

Abstract
Sphingolipid metabolism has been identified as a potential therapeutic target in cancer. Sphingosine-1-phosphate (S1P) is a potent bioactive sphingolipid metabolite produced by sphingosine kinases-1 and −2 (SPHK1 and SPHK2). Elevated SPHK1 has been found in numerous cancer types and been shown to contribute to survival, chemotherapeutic resistance and malignancy. However, its role in large granular Natural Killer (NK) large granular lymphocyte (LGL) leukemia has not been investigated. Here, we examine SPHK1 as a therapeutic target in LGL leukemia. We found that SPHK1 is overexpressed in peripheral blood mononuclear cells (PBMCs) from LGL leukemia patients which results in elevated S1P in the sera. The use of SPHK1 inhibitors, SKI-II or SKI-178, decreased leukemic NK cell viability and induced caspase-dependent apoptosis. SKI-II and SKI-178 restored the sphingolipid balance by increasing ceramide and decreasing S1P in leukemic NKL cells. SKI-II and SKI-178 also induced apoptosis in primary NK-LGLs from leukemia patients. Mechanistic studies in NK-LGL cell lines demonstrated that SKI-178 and SKI-II induced cell cycle arrest at G2/M. We found that SKI-178 induced phosphorylation of Bcl-2 at Ser70, and that this was dependent on CDK1. We further show that SPHK1 inhibition with SKI-178 leads to decreased JAK-STAT signaling. Our data demonstrate that SPHK1 represents a novel therapeutic target for the treatment of NK-LGL leukemia.
Keywords: apoptosis; large granular lymphocyte; LGL leukemia; natural killer; sphingosine kinase 1; SKI-178,SKI-II
Abbreviations
AC, acid ceramidase; CLPD-NK, chronic lymphoproliferative disorder of NK cells; JNK/SAPK, c-Jun NH2-terminal kinase/stress-activated protein kinase; LGL, large granular lymphocyte; NK, Natural Killer; PBMCs, peripheral blood mononuclear cells; RT-PCR, quantitative reverse transcription-polymerase chain reaction; S1P, Sphingosine-1-phosphate; SKI-II, Sphingosine kinase inhibit II; SPHK1, Sphingosine kinase-1; SPHK2, Sphingosine kinase-2; TNF-alpha, tumor necrosis factor alpha; WHO, World Health Organization.
Introduction
Large granular lymphocytes (LGLs) are cytotoxic immune cells derived from T (CD3+) or Natural Killer (NK, CD3−) lineages and normally represent 10–15% of PBMCs. LGL leukemia is a rare disorder characterized by clonal expansion of these cytotoxic lymphocytes.1 In 2008, the World Health Organization (WHO) distinguished 3 entities: T-cell large granular lymphocytic leukemia, chronic lymphoproliferative disorder of NK cells (CLPD-NK, provisional entity), and aggressive NK-cell leukemia. CLPD-NK and NK-cell leukemia are rare disorders of CD3−/CD56+ NK cells. Patients with aggressive NK-LGL present with a malignant clinical course and a median survival of only 2 months from diagnosis.2 Immunosuppressive regiments are utilized in LGL leukemia but such treatments are not curative.3 The aggressive NK-LGL variant has no effective treatment and is refractory to conventional chemotherapy. The lack of effective therapy and therapeutic targets results from an incomplete picture of survival mechanisms that contribute to the pathogenesis of leukemic LGLs.3
Our lab has shown that various survival pathways are constitutively active in leukemic LGLs4 including STAT3 which plays a central role in LGL leukemia pathogenesis through both constitutive and mutational activation.5-7 The sphingolipid rheostat controls cell fate through the balance between pro-apoptotic ceramide and pro-survival sphingosine-1-phosphate (S1P). Ceramide acts as a hub in the sphingolipid pathway and is metabolized to give diverse bioactive products.8 Ceramide accumulates during apoptosis induced through Fas, tumor necrosis factor α (TNF-α), various chemotherapeutic agents and radiation, and is implicated in the activation of several death-signaling pathways including c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK).9-11 Sphingosine is produced through enzymatic conversion of pro-apoptotic ceramide to sphingosine by various ceramidases, including acid ceramidase (AC), whereas SPHK1 catalyzes the phosphorylation of sphingosine to produce pro-survival S1P. S1P signals both intracellularly and through autocrine and paracrine mechanisms.8,12 Elevated AC and SPHK1 are associated with increased survival of cancer cells.11
Elevated SPHK1 levels have been found in numerous cancer types and dysregulation of this enzyme has been linked to hematological malignancy progression.8,12 SPHK1 is overexpressed in many solid tumors and hematologic cancers including acute myeloid leukemia.13 SPHK1 expression has been correlated with chemotherapeutic resistance,13,14 resistance to radiation8,15 and malignant characteristics of tumors.16 This has led to SPHK1 being considered as a novel therapeutic target. Pharmacological inhibitors of SPHK1 (e.g. SKI-II, SKI-178) or biological inhibition with SPHK1 siRNAs increase ceramide and decrease S1P levels resulting in induction of apoptosis and increased radiation and chemotherapy sensitivity in malignant cancer cells.17-19 Sphingosine Kinase Inhibitor II (SKI-II) is a non-selective inhibitor of SPHK1 and SPHK2 with anti-proliferative activity in a variety of cancer cell lines.18 It has been shown to be 2-fold more selective for SPHK2 than SPHK1. SKI-178 is a SPHK1 selective, non-lipid based inhibitor developed from the optimization of Sphingosine Kinase Inhibitor I (SKI-I).17 SKI-178 is a novel SPHK1 inhibitor that exhibits greater efficacy and specificity toward SPHK2.17
We showed previously that total ceramide levels are decreased in leukemic NK cells compared to normal NK cells.20 The use of SKI-II selectively induced apoptosis in T-LGL leukemia patient PBMCs but this was not further studied.21 We hypothesized that SPHK1 is over-expressed in NK-LGL leukemia cells representing a potential therapeutic target. Indeed, we found that there was increased SPHK1 mRNA and protein in NK-LGL patient samples. Pharmacologic intervention with SPHK inhibitors increased apoptosis and decreased cell viability in leukemic NK cells. Treatment with SPHK1 inhibitors increased ceramide levels and decreased S1P levels while inducing caspase-dependent apoptosis. Mechanistic studies showed that this occurs through a CDK1-mediated pathway and is cell cycle dependent. This work highlights SPHK1 as a potential therapeutic target in NK-LGL leukemia.
