

ABSTRACT
Tangeretin, a major phytochemicals in tangerine peels - an important Chinese herb, has been found to have anti-carcinogenic properties. To improve bioavailability and increase potency of tangeretin, its derivative, 5-acetyloxy-6,7,8,4′-tetramethoxyflavone (5-AcTMF), has been synthesized and shown potent inhibition of proliferation activity against human breast and leukemia cancer cell lines. In this study, we have further investigated the anticancer effects of 5-AcTMF on CL1-5 non-small cell lung cancer cells (NSCLC) both in vitro and in vivo and demonstrated that 5-AcTMF effectively inhibited cancer cell proliferation, induced G2/M-phase arrest associated with cdc2 and CDC25c and increased in the apoptotic cells associated with caspase activation, down regulation of Bcl-2, XIAP and Survivn, inducing release of cytochrome c into the cytosol and disruption of mitochondrial membrane potential. We also found that 5-AcTMF treatment of CL1-5 activated autophagy, indicated by triggered autophagosome formation and increased LC3-II levels and formation of LC3 puncta. Moreover, we also found that 5-AcTMF lowered phophoatidylinositol 3-kinase/AKT/mTOR signaling pathway. Over-expression of AKT by AKT cDNA transfection decreased 5-AcTMF mediated apoptosis and autophagy, supporting the induction of apoptosis and autophagy by inhibition of AKT pathway. In an animal study, 5-AcTMF effectively delayed tumor growth in a nude mouse model of CL1-5 xenografts without observed adverse effect. Immunohistochemistry Analysis indicated that 5-AcTMF induced CL1-5 cell apoptosis and autophagy in vivo. Taken together, these data demonstrate that 5-AcTMF is a novel small molecule agent that can inhibit NSCLC cell proliferation, and induce G(2)/M phase arrest and via the mitochondrial apoptotic pathway and autophagy.
KEYWORDS: AKT; apoptosis; autophagy; G2/M arrest; non-small cell lung cancer cell; 5-Acetyloxy-6,7,8,4′-tetramethoxyflavone
Abbreviations
- 5-AcTMF
5-Acetyloxy-6,7,8,4′-tetramethoxyflavone
- NSCLC
non-small cell lung cancer
- PARP
poly ADP ribose polymerase
- XIAP
X-linked inhibitor of apoptosis protein
- TAN
Tangeretin
- PMF
Polymethoxyflavones
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
- 3-MA
3-Methyladenine
- PBS
phosphate-buffered saline.
Introduction
`Lung cancer, one of the leading causes of cancer death worldwide. Statistic data heve shown that 5-year survival rate for patients with lung cancer was 13%.1 There are 3 types of lung cancer: non–small cell lung cancers (NSCLCs), small cell lung cancers (SCLCs) and lung carcinoid tumor. NSCLCs could be further subdivided into adenocarcinoma, squamous cell carcinoma and large cell carcinoma.2 Approximately 85% of lung cancer are NSCLCs and most of NSCLCs patients with advanced stage at diagnosis.3 Regarding the treatment in NSCLCs patients, the outcomes of chemotherapy and radiotherapy regimens are poor due to drug resistance and cell protective mechanisms.4
In searching for novel cancer medication, extensive research in the past decade has been performed and found that some natural phytochemicals showed inhibitory effects on carcinogenesis and tumor growth.5,6 Recent studies have demonstrated that the combination of cancer drugs, either commercially available or newly developed, with dietary phytochemicals exhibits stronger anti-cancer properties than that of a single agent. More importantly, synergistic effects have been found between some natural compounds and current anti-cancer drugs, which often yield less toxic to primary cells.7,8
Polymethoxyflavones (PMFs) are an important and unique class of flavonoids, found exclusively in citrus species. There are great interests in health promoting and medicinal properties of citrus PMFs, which have been shown to exhibit a myriad of biological activities including anti-cancer anti-angiogenesis, inhibition of inflammation, reduction of atherogenesis, and neuro-protective properties.9-13 Phytochemical compounds play important physiological and ecological roles. In addition, these compounds are also of commercial interests in the food and pharmaceutical industries because of multitude of applications.14
Tangeretin (5,6,7,8,4′-pentamethoxyflavone, TAN) is one of the dominant citrus peel PMFs found in tangerine, sweet orange, and other citrus peels.15 Recent studies have demonstrated that the property of TAN in suppressing triglyceride accumulation in mature 3T3-L1 adipocytes16 and showed anti-inflammatory effect in microglial cell.17 Besides, TAN also found to inhibit cancer cell growth by inducing G1 arrest or apoptosis in some types of cancer cells.18-20
However, due to the poor solubility and bioavailability of some biologically active phytochemicals such as TAN, acetylation modification is often employed to obtain a pro-drug derivative to increase in vivo absorption and efficacy of the interested natural products.21,22 By applying the same rational, we have prepared the acetyl derivative of tangeretin via 2-step reaction, i.e. 5-demethylation of TAN in an acid catalyzed alcohol solution to yield 5-demethyltangeretin and acetylation of 5-demethyltangeretin with acetic anhydride under basic conditions.23,24 The resulted 5-acetyloxy-6,7,8,4′-tetramethoxyflavone (5-AcTMF) have been found to exert in vitro anti-proliferative effects on human multiple myeloma U226 cells23 and breast cancer cells MCF-7.24 5-AcTMF also inhibited proliferation and induced mitochondria-associated apoptotic pathway in U226 cells. Additionally, in vitro study has shown that 5-AcTMF exhibits cytotoxicity against MCF-7 cells by inducting intrinsic apoptosis, indicating that 5-AcTMF possesses much stronger inhibitory effects on the growth of MCF-7 cancer cells compared with its parent compound tangeretin. However, up to date, there has no report on the antitumor effects of 5-AcTMF against human lung cancer cells and the underlying mechanism needs to be explored. Therefore, in this study, we investigated the anticancer effects of 5-AcTMF on NSCLC cell line in vitro and in vivo, and elucidated the possible molecular mechanisms responsible for its anticancer activity.
Result
5-AcTMF inhibited the proliferation of NSCLC cells
To investigate the inhibitory effect of 5-AcTMF on proliferation of human NSCLC cancer cells, 4 different human NSCLC cancer cells, CL1-5, H1299, H226, and A549 were incubated in the absence or presence of various concentrations of 5-AcTMF (0.625-10 μM) for 24 or 48 h, and cell viability was determined by the MTT assay. As shown in Fig. 1A, 5-AcTMF significantly inhibited the growth of all 4 NSCLC cell lines in a dose-and time-dependent manner. After 48 h of treatment, the IC50 values of 5-AcTMF against CL1-5, H1299, H226 and A549 cells were 3.2 ± 1.2 μM, 6.7 ± 0.9 μM, 10.2 ± 2.3 μM and 9.8 ± 1.6, respectively. 5-AcTMF appeared to be more potent in CL1-5 cells compared to H1299, H226 and A549 cells. 5-AcTMF also showed the morphological changes of cytoplasmic shrinkage and cellular flattening following 48 h of treatment (Fig. 1B).
Figure 1.

Cytotoxic effect of 5-AcTMF on 3 human NSCLC cancer cell lines. (A) CL1-5, H1299, H226 and A549 cells were treated with different concentrations for 24 or 48 h and the cytotoxicity were analyzed by MTT assay. The data represents the mean ± SEM of 3 experiments each conducted in triplicate.*p < 0.05, **p < 0.01, ***p < 0.001, compared with the DMSO-treated group (one-way ANOVA). (B) Morphological changes in CL1-5 cells after treatment with 5-AcTMF. Cells were treated with the indicated concentrations of 5-AcTMF. After a 48 h-incubation, morphological changes were observed under light microscopy. Magnification × 100.
5-AcTMF induced G2/M cell cycle arrest and apoptosis in CL1-5 NSCLC cells
Because 5-AcTMF inhibited NSCLC cancer cell growth, we next examine whether 5-AcTMF causes cell death by cell cycle arrest and/or apoptosis. CL1-5 cells were treated with 5-AcTMF at 2.5 μM and 5 μM for 24 or 48 h, and then the DNA content in cells was quantitated by flow cytometry of cells stained with propidium iodide (PI). Results showed that cells treated with 5-AcTMF led to accumulation of G2/M phase cells and was associated with a concomitant decrease of cells in the G1 phase (Fig. 2A). The percentage of Annexin V positive staining (Fig. 2B) and sub-G1 phase cells (Fig. 2A), which represents early and late apoptotic cells, respectively, also increased in a dose and time dependent manner in CL1-5 cells. Accordingly, cells were treated with 2.5 μM or 5 μM 5-AcTMF for 48 h and then the cleavage of caspase-3, -8, -9 and PARP were also assayed by western blotting analysis. As shown in Fig. 2C, 5-AcTMF increased the caspase-3, 9 and PARP cleavage in a dose dependent manner, whereas caspase-8 cleavage was not affected. Overall, these data illustrated that apoptosis and G2/M phase arrest, were involved in the response of CL1-5 NSCLC cells to 5-AcTMF treatment.
