Hs00560402 for Noxa, Hs00236329 for Bcl-XL, and Hs03043899 for Mcl-1 (Applied Biosystems)], cDNAs were amplified in a fluorescence thermocycler (Applied Biosystems 7500HT Fast Real-time PCR system) and were analyzed based on the expression level of GADPH with SDS2.2 software (Applied Biosystems).
Statistical analysis
Values represent the means ±SD for three separate experiments. The significance of differences between experimental variables was determined using the Student's t-test. Values were considered statistically significant at P < 0.05.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Sumitra Deb, Swati Deb, and the members of Steven Grant's lab for their valuable comments. This work was supported in part by the Massey Cancer Center Pilot Project grant (to GWK and HH). WN was partly supported by Nippon Medical School.
References
- 1.Jackman DM, Johnson BE. Small-cell lung cancer. Lancet 2005; 366:1385-96; PMID:16226617; http://dx.doi.org/ 10.1016/S0140-6736(05)67569-1 [DOI] [PubMed] [Google Scholar]
- 2.Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol 2007; 2:327-43; PMID:17409807; http://dx.doi.org/ 10.1097/01.JTO.0000263718.69320.4c [DOI] [PubMed] [Google Scholar]
- 3.William WN Jr, Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol 2011; 8:611-9; PMID:21691321; http://dx.doi.org/ 10.1038/nrclinonc.2011.90 [DOI] [PubMed] [Google Scholar]
- 4.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47-59; PMID:18097445; http://dx.doi.org/ 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 5.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell 2010; 37:299-310; PMID:20159550; http://dx.doi.org/ 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Ezra JM, Kornstein MJ, Grimes MM, Krystal G. Small cell carcinomas of the lung express the Bcl-2 protein. Am J Pathol 1994; 145:1036-40; PMID:7977636 [PMC free article] [PubMed] [Google Scholar]
- 7.Ikegaki N, Katsumata M, Minna J, Tsujimoto Y. Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res 1994; 54:6-8; PMID:8261463 [PubMed] [Google Scholar]
- 8.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al.. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677-81; PMID:15902208; http://dx.doi.org/ 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
- 9.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, et al.. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res 2007; 67:1176-83; PMID:17283153; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2203 [DOI] [PubMed] [Google Scholar]
- 10.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al.. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68:3421-8; PMID:18451170; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5836 [DOI] [PubMed] [Google Scholar]
- 11.Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, Oleksijew A, O'Connor JM, Wang B, Frost DJ, et al.. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res 2008; 14:3268-77; PMID:18519752; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4622 [DOI] [PubMed] [Google Scholar]
- 12.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, et al.. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 2011; 29:909-16; PMID:21282543; http://dx.doi.org/ 10.1200/JCO.2010.31.6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, et al.. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012; 18:3163-9; PMID:22496272; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauck P, Chao BH, Litz J, Krystal GW. Alterations in the Noxa/Mcl-1 axis determine sensitivity of small cell lung cancer to the BH3 mimetic ABT-737. Mol Cancer Ther 2009; 8:883-92; PMID:19372561; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-1118 [DOI] [PubMed] [Google Scholar]
- 15.Nakajima W, Hicks MA, Tanaka N, Krystal GW, Harada H. Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell Death Dis 2014; 5:e1052; PMID:24525728; http://dx.doi.org/ 10.1038/cddis.2014.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al.. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44:1104-10; PMID:22941188; http://dx.doi.org/ 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia 2007; 21:1773-82; PMID:17525724; http://dx.doi.org/ 10.1038/sj.leu.2404760 [DOI] [PubMed] [Google Scholar]
- 18.Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, Colomer D. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin Cancer Res 2011; 17:3956-68; PMID:21652541; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-3412 [DOI] [PubMed] [Google Scholar]
- 19.He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ, McDermott U, Rothenberg SM, Benes CH, Ellisen LW. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer Discov 2013; 3:324-37; PMID:23274910; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson RC, Vardinogiannis I, Gilmore TD. The sensitivity of diffuse large B-cell lymphoma cell lines to histone deacetylase inhibitor-induced apoptosis is modulated by BCL-2 family protein activity. PLoS One 2013; 8:e62822; PMID:23667527; http://dx.doi.org/ 10.1371/journal.pone.0062822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, Ralli R, Scott CL, Frenzel A, Villunger A, et al.. Deciphering the molecular events necessary for synergistic tumor cell apoptosis mediated by the histone deacetylase inhibitor vorinostat and the BH3 mimetic ABT-737. Cancer Res 2011; 71:3603-15; PMID:21398407; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3289 [DOI] [PubMed] [Google Scholar]
- 22.Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, Yue P, Robati M, Phipson B, Fairlie WD, et al.. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 2012; 119:5807-16; PMID:22538851; http://dx.doi.org/ 10.1182/blood-2011-12-400929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis 2012; 3:e366; PMID:22875003; http://dx.doi.org/ 10.1038/cddis.2012.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, et al.. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett 2014; 5:1088-93; PMID:25313317; http://dx.doi.org/ 10.1021/ml5001867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al.. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19:202-8; PMID:23291630; http://dx.doi.org/ 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 26.Awan FT, Byrd JC. New strategies in chronic lymphocytic leukemia: shifting treatment paradigms. Clin Cancer Res 2014; 20:5869-74; PMID:25294898; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, et al.. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007; 14:943-51; PMID:17205078; http://dx.doi.org/ 10.1038/sj.cdd.4402072 [DOI] [PubMed] [Google Scholar]
- 28.Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, et al.. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 2010; 11:1149-59; PMID:21094089; http://dx.doi.org/ 10.1016/S1470-2045(10)70261-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.