Results
SPHK1 is overexpressed in NK-LGL leukemia
To determine the therapeutic potential of targeting SPHK1 in LGL leukemia, we measured the gene expression level of SPHK1 in freshly isolated LGLs from NK-LGL leukemia patients (n = 8) and in age and gender-matched normal donor NKs (n = 8). Quantitative reverse transcription-polymerase chain reaction (RT-PCR) results demonstrated that SPHK1 mRNA levels were increased in NK-LGL patient cells (n = 8) relative to purified NK cells isolated from normal donors (P < 0.05) (Fig. 1A). Immunoblot analysis of SPHK1 protein in NK-LGL leukemia patient cells (n = 5) or purified NK cells from normal donors (n = 2) showed SPHK1 protein levels were > 3-fold increased in patients compared to normal. To determine if the overexpression of SPHK1 in LGL leukemia cells affects the levels of sphingosine and S1P, mass spectrometry measurement of sphingosine and S1P was performed on NK-LGL patient sera (n = 8) and compared with sera from normal donors (n = 8). Serum levels of sphingosine were not significantly different in LGL patients compared to normal donors. However, S1P levels were increased in LGL patients' sera (P < 0.05, Fig. 1C), thereby demonstrating in vivo sphingolipid alterations as a result of the increased SPHK1 mRNA and protein.
Figure 1.
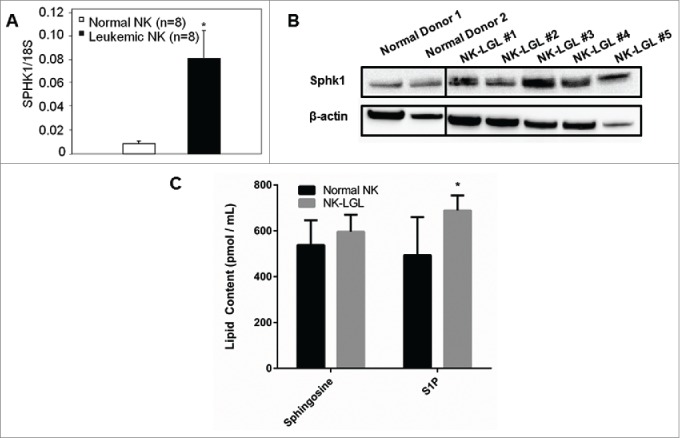
SPHK1 is overexpressed in leukemic NK cells and contributes to a dysregulated sphingolipid rheostat. (A) Quantitative real-time PCR was performed to measure levels of SPHK1 mRNA in PBMC from NK-LGL leukemia patients (CD3−CD56+ >80%, n = 8) or purified NK cells isolated from normal donors (n = 8). SPHK1 mRNA levels are expressed relative to 18S (Mean ± SEM) *, P < 0.05 (Mann-Whitney test). (B) Immunoblot analysis of SPHK1 protein in NK-LGL patient cells or purified NK isolated from normal donors. Loading of protein was confirmed by probing for β-actin. The vertical black line represents a break in the gel where an empty lane was present. (C) Levels of sphingosine and S1P were determined by mass spectrometry in sera from NK-LGL leukemia patients (n = 8) or normal donors (n = 8) (pmol / mL of sera). *, P < 0.05 indicates leukemic cells versus normal NK cells (Student's t-test).