Figure 2.
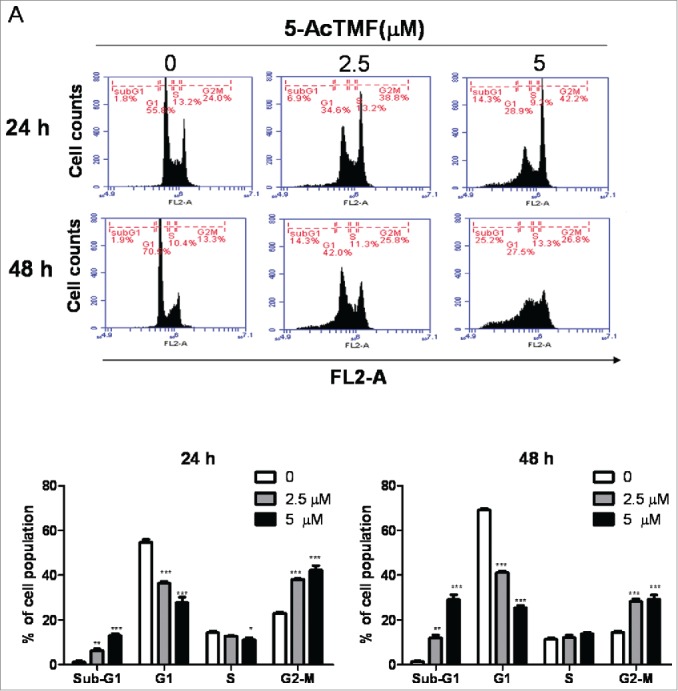
Effect of 5-AcTMF on cell cycle progression and apoptosis in CL1-5 NSCLC cells. (A) Cells were treated with 2. 5 or 5 μM 5-AcTMF for 24 or 48 h, and cell cycle distributions were then analyzed by flow cytometry. The means ± SEM of the experimental triplicates is presented in the bar graph. (B) Apoptosis in CL1-5 cells was examined after 24 or 48 h of treatment with 2.5 or 5 μM 5-AcTMF by annexin V-FITC/PI binding and analyzed by flow cytometry. The means ± SEM of the experimental triplicates are presented in the bar graph. (C) Cells were treated with or without 5-AcTMF (5 μM) for 48 h and then lyzed for protein extraction. The cleave-caspase3, 8, 9 and PARP were examined by western blot analysis. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of three independent experiments are presented in the bar graph. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the DMSO-treated group (one-way ANOVA).
Figure B.

(Continued).
5-AcTMF induces apoptosis primarily via the intrinsic mitochondrial pathway in CL1-5 NSCLC cells
Active caspase-9 and caspase-3 have been linked to the mitochondrial apoptosis pathway.26 Hence, to investigate whether mitochondrial pathway is involved in 5-AcTMF induced apoptosis of NSCLC cells, we analyzed the changes of the mitochondrial membrane potential (MMP, Δψm), release of cytochreome C and the level of Bcl-2 and IAP family proteins. Changes in MMP were assessed by JC-1 flow cytometry. As shown in Fig. 3A, 5-AcTMF caused a time dependent attenuation of red fluorescence and a concomitant increase in the green fluorescence in CL1-5 cells, indicating that 5-AcTMF triggered a reduction in MMP. Next, we measured the release of cytochrome c into the cytosol by using flow cytometry in fixed cells after 48 h of 5-AcTMF treatment. As shown in Fig. 3B, cytosolic cytochrome c was increased in 2.5 μM and 5 μM of 5-AcTMF treated compared with the vehicle group. Moreover, protein gel blot analysis showed that 48 h of 5-AcTMF treatment resulted in significant expression decrease of the anti-apoptotic proteins, such as BCL-2, XIAP and Survivin proteins. (Fig. 3C) in a dose dependent manner. The expression of pro-apoptotic Bax protein was not significantly affected, however the BCL-2/Bax ratio was decreased significantly. These data suggest that 5-AcTMF induced apoptosis of CL1-5 cells via a mitochondrial apoptotic pathway.
Figure 3.

Effect of 5-AcTMF on the mitochondrial pathway in CL1-5 NSCLC cells. Cells were treated with 5 μM 5-AcTMF for 24 or 48 h. (A) Disruption of mitochondrial membrane potential, B) Cytochrome C release were then analyzed by flow cytometry. The means ± SEM of the experimental triplicates is presented in the bar graph. (C) Cells were treated with or without 5-AcTMF (5 μM) for 48 hr and then lyzed for protein extraction. The BCL-2, Bax, XIAP and survivn were examined by protein gel blot analysis. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of 3 independent experiments are presented in the bar graph. *p<0.05, **p<0.01, ***p<0.001, compared with the DMSO-treated group (one-way ANOVA).
Cell cycle proteins are involved in 5-AcTMF induced G2/M phase arrest in CL1-5 NSCLC cells
To examine the mechanism responsible for induced cell cycle distribution by 5-AcTMF treatment, regulatory proteins involved in G2/M phase were tested by using western blotting. As shown in Fig. 4, 5-AcTMF treatment of CL1-5 cells resulted in a time dependent decrease of cdc25 expression, but increase of cdc2 phosphorylation (Tyr15) and cyclin B1, whereas the absolute protein level of cdc2, and negative regulators p21 was unaffected (Fig. 4).
Figure 4.
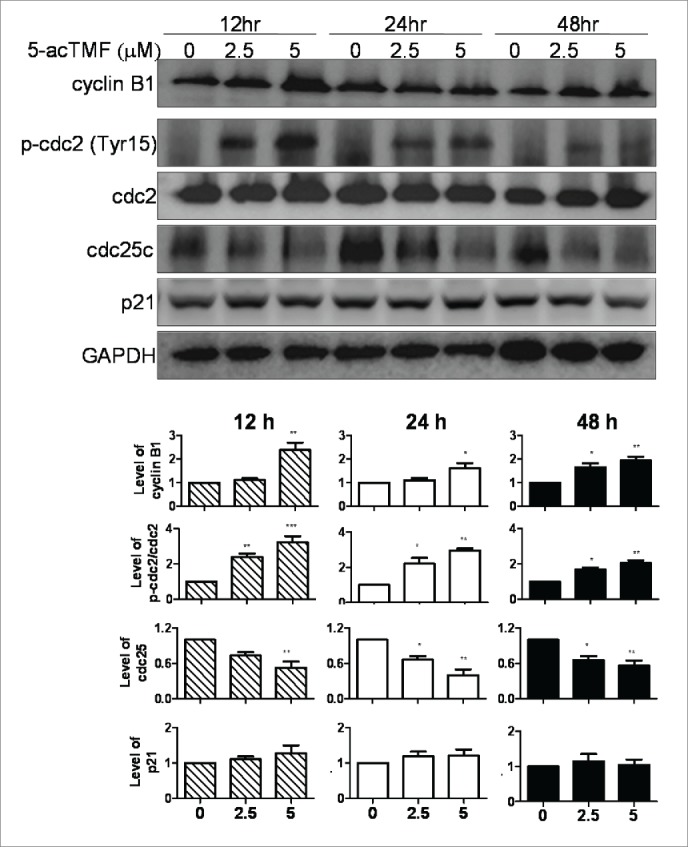
Effect of 5-AcTMF on the cell cycle checkpoint protein expression in the total cell lysate of CL1-5 cells. Cells were treated with or without 5-AcTMF (5 μM) for 12, 24, 48 h and then lyzed for protein extraction. The cyclin B1, phosphor-cdc2, total cdc2, cdc25 and p21 were examined by western blot analysis. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of 3 independent experiments are presented in the bar graph. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the DMSO-treated group (one-way ANOVA).