Pharmacologic inhibition of SPHK1 reduces viability and induces apoptosis in NK-LGL leukemia cells
To determine the role of SPHK1 in leukemic NK cell proliferation and survival, we used pharmacological inhibitors of SPHK1. We treated 2 different LGL cell lines (human NKL22 and rat RNK-1623) with various concentrations of SKI-178 and SKI-II and found that both inhibitors decreased cell viability of NKL and RNK16 (Fig. 2A). To determine whether SKI-II and SKI-178 induced apoptotic cell death in LGL leukemia cells, NKL and RNK16 cells were treated with the indicated doses of SKI-II or SKI-178 and assessed for apoptotic cell death using Annexin V-PE as a marker of apoptosis and 7-AAD as a marker of necrosis. Interestingly, we found that SKI-II and SKI-178 treatment induced apoptotic cell death but very little necrosis in leukemic NK cells (Fig. 2B). We also tested the antitumor effects of SKI-II and SKI-178 on primary human leukemic NK-LGL cells. These cells were freshly isolated from patient PBMCs (n = 5) and treated with SKI-178 (5 μM) or SKI-II (20 μM) (Fig. 2C). SKI-II and SKI-178 induced apoptotic cell death with similar efficacy as with the leukemic cell lines. Importantly, SKI-II and SKI-178 had no apoptotic effect on freshly isolated NK cells from normal donors (Fig. 2C), which is a key demonstration of tumor selectivity that supports potential future clinical development of these compounds.
Figure 2.
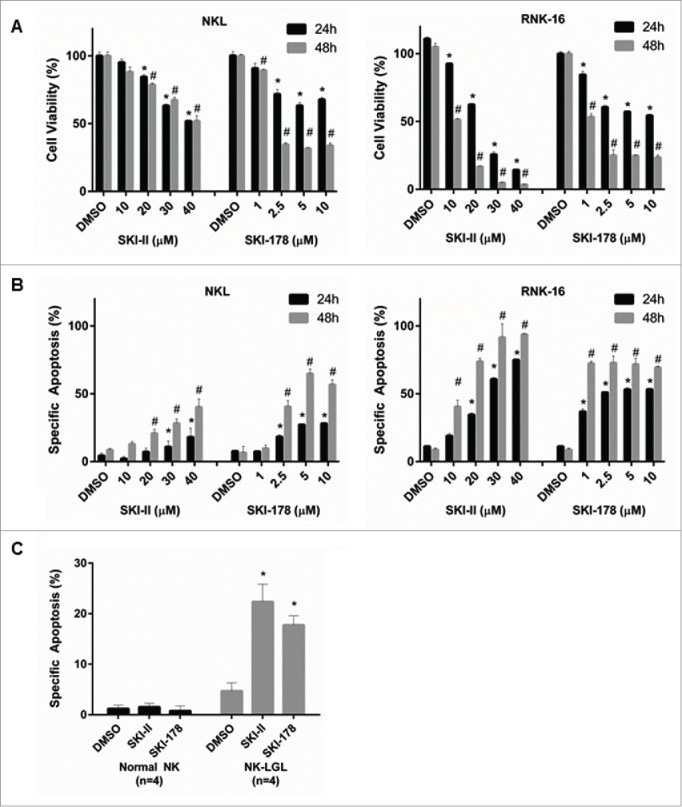
Pharmacologic inhibition of SPHK1 inhibited cell viability and induced apoptosis in leukemic NK cells. (A) NKL or RNK16 cells were treated with either SKI-II or SKI-178 at varying concentrations for 24 or 48 hours and cell viability was measured by MTS assay. (B) NKL or RNK16 cells were treated with either SKI-II or SKI-178 at varying concentrations for 24 or 48 hours and cells were stained with Annexin V and 7-AAD and analyzed by flow cytometry to detect apoptosis. (C) Four PBMC samples from NK-LGL leukemia patients (CD3−CD56+ cells > 80%) or 4 normal NK cell samples were treated with DMSO, 5 μM of SKI-178 or 20 μM of SKI-II for 24 hours and assayed for apoptosis as above. * (24h) and # (48h), P < 0.05, indicate significant differences of SKI-II or SKI-178 treated cells vs. DMSO treated cells (Student's t test).
Pharmacologic inhibition of SPHK1 restores the sphingolipid balance toward a pro-apoptotic state
SPHK1 is a critical regulator of the balance between pro-apoptotic ceramide and anti-apoptotic S1P. In previous studies with SKI-II and SKI-178, it was shown that treatment of cancer cells with these inhibitors resulted in altered bioactive sphingolipid levels.13,17 Here, we demonstrate that SPHK1 inhibition in NKL cells with either SKI-II or SKI-178 resulted in changes in intracellular sphingolipid metabolites. Levels of S1P were significantly decreased after 2h of SKI-II (25 μM) or SKI-178 (5 μM) treatment relative to vehicle control (Fig. 3A). Sphingosine levels remained unchanged in SKI-178 treated cells but were significantly elevated in SKI-II treated cells (Fig. 3A). Total ceramide levels were increased 2.5 to 3-fold after SKI-II and SKI-178 treatment. Ceramide subspecies C16:0, C24:0 and C24:1 were increased while the low levels of C18:1 remained unchanged (Fig. 3B).
Figure 3.
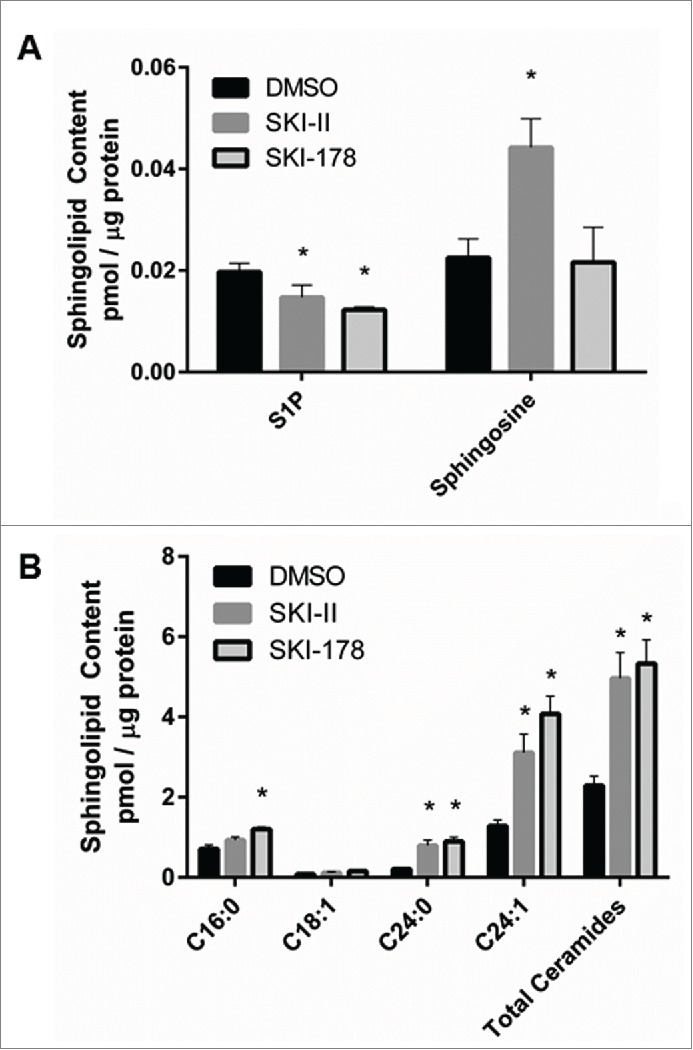
Pharmacologic inhibition of SPHK1 promotes the sphingolipid balance toward a pro-apoptotic state. NKL cells were treated in triplicate with 5 μM of SKI-178, 20 μM of SKI-II or DMSO vehicle control for 2 hours and lipids were extracted from 5 million cells. S1P and sphingosine (A) and ceramide (B) species, were quantified by tandem mass spectrometry, represented as pmol / μg of protein. *P < 0.05 indicates significant differences compared to DMSO treated cells (Student's t test).
G2/M arrest is induced in SKI-178 treated cells
Ceramide has been shown to induce cell cycle arrest at the G2/M phase in cancer cells.24-26 Therefore, NKL cells were treated with SKI-178 or vehicle for 12 or 16h and DNA content was analyzed using PI staining and flow cytometry. Vehicle treated controls had equivalent distribution among G1, S and G2/M phases at 12 and 16h (Fig. 4A). However, the majority of SKI-178-treated cells had G2/M DNA content (Fig. 4B). These results suggest that SKI-178 induces cell cycle arrest. Phosphorylation of histone H3 at Ser10 has been shown to be a marker of chromosome condensation during both apoptosis as a result of cell cycle arrest27,28 and mitosis.29 Phospho-histone H3 (Ser10) reached a maximum in SKI-178 treated cells at 16h, but minimal phosphorylation was seen in vehicle treated cells (Fig. 4C). This suggests those cells undergoing SKI-178 treatment are entering apoptosis or are arrested at G2/M.
Figure 4.

Leukemic NK cells exhibit G2/(M)arrest after inhibition of SPHK1. NKL cells were treated with SKI-178 for the indicated time and cell cycle analysis performed. (A) Flow cytometry analysis of cell cycle distribution in NKL cells treated with SKI-178 (5 μM) or DMSO control for indicated time intervals. The histograms are representative of 3 independent experiments. (B) Percentage of NKL cells in each stage of the cell cycle after 12 or 16h treatment with SKI-178 (5 μM) or vehicle (DMSO). *P < 0.05 indicates significant differences compared to DMSO treated cells (Student's t test). (C) Immunoblot analysis of phospho-histone H3 and histone H3 at the indicated time intervals after treatment with SKI-178 (5 μM) or vehicle (DMSO).