5-AcTMF induces autophagy in CL1-5 NSCLC cells
Growing evidence indicates that autophagy and apoptosis are the major pathway of programmed cell death and they can cooperate, antagonize or assist with each other.27 Having observed the 5-AcTMF induced apoptosis in CL1-5 cells (Fig. 2 and Fig. 3), we further assessed whether 5-AcTMF induces autophagy in CL1-5 cells of NSCLC. Firstly, autophagy was evaluated by acridine orange staining (AO) in FACS analysis. In acidic compartments (autophagolysosomes), the protonated form of AO accumulates and forms aggregates, which are characterized by yellow-orange fluorescence (FL3). Staining of normal cells with AO, a weak base, characterizes green fluorescence (FL1). As shown in Fig. 5A, 5-AcTMF treatment resulted in the appearance of yellow-orange (FL-3) fluorescence after 48 h. A more specific measurement of autophagy can be accomplished by changing the microtubule-associated protein light chain 3 (LC3), namely, the conversion of LC3-I (18 kD) to LC3-II (16 kD) by protein gel blot analysis because the amount of LC3-II correlated well with the number of autophagosomes. As shown in Fig. 5B, after treatment with 2.5 μM and 5 μM of 5-AcTMF for 48 h, the ratio of LC-II/LC3-I as well as LC3-II level were increased in CL1-5 in time dependent manner. Moreover, the formation of punctate spots with GFP-LC3 fusion protein is a well-characterized marker for visualizing autophagosomes. In this study, we also observed the visible cytoplasmic puncta of fluorescence pattern of LC3-GFP when CL1-5 cells were transfected with the LC3-GFP plasmid then treated with 5-AcTMF for 48 hr (Fig. 5C). Taken together, these results suggested that 5-AcTMF induce autophagy in CL1-5 lung cancer cells.
Figure 5.

5-AcTMF induces autophagy in CL1-5 lung cancer cells. Cells were treated with 2.5 μM and 5 μM 5-5-AcTMF for indicated times. (A) After which the cells were stained with acridine orange staining and then analyzed by flow cytometry. . The means ± SEM of the experimental triplicates are presented in the bar graph at the bottom. (B) The protein expression of LC3. Cells were treated with 2.5 or 5 μM 5-AcTMF for 48 h and then lyzed for protein extraction. The LC3-II was examined by protein gel blot analysis. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of 3 independent experiments are presented in the bar graph. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the DMSO-treated group (one-way ANOVA). C) Cells were transfected with expression plasmid for GFP-LC3. Cells were treated with 2.5 and 5 μM 5-AcTMF for 48 h. Punctate pattern of green fluorescence was observed by transmission electron microscopy.
Inhibition of autophagy suppresses 5-AcTMF mediated CL1-5 cells apoptotic cell death
Autophagy inhibition by a specific autophagy inhibitor, such as 3-methyladenine (3-MA), can promote apoptosis in human cancer cells.28-29 Thus, we have investigated the effect of 3-MA in 5-AcTMF induced cells and found the formation of AVO-positive autophagosomes being noticeably suppressed by 3-MA treatment (Fig. 6A). In addition, the co-treatment of 5-AcTMF with 3-MA significantly inhibited not only LC3-II accumulation but also PARP cleavages (Fig. 6C). Then we checked the effect of 3-MA on the apoptosis induction using Annexin V/PtdIns double staining. Annexin V/PI-positive cells were markedly decreased in cells co-treated with 5-AcTMF and 3-MA, compared with those treated with 5-AcTMF alone (Fig. 6B). In a cell viability assay, 3-MA was found to reduce the 5-AcTMF cytotoxicty (Fig. 6D). These results obviously revealed that autophagy inhibition by 3-MA could suppress 5-AcTMF induced apoptosis in CL1-5 cells.
Figure 6.

Inhibition of autophagy by 3-MA suppresses CL1-5 inhibited apoptotic cell death.(A) CL1-5 cells were pre-incubated with or without 3-MA (5 mM) for 3 h and then incubated with 5-AcTMF (5 µM) for 48 h, after which the cells were stained with acridine orange staining or B) FITC-conjugated Annexin V/PtdIns and then analyzed by flow cytometry. The means ± SEM of the experimental triplicates are presented in the bar graph. C) Western blot analysis was performed with antibodies against LC3, and cleaved PARP, respectively. GAPDH was used as loading control. The means ± SEM of 3 independent experiments are presented in the bar graph. (D) Cells were treated with 3-MA and 5-AcTMF FOR 48 h under the same conditions as mentioned before and then cell viability was evaluated by MTT assay. The data represents the mean ± SEM of 3 experiments each conducted in triplicate. nsP > 0.05, *p < 0.05, **p < 0.01, (Student's t-test) was comparison between 5-AcTMF and 3-MA+5-AcTMF groups.
The role of PI3K/AKT/mTOR in 5-AcTMF –mediated apoptosis and autophagy in CL1-5 NSCLC cells
Over the last decade, extensive work has been done to demonstrate that PI3K/AKT/mTOR and MAPK signaling pathways played important roles in regulating cell proliferation, cell growth arrest, apoptosis and autophagy.30,31 To clarify whether 5-AcTMF exerted its antitumor effects by changes in PI3K/AKT/MOTR and MPAK pathway, the active phosphorylation status of AKT, mTOR, MAPK, ERK, JNK and p38 was performed by western blotting. As shown in Fig. 7, treatment of CL1-5 with 5 μM of 5-AcTMF remarkably inhibited the phosphorylation of PI3K (Tyr458), AKT (Ser473) and mTOR (Ser2481) compared to control cells, in which 5-AcTMF did not cause significant changes in the ERK, p38 and JNK MAP kinase phosphorylation (supplemental Fig. 1).
Figure 7.
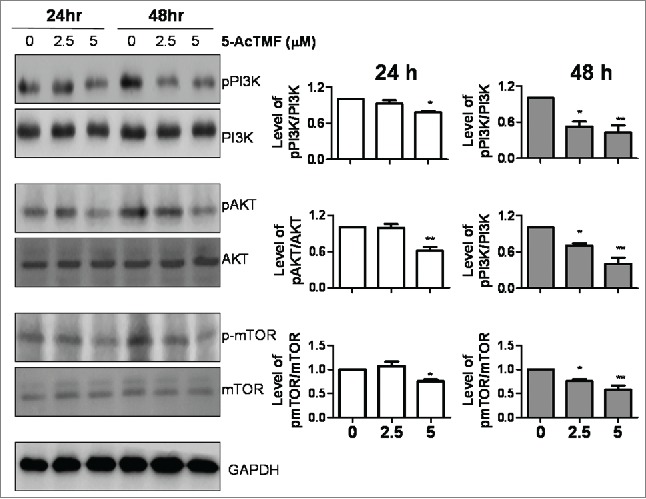
Effect of 5-AcTMF on PI3K/AKT/mTOR pathway in CL1-5 cells. Cells were treated with 2.5 μM and 5 μM 5-AcTMF for 24 or 48 h, and then lyzed for protein extraction and subjected to western blot analysis using antibodies against indicated proteins. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of 3 independent experiments are presented in the bar graph. *p < 0.05, **p < 0.01, compared with the DMSO-treated group (one-way ANOVA).
To confirm the involvement of PI3K/AKT/mTOR pathway in 5-AcTMF induced growth inhibition, we transfected CL1-5 with a constitutively active form of active AKT cDNA. AKT over-expressed cells were treated with 5-AcTMF and the induction of apoptosis, cell cycle distribution and autophagy were assayed. Cells transfected with active AKT cDNA were considerably more resistant to 5-AcTMF induced autophagy and apoptosis than that with the cDNA in the control group. As shown in Fig. 8, active AKT cDNA transfectants suppressed the 5-AcTMF induced apoptosis and autophagy, evidenced by decreased number of acidic vesicular organelles (Fig. 8A) and Annexin V positive cells (Fig. 8B), and by reduced expression of LC3-II and cleaved PARP (Fig. 8C). In a cell viability assay, AKT cDNA transfectants reduced the 5-AcTMF cytotoxicity (Fig. 8D). However, AKT cDNA transfectants did not affect the 5-AcTMF induced G2/M arrest (supplemental Fig. 1). Therefore, these results suggest that the PI3K/AKT/mTOR pathway be required in the regulation of apoptosis and autophagy in responses to 5-AcTMF in CL1-5 cells.
Figure 8.