SKI-178 induces caspase-cleavage dependent apoptosis, mitochondrial depolarization and Bcl-2 phosphorylation
Caspase-3 and PARP cleavage are hallmarks of apoptosis.30,31 Both caspase-3 and PARP are cleaved in a dose- and time-dependent manner in NKL cells (Figs. 5A and B respectively) and RNK16 cells (Supplement Fig. S1A) treated with SKI-178. These results were also seen with SKI-II (Supplement Fig. S1B). These effects are first detected at 16h, which is the same time point where the maximum phosphorylation of Histone H3 occurs during G2/M arrest. Similar changes were detected in primary NK-LGL patient samples after SKI-178 treatment (Fig. 5C). To determine if apoptosis in SKI-178 treated cells is caspase-dependent, NKL cells were pre-treated with z-VAD-fmk, a pan-caspase inhibitor. z-VAD-fmk partially abrogated the decrease in cell viability (Supplement Fig. S1C) and the increase in apoptosis (Fig. 5D) that is induced by SKI-178 treatment at both 24 and 48h (P < 0.05). Mitochondrial depolarization is an initial event in the induction of programmed cell death and results in a cleavage cascade contributing to caspase-3 and PARP cleavage. Mitochondrial depolarization can be detected using JC-1, a dye that accumulates in the active mitochondria and exhibits potential-dependent fluorescent emission. Both SKI-II (data not shown) and SKI-178 induced mitochondrial depolarization in NKL cells, as shown by the red to green shift in fluorescence in NKL cells after 12h (Fig. 5E and Supplement Fig. S1D). Treatment with SKI-178 results in phosphorylation of Bcl-2 at Ser70 at 16h and cleavage at 24h (Fig. 5F). Bcl-2 is an anti-apoptotic protein that prevents mitochondrial dysfunction.32 The levels of Mcl-1 and Bcl-XL, 2 other anti-apoptotic Bcl-2 family members shown to play survival roles in hematologic cancers,33 did not show significant changes following SKI-178 treatment. Therefore, SKI-178 treatment leads to mitochondrial depolarization and apoptosis induction that correlated with elevated p-Bcl-2.
Figure 5.
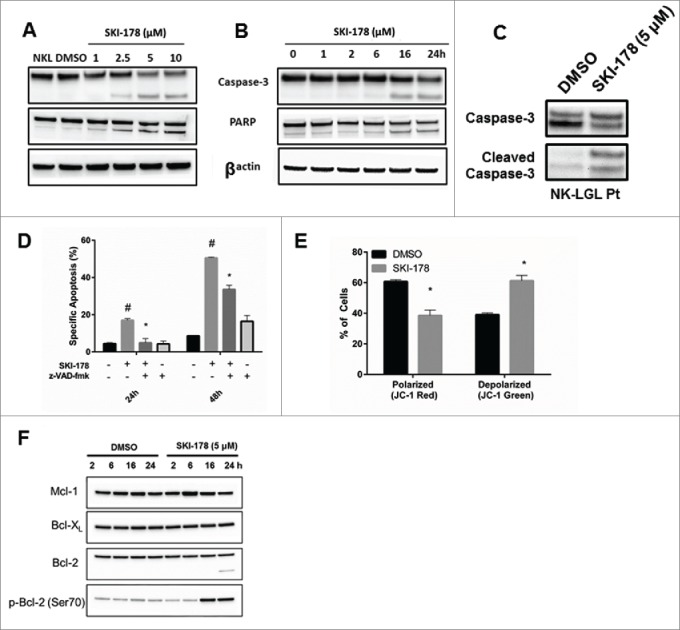
Apoptosis induced through pharmacologic inhibition of SPHK1 is caspase-dependent and targets the mitochondria. (A) Dose-dependent treatment of NKL cells with SKI-178 for 24 hours. Immunoblot analysis shows caspase-3 and PARP cleavage. Equal loading of protein was confirmed by probing for β-actin. (B) Time-dependent treatment of NKL with 5 μM of SKI-178. Immunoblot analysis shows caspase-3 and PARP cleavage. Equal loading of protein was confirmed by probing for β-actin. (C) Primary PBMCs from an NK-LGL patient were treated with DMSO or SKI-178 (5 μM) for 24 hours. Immunoblot analysis shows caspase-3 cleavage. (D) NKL cells were pre-treated with the pan-caspase inhibitor z-VAD-fmk (25 μM) for 30 minutes and then treated with SKI-178 (5 μM). Cells were assayed for apoptosis at 24 or 48 hours with Annexin V and 7-AAD staining and analyzed by flow cytometry. Mean ± SEM shown. *, P < 0.05, indicates significant difference between z-VAD-fmk pre-treatment along with SKI-178 versus SKI-178-only treated cells. (E) NKL cells were treated with DMSO or SKI-178 (5 μM) for 16 hours and then harvested. The mitochondrial membrane potential, ΔΨM, was investigated using JC-1 dye and analyzed using flow cytometry. Bar graph illustrates percentage of cells with high mitochondrial polarization (JC-1 red) and percentage cells with mitochondrial depolarization (JC-1 green). *, P < 0.05, indicate significant differences between DMSO and SKI-178 treated cells (Student's t-test). Mean ± SEM shown. (F) Immunoblot analysis of each of the pro-survival Bcl-2 family members. NKL cells were treated with DMSO or SKI-178 (5 μM) for the indicated time.
Apoptosis induced by SKI-178 is associated with inactivation of ERK1/2 and activation of p38 and JNK
Abundant evidence suggests a role for mitogen-activated protein kinases (ERK1/2, JNK, p38 MAPK) in determining leukemic cell fate.34 The treatment of NKL cells with SKI-178 resulted in a rapid decrease in phosphorylation of ERK1/2. This deactivation of ERK1/2 was sustained for up to 24 hours (Fig. 6A). An increase in phosphorylation of p38 MAPK was detected at 16 and 24 hours. Additionally, JNK phosphorylation and c-JUN phosphorylation occurred in parallel and were elevated with SKI-178 treatment at all-time points (Fig. 6A). These effects were dose-dependent in both NKL and RNK-16 cells (Fig. 6B)
Figure 6.

Inhibition of SPHK1 alters multiple survival pathways in leukemic NKL cells. (A) NKL cells were treated for the indicated time with either DMSO or SKI-178 (5 μM) and then harvested for protein. Equal amounts of protein lysate were analyzed by western blotting with antibodies against P-JNK, JNK, P-c-JUN, P-p38 MAPK, p38 MAPK, P-ERK1/2 and ERK1/2. Equal loading confirmed by total JNK, p38 and ERK1/2 levels and β-Actin. (B) Dose-dependent treatment of NKL or RNK-16 cells with SKI-178 for 24 hours. Equal amounts of protein lysate were analyzed by protein gel blotting with antibodies against P-JNK, JNK, P-p38 MAPK, p38 MAPK, P-ERK1/2 and ERK1/2. Equal loading of protein was confirmed by probing for β-actin. C and D) NKL cells were pretreated for 1 hour with a JNK inhibitor, SP600125 (SP) at 10 μM, a CDK1 inhibitor, Ro3306 at 10 μM, or a second JNK inhibitor, JNK inhibitor XVI at 10 μM. The cells were then incubated with SKI-178 (5 μM) or DMSO for 48h and cell viability assessed by MTS assay (C) or Annexin V/7-AAD apoptosis by flow cytometry (D E) NKL cells were pretreated with SP (10 μM), Ro3306 (10 μM) or JNK inhibitor XVI (10 μM) for 1 hour and then incubated with SKI-178 (5 μM) or DMSO for 24 hours. Cells were harvested for protein lysate and equal amounts of protein were analyzed by western blotting with antibodies against P-Bcl-2 (Ser70) and Bcl-2. Equal loading was confirmed by β-actin. *P < 0.05 indicates significant differences as shown (Student's t test).
To elucidate the functional significance of these pathways, we pre-treated NKL cells with inhibitors of these pathways followed by SKI-178 treatment and assessed cell viability and apoptosis. Inhibition of p38 with SB203580 had no effect on SKI-178 treatment (data not shown). Pre-treatment with the non-specific JNK inhibitor SP60012535 provided modest resistance to the decreased cell viability observed with SKI-178 (Fig. 6C) and did not inhibit the apoptotic effects (Fig. 6D). SP600125 has been shown to also block CDK1 in addition to JNK.35 Pre-treatment with a CDK1 inhibitor, Ro3306, significantly attenuated the effects of SKI-178 on both cell viability (Fig. 6C) and apoptosis (Fig. 6D). To determine if the effects of SKI-178 were JNK or CDK1 specific, a second JNK inhibitor, JNK inhibitor XVI, was used. Treatment with this highly specific JNK inhibitor36 had no significant effects on the impact of SKI-178 on cell viability or apoptosis (Fig. 6C and Fig. 6D). Taken together, these data suggest a key role for CDK1 in SKI-178 mediated viability reduction and increased apoptosis induction in NKL cells.
The kinase that phosphorylates Bcl-2 at Serine70 is debated in the literature. It has been suggested that CDK137 or JNK38 mediate this event. Using the inhibitors previously mentioned, we determined that CDK1 inhibition before SKI-178 treatment fully inhibited Bcl-2 phosphorylation and cleavage (Fig. 6E). The effects of JNK inhibition with SP600125 or JNK Inhibitor XVI did not reverse the phosphorylation of Bcl-2. Taken together, these data indicate that CDK1 inhibition blocks Bcl-2 phosphorylation and has the ability to rescue cells from SKI-178-induced effects.
SKI-178 targets JAK-STAT signaling
To explore other potential molecular mediators of SKI-178 effects in an unbiased manner, we used a high-throughput Reverse Phase Protein Microarry (RPMA) to analyze signaling pathways that are altered upon SKI-178 treatment of NKL cells. This verified the above mentioned changes in P-Bcl-2, P-JNK, P-p38 MAPK and P-ERK (Supplement Fig. S2). Interestingly, treatment of NKL cells with SKI-178 led to decreased phosphorylation of both JAK and STAT proteins (Fig. 7A and B). The JAK/STAT pathway is dysregulated in both T- and NK-LGL leukemia.5,39 The RPMA targeting of JAK/STAT signaling was verified by immunoblotting analysis and illustrates that P-STAT3, P-STAT5, P-JAK1 and P-JAK2 decrease dose-dependently with SKI-178 treatment (Fig. 7C and D). The importance of JAK/STAT signaling in LGL leukemia survival and pathogenesis and the integration of JAK/STAT signaling inhibition with a sphingosine kinase inhibitor provides further evidence of the promising potential of SPHK1 as a therapeutic target.
Figure 7.