Role of AKT on 5-AcTMF -induced autophagy and apoptosis in CL1-5 cells. A) Active AKT cDNA or pBabe cDNA (group) transfefction cells were treated with 5 μM of 5-AcTMF for 48 h, after which the cells were stained with acridine orange staining and then analyzed by flow cytometry. The means ± SEM of the experimental triplicates are presented in the bar graph at the bottom. (B) Active AKT cDNA or pBabe transfefction cells were treated with 5 μM 5-AcTMF for 48 h annexin V-FITC/PI binding were analyzed by flow cytometry. The means ± SEM of the experimental triplicates is presented in the bar graph (C) Cells were treated with 5 μM 5-AcTMF for 48 h and then lyzed for protein extraction and subjected to protein gel blot analysis using antibodies against indicated proteins. GAPDH was used as a loading control. Densitometric analysis was performed using Image J software. The means ± SEM of 3 independent experiments are presented in the bar graph. (D) Active AKT cDNA or pBabe transfefction cells were treated with 5-AcTMF FOR 48 h under the same conditions as mentioned before and then cell viability was evaluated by MTT assay. The data represents the mean ± SEM of 3 experiments each conducted in triplicate. nsP > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t-test) was comparison between pBabe+5-AcTMF and Active AKT+5-AcTMF groups.
Figure 8.
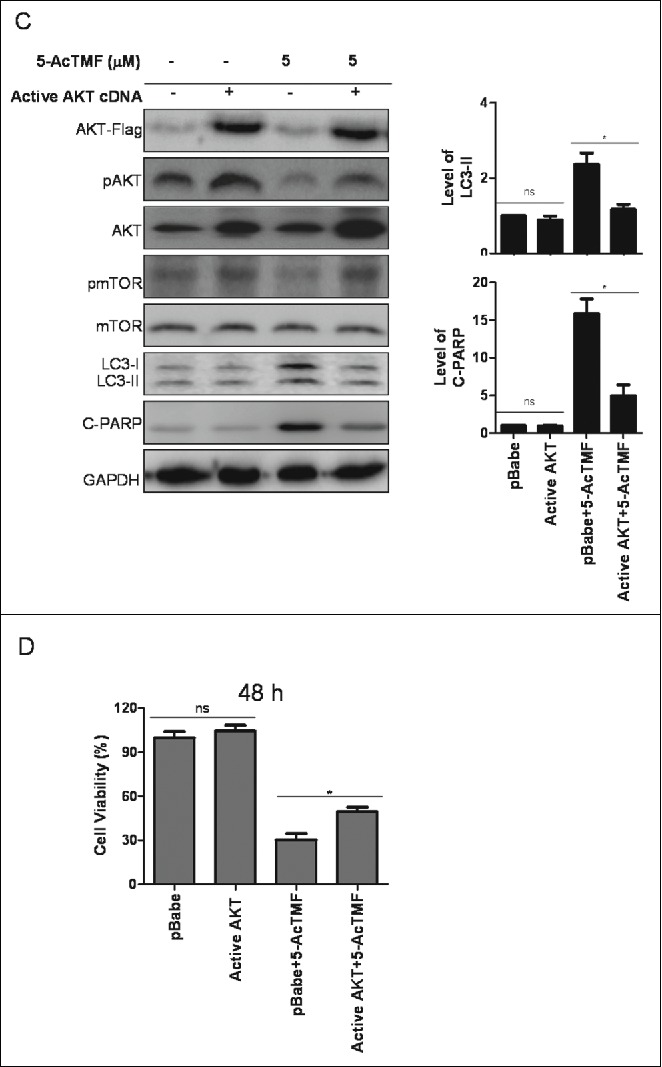
(Continued).
5-AcTMF inhibits tumor growth of CL1-5 in nude mice
To validate our in vitro results, we performed experiments to analyze whether 5-AcTMF inhibits the growth of CL1-5 cancer xenograft in mice. CL1-5 cells (1 × 107) were injected s.c into the backs of BALB/c athymic nude mice. Tumor growth inhibition was most evident in mice adopted 20 mg/kg of 5-AcTMF or vehicle control via i.p injection. As shown in Fig. 9A and B, 5-AcTMF significantly inhibited tumor growth as measured on day 28 (43% inhibition, p< 0.05 vs. control). In addition, there was no observed sign of toxicity, as judged by parallel monitoring of body weight (Fig. 9C) and tissue dissections of lungs, livers and kidneys (Fig. 9D), in 20 mg/kg of 5-AcTMF treated mice.
Figure 9.
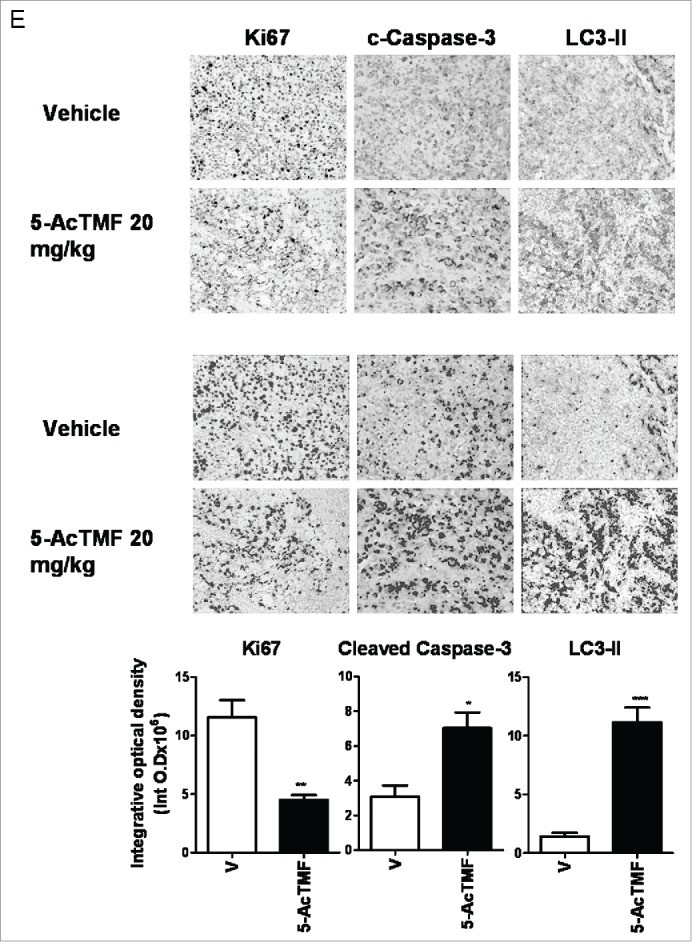
Effect of 5-AcTMF on tumor growth of CL1-5 in nude mice. (A) Mean of tumor volume measured at the indicated number of days after implant. *p<0 .05, (Student's t-test) was comparison between vehicle and 5-AcTMF groups. (B) Representative tumors from the vehicle control and 5-AcTMF (20 mg/kg) -treated groups (day 28). C) Mean of body weight measured at the indicated number of days after implant. The data shown here are one of 2 different experiments and similar results were obtained. Tumor volume and body weight graph points represent mean values ± SEM (n = 6) for each experiment. D) Tissue sections of livers, lungs, and kidneys of plumbagin-treated nude mice stained with H&E. (E) Quantification of Ki67, cleaved cleaved caspase-3 and LC3-II. After 28 d treatment, the tumors were removed and stained as described in material and methods section. Immunohistochemically positive threshold selection was performed at by Image J software (red selection in Fig. 9E), and 3 samples and 5 randomly chosen fields/sample were evaluated. Bars represent mean values of 3 samples and 5 randomly chosen fields/samples were evaluated. Bar graphs are expressed as means± SEM. nsP > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t-test) was comparison between vehicle and 5-AcTMF groups.
Figure 9.

(Continued).
To characterize the molecular mechanisms underlying the observed effects in the xenograft model, we studied whether the marker for cell proliferation (Ki67), apoptosis (cleaved caspase-3), or autophagy (LC3-II) was regulated in tumor by using immunohistochemistry in each treatment group. As shown in Fig. 9E, expression of c-caspase-3 and LC3-II was increased in the tumor treated by 5-AcTMF in mice xenograft model. In contrast, the Ki67 expression in the 5-AcTMF treated group is less than those in the vehicle control group. These data suggested that both apoptosis and autophagy effects might be correlated with 5-AcTMF antitumor effects in vivo.
Discussion
Recent studies have indicated that 5-AcTMF suppressed tumor growth of human multiple myeloma (U266) cells 23 and human MCF-7 breast cancer,24 but did not elucidate its anti-cancer mechanism. In the present study, we have evaluated the antitumor effects of 5-AcTMF in CL1-5 NSCLC lung cancer cell line and found that 5-AcTMF suppressed cancer cell growth through the mechanisms of G2/M cell cycle arrest, autophagy and apoptosis.
Apoptosis plays an important role of cell death in various biological processes including normal cell proper development and removal of unwanted harmful cells,32 thus is considered a regulated form of cell death. Desregulated apoptosis results in several pathological conditions including autoimmune disorders, neurodegenerative diseases and cancer.33,34 Thus, the development of most anti-cancer drugs aims to induce tumor cell apoptosis. Apoptosis is recognized for its ability to modulate cell death and has become one of the general standards to define or screen the potency of novel anti-cancer agents. Regarding caspase-mediated apoptosis, 2 main signaling pathways have been identified, death receptor - the extrinsic pathway and the mitochondrial intrinsic pathway.32,35 The death receptor pathway mediated by active/cleaved caspase-8 plays a key role in maintaining tissue homeostasis, especially the immune system. The mitochondrial pathway mediated by active/cleaved caspase-9 is responsible for response to extracellular cues and internal insults such as DNA damage and increased cytosolic Ca2+ or ROS generation.36 Polymethoxyflavone mediated apoptosis on broad spectrum of cancer cells through ROS generation, increased cytosolic Ca2+, activation of intrinsic or extrinsic apoptosis pathway have been documented in recent report. For instance, casticin, one of the polymethoxyflavones, can induce human cervical cancer HeLa cell apoptosis through ROS-mediated mitochondrial pathway.37 Furthermore, 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone (5-OH-HxMF) and 3′-hydroxy-5,6,7,4′-tetramethoxyflavone (3′-OH-TtMF), the polymethoxyflavones derived from Citrus sinensis L., induced Ca2+-mediated apoptosis by activation of μ-calpain and caspase12.38 Moreover, the lead compound of 5-AcTMF, tangeretin, can induce cell death of AGS human gastric cancer cell death through triggering extrinsic apoptotic pathway via activating FasL-mediated death receptor pathway and inducing intrinsic apoptotic pathway through upregluating Bax that contribute to caspase cascade activation.19 Previous studies have demonstrated that 5-AcTMF could induce apoptosis through mitochondrial membrane depolarization in a human MCF-7 breast cancer cell lines and U266 human multiple myeloma, with up-regulation of Bax and down-regulation of Bcl-2 proteins, and the activation of caspase-3.30,31 In addition, our study findings demonstrated that 5-AcTMF induced apoptosis of CL1-5 which through up-regulating cleaved caspase-3, caspase-9 and PARP, down regulating Bcl-2, srvivin, and XIAP, suggesting that the activity of 5-AcTMF might be responsible for cell death through the intrinsic mitochondrial apoptotic pathway in NSCLC cells. However, the upstream pathway of intrinsic mitochondrial apoptotic pathway, such as increased cytosolic Ca2+ or ROS generation, is still unclear in our present data. This study undertakes a further investigation in the future.
It was well known that the regulation of cell growth and proliferation of mammalian cells are mediated through cell cycle progression. Recently, studies have shown an association between cell cycle regulation and cancer and inhibition of the cell cycle has become an appreciated target for management of cancer.39 Previous study showed that tangeretin induced cell-cycle G1 arrest through inhibiting the activity of cyclin-dependent kinases 2 and 4, and through elevating Cdk inhibitors p21 and p27 in human colorectal carcinoma cells.18 One of the previous findings is that G2/M arrest in colon cancer is triggered by 5-demethyltangeretin through induction of p53 and p21 activation and the reduction of Cdc2 and cyclin B1 expression.40 To our knowledge, there is no study to address the role of 5-AcTMF in the regulation of cell cycle. Hence, we were interested in finding the impact of the anti-cancer potency of 5-AcTMF on cell cycle regulating effect. In current studies, the effect of 5-AcTMF on cell cycle progression was examined by flow cytometry. Our findings showed that 5-AcTMF arrested the growth of CL1-5 cells at the G2/M phase. Besides, 5-AcTMF leads to downregulation of cdc25c and upregulation of cyclin B1, resulted in a G2/M cell cycle arrest in CL1-5 cells and eventually lead to apoptotic cell death. These results indicated that the regional modification of tangeretin at its 5-position can potentially cause TAN to have different effects in cell cycle regulation of NSCLCs.
P53 is a well-known tumor suppressor protein which acts through a number of regulatory pathways to inhibit tumor growth, such as repair damaged DNA, cell cycle checkpoints, autophagy and apoptosis.41,42 However, the p53 gene is often in the stage of mutation or deletion or otherwise functionally inactivation of human tumors.43 Thus, the development of anti-cancer agents that can kill p53-mutated or null cells is an important context. It has been reported that the poor efficacy of many chemotherapeutic agents is thought to be partially attributed to the lack of functioning p53 for optimal activity in inducing cancer cell death.44 Besides increasing bioavailability and efficacy via targeted modification of tangeretin to 5-AcTMF, more significant discovery of this study is that 5-AcTMF not only suppressed the growth of p53 wild type cell A549, but also the growth of p53 mutant CL-5, H1299 and H226 cell strands. The results further suggest that 5-AcTMF might suppress cancer cell growth via both p53 dependent and independent pathways.
Many recent studies have demonstrated that inhibition of autophagy by pharmacologic inhibitors, such as 3-Methyladenine (3-MA), wortmannin chloroquine (CQ), or knockdown of autophagy-related genes, such as Beclin-1, ATG5, and ATG7, promoted apoptotic cell death induced by chemotherapeutic agents, showing that autophagy may act as a protective mechanism in cancer cells elicited by anti-cancer drugs.45-47 In addition, our previous study has reported that flavonoid 5′-demethylnobiletin mediated apoptosis in CL1-5 cells was promoted by pretreatment with 3-MA.48 However contrary to these reports, in the present study, we observed that autophagy inhibition by 3-MA suppressed 5-AcTMF induced apoptosis in CL1-5 cells, as indicated by decreased levels of Annexin V and PtdIns staining and by PARP cleavage, which was consistent with the decrease in cell viability, suggesting that autophagy also can serve as a cytotoxic mechanism against apoptotic cell death induced by cytotoxic chemotherapy.
PI3K/AKT mTOR signaling has been identified as a key role in the pathogenesis of various forms of human cancer including lung cancer and plays a variety of physiologic roles in cell proliferation, growth, and survival.49,50 Activated AKT, in turn, phosphorylates a variety of downstream target including forkhead transcription factor (FKHR), glycogen synthase kinase (GSK)–3 and mammalian target of rapamycin (mTOR), and consequently to repress cell death and promote cell survival.51 In addition, inhibition of the AKT/mTOR pathway has been shown to trigger autophagy, and apoptosis in some types of cancer, such as malignant gliomas, cervical carcinoma, gastric cancer, breast cancer and melanoma.52,53 Our results have shown that 5-AcTMF treatment decreased the level of phosphor-PI3K, and also the AKT activation. In addition, 5-AcTMF also reduced the phosphorylated forms of mTOR. Moreover, transfection with active AKT cDNA significantly decreased 5-AcTMF mediated autophagy and apoptosis, suggesting that the anticancer effect of 5-AcTMF is mediated, at least, in part through PI3K/AKT/mTOR inhibition in human NSCLC cancer cells.
In summary, our study has elucidated a novel anti-cancer mechanism of 5-AcTMF in CL1-5 lung cancer cells to induce G2/M arrest, apoptosis and autophagy in vitro. In addition, our study also found that 5-AcTMF can inhibit CL1-5 tumor growth in nude mice by i.p. injection. Biopsy analysis has verified the induction mechanism of autophagy and apoptosis in the tumors studied. Attention must also be paid to that a more thorough understanding of the anti-cancer molecular mechanism of 5-AcTMF and its effect on different cancer models will require further study. Moreover, the modification of tangeretin has significantly increased the bioavailability and efficacy comparing to tangeretin as shown here and other studies. In conclusion, the current study attested a new mechanistic pathway in evaluating the anti-cancer property of 5-AcTMF and provided convincible data to merit a further research and application of 5-AcTMF and its related compounds in NSCLC drug discovery.
Material and methods
Cell culture and reagents
The CL1-5, H1299 and H226 lung adenocarcinoma cell lines were provided by Dr. Jeremy J. W. Chen (University of National Chung Hsing University) and were maintained in Ham's DMEM medium (Sigma-Aldrich). The A549 was obtained from American type culture collection and was cultured in F12K medium (Sigma-Aldrich). Both Ham's DMEM medium and F12K medium contain 10% heat-inactivated fetal bovine serum (Gibco Life Technologies), 100 U/ml penicillinan and 50 μg/ml streptomycin (Gibco Life Technologies). Cells were maintained at 37°C in a 5%CO2/95% air humidified incubator. 5-AcTMF was prepared as previously described (24) and dissolved at a concentration of 10 mM in dimethyl sulphoxide (DMSO) as a stock solution. Working solution was freshly prepared by dilution with medium to desired concentrations. 3-Methyladenine (3-MA) was purchased from Sigma-Aldrich.