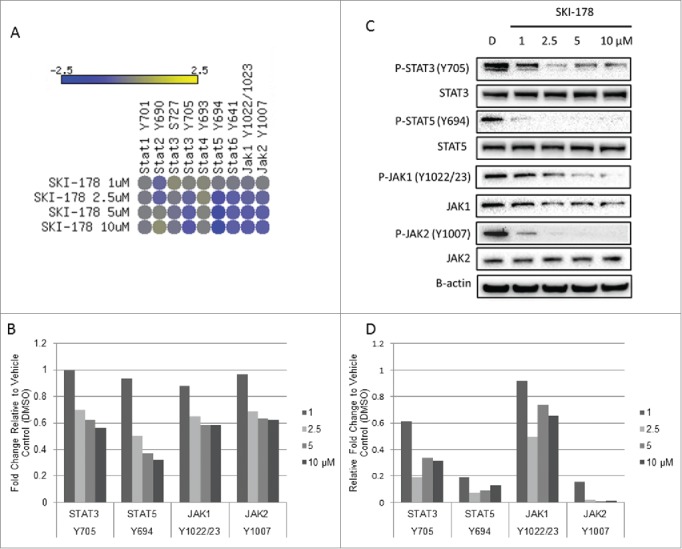
SKI-178 targets JAK-STAT signaling. (A) NKL cells were treated for 16 hours with the indicated doses of SKI-178 and analyzed using a reverse phase protein array. The heat map shown illustrates the phosphorylation states of JAK/STAT family members. Data is represented as Log2 of the fold change relative to vehicle control (DMSO) (B) Representative bar graph illustrating relative fold change of SKI-178 treated NKL cells to vehicle control (DMSO) (C) The reverse phase protein array data was verified by immunoblotting for the indicated proteins. NKL cells were treated for 16 hours with the indicated doses of SKI-178. Equal amounts of protein lysate were analyzed by immunoblotting with antibodies against P-STAT5 (Y694), STAT5, P-STAT3 (Y705), STAT3, P-JAK1 (Y1022/23), JAK1, P-JAK2 (Y1007) and JAK2. Equal loading was confirmed by β-actin. (D) Quantification of immunoblots in Figure 7C. Protein was normalized to β-actin and then relative expression levels were determined compared to vehicle control (DMSO).
Discussion
Our results show that SPHK1 is a novel therapeutic target in NK-LGL leukemia. The sphingolipid rheostat has been shown to be dysregulated in many types of cancers, with S1P production leading to abnormal cell growth and survival signaling.40 Here, demonstrated that SPHK1 is overexpressed in NK-LGL leukemia patients compared to normal donors, which resulted in increased serum S1P levels. Furthermore, targeting of SPHK1 with pharmacologic inhibitors, SKI-II and SKI-178, decreased leukemic NK cell viability and induced apoptotic cell death. These data demonstrate the role of SPHK1 in the pathogenesis of NK-LGL leukemia. The identification and validation of SPHK1 as a potential therapeutic target in NK-LGL leukemia is significant because current therapeutic regimens use methotrexate, cyclophosphamide or cyclosporine; all non-specific immunosuppressive agents which do not selectively kill LGL leukemia cells. Current therapeutic treatment regimens only result in modest 40% success rates and do not result in cure of this disease.3,41
We showed that the use of the sphingosine kinase inhibitors (SKI-II and SKI-178) potently induced apoptosis in two NK-LGL leukemia cell lines and primary LGLs from patients. The mechanisms by which SPHK1 inhibitors induce apoptosis and decrease the proliferative ability of leukemic NKs appear to be the result of multiple connected actions. SPHK1 inhibitor treatment in leukemic cell lines was sufficient to activate executioner caspases and the cleavage of PARP.13 We also observed depolarization of the mitochondria, caspase-3 and PARP cleavage, all hallmarks of apoptosis. Pharmacologic targeting of SPHK1 by SKI-II or SKI-178 inhibited production of pro-survival S1P. S1P can act intracellularly and can be released from cells to act through its cell surface receptors, S1PR1–5, of which S1PR5 was discovered in our lab as an overexpressed EST in LGL leukemia.40,42,43 S1P binding to S1PRs is associated with activation of survival pathways including ERK1/2.13,44 We observed rapid decreases in ERK1/2 phosphorylation after treatment with SPHK1 inhibitors. ERK1/2 can phosphorylate SPHK1, leading to increased production of S1P, which in turn further activates ERK1/2 resulting in a positive feedback loop.45 Ceramide accumulation has been shown to be a precursor to apoptosis induction in leukemic cells.46-48 We observed impressive increases in ceramide accumulation after inhibition of SPHK1, which suggests that elevated SPHK1 activity is a key ceramide detoxification pathway in NK-LGL. Ceramide can execute its apoptotic effects through multiple molecular pathways.9,11 Important immediate downstream effectors of ceramide accumulation include serine/threonine protein phosphatases PP1 and PP2A,47 which can also negatively regulate the ERK signaling pathway.49 The disruption of this loop, coupled with the observed increases in ceramide production that result from targeting of SPHK1 with pharmacologic inhibitors, demonstrates that SPHK1 inhibition can integrate multiple molecular targets to reduce survival of leukemic LGLs.