Cell viability assay
Cell viability was performed using a colorimetric assay for 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT, Sigma-Aldrich) assay. The cultures were seeded in 24 well- plates for overnight and then were treated with various concentrations of 5-AcTMF for additional 24 or 48h. After treatment, 200 μl MTT reagent (5mg/ml) was added and the cells were further incubated for 4 h at 37°C. Subsequently, the supernatant was aspirated and 600 μl DMSO was added to dissolve farmazan crystals. The absorbance was measured at 540 nm using the microplate reader (Tecan Sunrise). IC50 was calculated by polynominal regression analysis using Microsoft Excel software and the mean optical density (OD) ± SEM for each group of the replicates calculated. Percent of cell viability to treatments was calculated as follows: % Inhibition = 100 - (Test OD/DMSO -treated OD) × 100).
pBabe.puro derived retroviral particle production and infection
pBabe.puro-Myc-Flag-PKBA/AKT, a constitutively active mutant of PKB1/AKT cloned in the retroviral expression vector pBabe.puro was purchased from Addgene (Addgene plasmid 15294). HEK-293T cells were transiently transfected for 24 h by the use of jetPEM™ transfection reagent (polyplus) as indicated by the manufacturer, with 2.5 μg of pBabe-puro-based plasmids along with the plasmids for producing viral particles, includes gag-pol and VSV-G proteins. Viral particles were harvested from the media containing transfected HEK293T cells and immediately clarified by centrifugation. For performing viral infection, CL1-5 cells (5 × 105) were incubated for 48 h with viral particles enriched media supplemented with 8 μg/ml of polybrence (Sigma-Aldrich) to promote infection efficiency and cells were harvested after transfection and reseeded onto a culture plate for further experiments.
Annexin V-FITC binding assay
An apoptosis assay was performed according to the manufacturer's instruction using the Annexin V-FITC Apoptosis Detection Kit (BioVision). The cells were treated with DMSO or 5-AcTMF for 24 or 48 h. Cells were collected by trypsinization, washed twice with PBS and then re-suspended in 500 μl of 1x binding buffer. Cell suspensions were then incubated with 5μl of annexin V-FITC and 5 μl of propidium iodide (PI) for 15 min at room temperature in the dark. The cells were immediately analyzed by Accuri 5 flow cytometer (Accuri Cytometers, Inc.) and analyzed for cell cycle phases with C6 Accuri system software ((Accuri Cytometers, Inc.).
Cell cycle analysis
Cells were incubated with 0.1 % DMSO or 2.5 or 5 μM of 5-AcTMF for the indicated times. After treatment, cells were trypsinized and fixed with cold 80% ethanol, and then stored at −20°C overnight. Then, cells were washed with phosphate-buffered saline (PBS) and incubated with 50 μg/ml PtdIns and 25 μg/ml RNase A (BD bioscience) for 30 min at room temperature in the dark. These stained cells were subjected to cell cycle analysis using a Accuri 5 flow cytometer and analyzed for cell cycle phases with C6 Accuri system software.
Western blot analysis
The expression levels of cellular proteins were extracted and determined using protein gel blotting analysis. Briefly, equal amounts of lysates were electrophoresed on 8–12% sodium dodecyl sulfate polyacrylamide gel electro-phoresis (SDS-PAGE) and transferred onto PVDF membranes (Millipore) and blocked with 5% skim milk in TBST buffer for 1 h. Membranes were then probed with indicated primary antibodies overnight at 4°C. After TBST washing, the blots were probed with respective horseradish peroxidase-labeled secondary antibodies (Jackson ImmunoResearch Laboratories Inc.) overnight at 4°C. Visualization was performed using the LAS3000 AutoChemi system (Fujifilm) with enhanced chemiluminescence substrate (Perkin Elmer). Densitometric analysis was performed with ImageJ software (National Institute of Health). In this study, the primary antibodies were obtained from Cell Signaling Technology Inc. (cleaved caspase-3,8,9, cleaved PARP, XIAP, survivin, Bcl-2, p-ERK, ERK, p-p38, JNK, p-PI3K, p-Akt, p53, p21), from Santa Cruz Biotechnology Inc. (Santa Cruz, CA)(cyclin B1, p-cdc2, cdc25c, β-actin, Bcl-xL, PI3KIII, p-mTOR, mTOR, Bax, p-Akt), from Epitomics (cdc2, survivin, p38, Akt) ,from Millipore (anti-p-JNK) and from abgent (LC3).
Measurement of mitochondrial membrane potential (MMP)
The cells were treated with 0.1 % DMSO or various final concentrations of 5-AcTMF for the indicated times. Ten minutes prior to flow cytometry analysis, JC-1 reagent (Invitrogen) was added to cell culture at a final concentration 10 μg/mL at 37°C. Cells were washed twice in PBS and cells with FL-1 (530 nm) versus FL-2 (580 nm) dot plot were quantified by Accuri 5 flow cytometer and analyzed with C6 Accuri system software.
Determination of cytochrome c release from mitochondria by flow cytometry
To examined cytochrome c release from mitochondria, we used direct staining with FITC–anticytochrome c antibody (Santa Cruz Biotechnology) according to the method described previously (25). Briefly, cells were harvested, and treated with 50 μl digitonin (1mg/ml in PBS with 100 mM KCl) for 5 min on ice and then fixed for 20 minutes at room temperature with 4 % paraformaldehyde in phosphate-buffered saline (PBS). After incubation, cells were washed 3 times with PBS and permeated with permeation buffer (0.05% Triton X-100 in PBS and 2% bovine serum albumin [BSA]) for 1 h containing 10 μL FITC–anticytochrome c antibody. Staining was carried out at 4°C for 30 minutes. Unbound antibody was washed with PBS and cells analyzed by Accuri 5 flow cytometer and analyzed for cell cycle phases with C6 Accuri system software.
Flow cytometric quantification of acidic vesicular organelles (AVOs)
Cell were stained with acridine orange (1 μg/ml) (Sigma-Aldrich) at 37°C for 20 min. After removing solution, PBS was added to the dish and analyzed on a Accuri 5 flow cytometer and analyzed for cell cycle phases with C6 Accuri system software.
Analysis of cell with GFP-LC3
Cells were seeded into the Lab-Tek® 8-well chamber slide (Thermo Fisher Scientific, Inc.) at 30 % confluence 100 μL Lyovec (InvivoGen) was mixed with 2 μg p-GFP-LC3 plasmid at room temperature for 20 min to form the transfection complex. The complex was added to cells for 24 h. Transfected cells were incubated with 0.1 % DMSO or 5-AcTMF (2.5 or 5 μM) for an additional 24 h. After incubation, the chamber slides were washed with PBS to remove detection reagent, and fixed with 4% paraformaldehyde for 30 min at room temperature. Nuclei were stained with DAPI solution (1 ug/ml) for 30 min at 37°C. After removing the staining solution, punta formation of LC3 was observed under the confocal microscope. (Leica SP5, Leica Microsystems Vertrieb GmbH).
In vivo antitumor activity
All the animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University. CL1-5 cells were mixed with Matrigel (BD Biosciences) at a 1:1 ratio. Cells (1 × 107) were inoculated subcutaneously in the nude mice's back and tumors were allowed to grow for 10 d until they reached 2 mm3 and then treated with either vehicle (10% DMSO + 90% glyceryl trioctanoate) (Sigma-Aldrich) or 5-AcTMF 20 mg/kg (dissolved in DMSO and glyceryl trioctanoate (10:90 v/v)). Mice were treated daily with an i.p. injection using 100 μL total volumes. Body weight was measured and the physical condition of the mice was also carefully observed daily. Tumor volume was calculated by using the formula: length × width × thickness × 0.5 and expressed in mm3 before each treatment. Tumor-bearing mice were sacrificed after 28 d. Tumor growth inhibition was calculated by using the formula: 100% × (Tvolcontrol -TVoltreated)/Tvolcontrol, where Tvolcontrol is final tumor volume - initial tumor volume. Tumors, as well as other vital organs of the treated and control mice, were harvested, fixed in 4% formaldehyde for 24 h before being transferred to 70% ethanol. Tumor samples were subsequently paraffin-embedded, and 6 μM-thick sections were cut and baked onto microscope slides for histologic study.
Immunohistochemical analysis
At experimental day 28, the tumors were harvested, fixed in 4% formaldehyde for 24 h before being transferred to 70% ethanol. Tumor samples were subsequently paraffin-embedded, and 6 μM-thick sections were cut and baked onto microscope slides. For immunohistochemical analyses, sections were treated with boric-acid/EDTA buffer for antigen-retrieval, at 95–98°C for 20 min by microwave. Endogenous peroxidase activity was inhibited with 0.02% H2O2 for 10-15 min at room temperature, and sections were washed with PBS and incubated with primary rabbit antibodies against mouse Ki67 (clone 16A8, Biolegend), cleaved LC3-II (clone AP1805a, Abgent) and cleaved caspase-3 (clone Asp175, cell signaling) 4°C overnight, followed by HRP-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories Inc.) for 30 min at room temperature and then visualized using a colorimetric method (DAB kit; DAO Envision-HARP, Carpentaria, CA, USA). Nuclei were stained lightly with hematoxylin. The slides were viewed with a microscope (CKX41; Olympus). For quantification of of Ki-67, LC3-II, and active caspase-3 integrative optical density (Int O.D) in tumors, up to 4 random fields for each tumor section at × 100 magnification was calculated using Image J software. For each image, and particles created by the thresholds were analyzed for total area.
Statistical analysis
Data are expressed as the mean ±SEM. All Statistical analyses were performed using the GraphPad Prism Version 5.0 (San Diego, CA). The statistical significance of differences between groups was examined by one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison procedure or student's t test. Values of p less than 0.05 were considered statistically significantly different.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was funded by grants from Taichung Veterans General Hospital - National Chung-Hsing University, Taiwan (TCVGH-NCHU1027608), National Science Council, Taiwan (NSC-102-2320-B-005-007-MY3) and the Education Administration of Hubei Province, China (2014BHE036).
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.The World Health Organization histological typing of lung tumours. Ed. 2. Am J Clin Pathol 1982; 72:123-36; PMID:7064914 [DOI] [PubMed] [Google Scholar]
- 3.Le Péchoux C. Role of postoperative radiotherapy in resected non-small cell lung cancer: a reassessment based on new data. Oncologist 2011; 16:672-81; PMID:21378080; http://dx.doi.org/ 10.1016/S0140-6736(10)60059-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.NSCLC Meta-Analyses Collaborative Group, Arriagada R, Auperin A, Burdett S, Higgins JP, Johnson DH, Le Chevalier T, Le Pechoux C, Parmar MK, Pignon JP, et al.. Adjuvant chemotherapy, with or without postoperative radiotherapy, in operable non-small cell lung cancer: two meta-analyses of individual patient data. Lancet 2010; 375:1267-77; PMID:20338627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosseini A, Ghorbani A. Cancer therapy with phytochemicals: evidence from clinical studies. Avicenna J Phytomed 2015; 5:84-97; PMID:25949949 [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Khor TO, Shu L, Su Z, Fuentes F, Lee JH, Kong AN. Plants against cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Medicinal Chem 2012; 12:1281-1305; PMID:22583408; http://dx.doi.org/ 10.2174/187152012803833026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uesato S, Yamashita H, Maeda R, Hirata Y, Yamamoto M, Matsue S, Nagaoka Y, Shibano M, Taniguchi M, Baba K, et al.. Synergistic antitumor effect of a combination of paclitaxel and carboplatin with nobiletin from Citrus depressa on non-small cell lung cancer cell lines. Planta Med 2014; 80:452-7; PMID:24687742; http://dx.doi.org/ 10.1055/s-0034-1368321 [DOI] [PubMed] [Google Scholar]
- 8.Yunos NM, Beale P, Yu JQ, Huq F. Synergism from the combination of oxaliplatin with selected phytochemicals in human ovarian cancer cell lines. Anticancer Res 2011; 31:4283-9; PMID:22199293 [PubMed] [Google Scholar]
- 9.Lai CS, Li S, Miyauchi Y, Suzawa M, Ho CT, Pan MH. Potent anti-cancer effects of citrus peel flavonoids in human prostate xenograft tumors. Food Funct 2013; 4:944-9; PMID:23673480; http://dx.doi.org/ 10.1039/c3fo60037h [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Wang J, Fang L, Zheng Z, Zhi D, Wang S, Li S, Ho CT, Zhao H. Anticancer activities of citrus peel polymethoxyflavones related to angiogenesis and others. Biomed Res Int 2014; 2014:453972; PMID:25250322; http://dx.doi.org/ 10.1155/2014/453972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S, Lambros T, Wang Z, Goodnow R, Ho CT. Efficient and scalable method in isolation of polymethoxyflavones from orange peel extract by supercritical fluid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 846:291-7; PMID:17035106; http://dx.doi.org/ 10.1016/j.jchromb.2006.09.010 [DOI] [PubMed] [Google Scholar]
- 12.Chiu SP, Wu MJ, Chen PY, Ho YR, Tai MH, Ho CT, Yen JH. Neurotrophic action of 5-hydroxylated polymethoxyflavones: 5-demethylnobiletin and gardenin A stimulate neuritogenesis in PC12 cells. J Agric Food Chem 2013; 61:9453-63; PMID:24003765; http://dx.doi.org/ 10.1021/jf4024678 [DOI] [PubMed] [Google Scholar]
- 13.Li S, Pan MH, Lai CS, Lo CY, Dushenkov S, Ho CT. Isolation and syntheses of polymethoxyflavones and hydroxylatedpolymethoxyflavones as inhibitors of HL-60 cell lines. Bioorg Med Chem 2007; 15:3381-9; PMID:17391969 [DOI] [PubMed] [Google Scholar]
- 14.Ortuño A, Báidez A, Gómez P, Arcas MC, Porras I, GarcíaLidón A, Del Río JA. Citrus paradisi and Citrus sinensis flavonoids: their influence in the defence mechanism against Penicillium digitatum. Food Chemistry 2006; 98:351-58; http://dx.doi.org/ 10.1016/j.foodchem.2005.06.017 [DOI] [Google Scholar]
- 15.Benavente-García O, Castillo J. Update on uses and properties of citrus flavonoids: new findings in anticancer, cardiovascular, and anti-inflammatory activity. J Agric Food Chem 2008; 56:6185-6205;PMID:18593176; http://dx.doi.org/ 10.1021/jf8006568 [DOI] [PubMed] [Google Scholar]
- 16.Miyata Y, Tanaka H, Shimada A, Sato T, Ito A, Yamanouchi T, Kosano H. Regulation of adipocytokine secretion and adipocyte hypertrophy by polymethoxyflavonoids, nobiletin and tangeretin. Life Sci 2011; 88:613-8; PMID:21295043; http://dx.doi.org/ 10.1016/j.lfs.2011.01.024 [DOI] [PubMed] [Google Scholar]
- 17.Shu Z, Yang B, Zhao H, Xu B, Jiao W, Wang Q, Wang Z, Kuang H. Tangeretin exerts anti-neuroinflammatory effects via NF-κB modulation in lipopolysaccharide-stimulated microglial cells. Int Immunopharmacol 2014; 19:275-82; PMID:24462494; http://dx.doi.org/ 10.1016/j.intimp.2014.01.011 [DOI] [PubMed] [Google Scholar]
- 18.Morley KL, Ferguson PJ, Koropatnick J. Tangeretin and nobiletin induce G1 cell cycle arrest but not apoptosis in human breast and colon cancer cells. Cancer Lett 2007; 251:168-78; PMID:17197076; http://dx.doi.org/ 10.1016/j.canlet.2006.11.016 [DOI] [PubMed] [Google Scholar]
- 19.Dong Y, Cao A, Shi J, Yin P, Wang L, Ji G, Xie J, Wu D. Tangeretin, a citrus polymethoxyflavonoid, induces apoptosis of human gastric cancer AGS cells through extrinsic and intrinsic signaling pathways. Oncol Rep 2014; 31:1788-94; PMID:24573532; http://dx.doi.org/ 10.3892/or.2014.3034 [DOI] [PubMed] [Google Scholar]
- 20.Hirano T, Abe K, Gotoh M, Oka K. Citrus flavone tangeretin inhibits leukaemic HL-60 cell growth partially through induction of apoptosis with less cytotoxicity on normal lymphocytes. Br J Cancer 1995; 72:1380-8; PMID:8519648; http://dx.doi.org/ 10.1038/bjc.1995.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura S, Fujimoto K, Matsumoto T, Ohta T, Ogawa K, Tamur H, Matsuda H, Yoshikawa M. Structures of acylated sucroses and an acylated flavonol glycoside and inhibitory effects of constituents on aldose reductase from the flower buds of Prunus mume. J Nat Medicines 2013; 67:799-806; PMID:23456234; http://dx.doi.org/ 10.1007/s11418-013-0750-7 [DOI] [PubMed] [Google Scholar]
- 22.Rubio S, Quintana J, Eiroa JL, Triana J, Estevez F. Acetyl derivative of quercetin 3-methyl ether-induced cell death in human leukemia cells is amplified by the inhibition of ERK. Carcinogenesis 2007; 28:2105-13; PMID:17548901; http://dx.doi.org/ 10.1007/s11418-013-0750-7 [DOI] [PubMed] [Google Scholar]
- 23.Zhi D, Liu S, Li L, Wang L, Wang J, Ma J, Wang S, Zhao H, Ho CT, Wang Y, et al.. 5-Acetyl-6,7,8,4′-tetramethylnortangeretin induces apoptosis in multiplemyeloma U266 cells. Food Sci Hum Wellness 2014; 3:197-203; http://dx.doi.org/ 10.1016/j.fshw.2014.12.003 [DOI] [Google Scholar]
- 24.Wang J, Duan Y, Zhi D, Li G, Wang L, Zhang H, Gu L, Ruan H, Zhang K, Liu Q, et al.. Pro-apoptotic effects of the novel tangeretin derivate 5-acetyl-6,7,8,4′-tetramethylnortangeretin on MCF-7 breast cancer cells. Cell Biochem Biophys 2014; 70:1255-63; PMID:24938898; http://dx.doi.org/ 10.1007/s12013-014-0049-7 [DOI] [PubMed] [Google Scholar]
- 25.Christensen ME, Jansen ES, Sanchez W, Waterhouse NJ. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarisation and cytochrome c release. Methods 2013; 61:138-45; PMID:23545197; http://dx.doi.org/ 10.1016/j.ymeth.2013.03.020 [DOI] [PubMed] [Google Scholar]
- 26.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15:2922-33; PMID:11711427 [PubMed] [Google Scholar]
- 27.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/ 10.1038/nrm3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends cell Biol 2011; 21:387-92; PMID:21561772; http://dx.doi.org/ 10.1016/j.tcb.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aredia F, Giansanti V, Scovassi AI. Autophagy and cancer. Cell 2012; 1:520-34; http://dx.doi.org/ 10.3390/cells1030520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013; 10:143-53; PMID:23400000 [DOI] [PubMed] [Google Scholar]
- 31.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 2014; 344:174-9; PMID:24333738; http://dx.doi.org/ 10.1016/j.canlet.2013.11.019 [DOI] [PubMed] [Google Scholar]
- 32.Goldar S, Khaniani MS, Derakhshan SM, Baradaran B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J Cancer Prev 2015; 16:2129-44; PMID:25824729; http://dx.doi.org/ 10.7314/APJCP.2015.16.6.2129 [DOI] [PubMed] [Google Scholar]
- 33.Sankari SL, Masthan KM, Babu NA, Bhattacharjee T, Elumalai M. Apoptosis in cancer–an update. Asian Pac J Cancer Prev 2012; 13:4873-8; PMID:23244073; http://dx.doi.org/ 10.7314/APJCP.2012.13.10.4873 [DOI] [PubMed] [Google Scholar]
- 34.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 2011; 351:41-58; PMID:21210296; http://dx.doi.org/ 10.1007/s11010-010-0709-x [DOI] [PubMed] [Google Scholar]
- 35.Khan KH, Blanco-Codesido M, Molife LR. Cancer therapeutics: targeting the apoptotic pathway. Crit Rev Oncol Hematol 2014; 90:200-19; PMID:24507955; http://dx.doi.org/ 10.1016/j.critrevonc.2013.12.012 [DOI] [PubMed] [Google Scholar]
- 36.Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 2006; 13:1423-33; PMID:16676004; http://dx.doi.org/ 10.1038/sj.cdd.4401950 [DOI] [PubMed] [Google Scholar]
- 37.Chen D, Cao J, Tian L, Liu F, Sheng X. Induction of apoptosis by casticin in cervical cancer cells through reactive oxygen species-mediated mitochondrial signaling pathways. Oncol Rep 2011; 26:1287-94; PMID:21725610; http://dx.doi.org/ 10.3892/or.2011.1367 [DOI] [PubMed] [Google Scholar]
- 38.Sergeev IN, Li S, Colby J, Ho CT, Dushenkov S. Polymethoxylated flavones induce Ca(2+)-mediated apoptosis in breast cancer cells. Life Sci 2006; 80:245-53; PMID:17046027; http://dx.doi.org/ 10.1016/j.lfs.2006.09.006 [DOI] [PubMed] [Google Scholar]
- 39.Aarts M, Linardopoulos S, Turner NC. Tumour selective targeting of cell cycle kinases for cancer treatment. Curr Opin Pharmacol 2013; 13:529-35; PMID:23597425; http://dx.doi.org/ 10.1016/j.coph.2013.03.012 [DOI] [PubMed] [Google Scholar]
- 40.Charoensinphon N, Qiu P, Dong P, Zheng J, Ngauv P, Cao Y, Li S, Ho CT, Xiao H. 5-demethyltangeretin inhibits human nonsmall cell lung cancer cell growth by inducing G2/Mcell cycle arrest and apoptosis. Mol Nutr Food Res 2013; 57:2103-11; PMID:23926120; http://dx.doi.org/ 10.1002/mnfr.201300136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Q, Cho WC. Battle against cancer: an everlasting saga of p53. Int J Mol Sci 2014; 15:22109-27; PMID:25470027; http://dx.doi.org/ 10.3390/ijms151222109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Speidel D. The role of DNA damage responses in p53 biology. Arch Toxicol 2015; 89:501-17; PMID:25618545; http://dx.doi.org/ 10.1007/s00204-015-1459-z [DOI] [PubMed] [Google Scholar]
- 43.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 2000; 77:81-137; PMID:10549356; http://dx.doi.org/ 10.1016/S0065-230X(08)60785-X [DOI] [PubMed] [Google Scholar]
- 44.Corazzari M, Lovat PE, Oliverio S, Di Sano F, Donnorso RP, Redfern CP, Piacentini M. Fenretinide: a p53-independent way to kill cancer cells. Biochem Biophys Res Commun 2005; 331:810-5; PMID:15865936; http://dx.doi.org/ 10.1016/j.bbrc.2005.03.184 [DOI] [PubMed] [Google Scholar]
- 45.Lee JW, Kim KS, An HK, Kim CH, Moon HI, Lee YC. Dendropanoxide induces autophagy through ERK1/2 activation in MG-63 human osteosarcoma cells and autophagy inhibition enhances dendropanoxide-induced apoptosis. PLoS One 2013; 8:e83611; PMID:24358301; http://dx.doi.org/ 10.1371/journal.pone.0083611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem Pharmacol 2011; 82:427-34; PMID:21704023; http://dx.doi.org/ 10.1016/j.bcp.2011.06.015 [DOI] [PubMed] [Google Scholar]
- 47.Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends cell Biol 2011; 21:387-92; PMID:21561772; http://dx.doi.org/ 10.1016/j.tcb.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen YK, Wang HC, Ho CT, Chen HY, Li S, Chan HL, Chung TW, Tan KT, Li YR, Lin CC. 5-Demethylnobiletin promotes the formation of polymerized tubulin, leads to G2/M phase arrest and induces autophagy via JNK activation in human lung cancer cells. J Nutr Biochem 2015; 26:484-504; PMID:25765513; http://dx.doi.org/ 10.1016/j.jnutbio.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 49.Sun Z, Wang Z, Liu X, Wang D. New development of inhibitors targeting the PI3K/AKT/mTOR pathway in personalized treatment of non-small cell lung cancer. Anticancer Drugs 2015; 26:1-14; PMID:25304988; http://dx.doi.org/ 10.1097/CAD.0000000000000172 [DOI] [PubMed] [Google Scholar]
- 50.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005; 16:797-803; PMID:16096426; http://dx.doi.org/ 10.1097/01.cad.0000173476.67239.3b [DOI] [PubMed] [Google Scholar]
- 51.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4:988-1004; PMID:16341064; http://dx.doi.org/ 10.1038/nrd1902 [DOI] [PubMed] [Google Scholar]
- 52.Sun H, Wang Z, Yakisich JS. Natural products targeting autophagy via the PI3K/Akt/mTOR pathway as anticancer agents. Anticancer Agents Med Chem 2013; 13:1048-56; PMID:23293890; http://dx.doi.org/ 10.2174/18715206113139990130 [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Chen LX, Ouyang L, Cheng Y, Liu B. Plant natural compounds: targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif 2012; 45:466-76; PMID:22765290; http://dx.doi.org/ 10.1111/j.1365-2184.2012.00833.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.