Whereas S1P promotes cell growth and proliferation and survival, ceramide and sphingosine can mediate cell cycle arrest and induce apoptosis. The downregulation of SPHK1 has previously been shown to induce cell cycle arrest and apoptosis through the activation of the mitochondrial cell death pathway.16,50 We observed arrest of leukemic NK cells at the G2/M stage of the cell cycle following treatment with SKI-178. This effect is comparable to what we have observed in SKI-178 treated AML cells lines.51 We found that Bcl-2 phosphorylation at Ser70 increased following inhibition of SPHK1, as well as Bcl-2 cleavage. The phosphorylation of Bcl-2 and subsequent cleavage were associated with mitochondria-dependent apoptosis.38,52 Interestingly, the phosphorylation of Bcl-2 at Ser70 occurs under normal cell cycle conditions at the G2/M stage.38 The extended time spent in G2/M has been suggested to result in even further increases of Bcl-2 phosphorylation, leading to the dissociation of Bax from Bcl-2 and its ensuing entry into the mitochondria leading to depolarization.32,37 Interestingly, we found that it was CDK1, but not JNK, that contributes to the phosphorylation of Bcl-2. Additionally, phosphorylation of histone H3 increases after SPHK1 inhibition. This is similar to what we have recently observed in AML cells.51 Normally, this occurs during mitosis and accompanies chromosome condensation. However, during apoptotic cell death, chromosome condensation is one of the first changes and this suggests a similar role for histone H3 phosphorylation during apoptosis.28 These molecular events may represent useful biomarkers of response for future clinical application of SKI-178.
JAK/STAT signaling plays an integral role in LGL leukemia. The identification of both constitutive activation of STAT3 in all LGL leukemia patient cells, as well as up to 40% of patients harboring activating mutations in STAT3 suggests a central role for STAT signaling in LGL leukemia pathogenesis.5-7,53 Our identification of JAK/STAT inhibition through the use of RPMA analysis of SKI-178 treated NKL cells suggests that SPHK1 inhibition can integrate multiple molecular signaling pathways in LGL leukemia. S1P and the S1P receptors have been shown to be critical for persistent STAT3 activation in tumors.54,55 Our finding that SPHK1 inhibition leads to decreased phosphorylation of JAK1/JAK2/STAT3/STAT5 suggests that a SPHK1/S1P/S1PR axis is central to the persistent activation of STAT signaling in LGL leukemia.
Our finding that SKI-II and SKI-178 potently induced apoptosis in patient leukemic LGLs but had little effect on normal NKs illustrates selective S1P addiction in leukemic cells. Thus, SPHK1 inhibitors warrant further investigation and consideration as potential therapeutics for NK-LGL leukemia.
Materials and Methods
Reagents and cell culture
Sphingosine kinase inhibitor 2 (SKI-II) was purchased from SelleckChem. SKI-178 was purchased from Merck Millipore. z-VAD-fmk was purchased from Invivogen. Antibodies for β-actin, Caspase-3, PARP, Bcl-2, p-Bcl-2 (Ser70), JNK, p-JNK(Thr183/Tyr185), SPHK1, c-Jun, p-c-Jun, p-Histone-H3, p38 and p-p38 (Thr180/Tyr182) were purchased from Cell Signaling Technology Inc.. Human NKL cells (kindly provided by Dr. Howard Young at National Cancer Institute) were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) plus 100 IU/ml IL-2 (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann – La Roche Inc.).56 Rat RNK-16 cells (kindly provided by Dr. Craig Reynolds at National Cancer Institute) were cultured in RPMI-1640 supplemented with 10% FBS, non-essential amino acids (Sigma), 1% sodium pyruvate (Sigma) and 25 μM 2-mercaptoethanol. All cells were maintained in a 37°C humidified 5% CO2 atmosphere incubator. Cell lines were authenticated by Genetica using STR profiling.
Patient characteristics and preparation of peripheral blood mononuclear cells (PBMC)
All patients met the criteria of chronic NK leukemia with increased numbers (>80 %) of CD3−CD56+ or CD3−CD16+ NK cells in the peripheral blood. Peripheral blood samples were obtained and informed consents signed for all LGL leukemia patients according to protocols approved by the Institutional Review Boards of Penn State Hershey Medical Center and the University of Virginia Medical Center. Normal NK cells from age- and gender-matched healthy donors were obtained from the Blood Bank of the Milton S. Hershey Medical Center and were isolated by selection using StemCell Technologies, Inc. EasySep Human NK cell Enrichment Kit. PBMC were isolated by Ficoll-Hypaque gradient separation as described previously.20 Cell viability was determined by trypan blue exclusion assay with more than 95% viability in all samples.
Real-time quantitative PCR
Quantitative real-time PCR was performed using primer sets specific for human SPHK1 and an internal standard 18S rRNA in an ABI PRISM 7900 sequence detector (Foster City, CA). Five × 106 PBMC from three aggressive NK-LGL leukemia patients or five chronic NK-LGL leukemia patients (CD3−CD56+ NK cells >80%) and freshly purified CD3−CD56+ NK cells from 8 age- and gender- matched normal healthy controls were subjected to total RNA extraction using TRIzoL Reagent. First strand cDNA was synthesized from 2 μg purified total RNA using random hexamers and MMLV reverse transcription reagents in a total volume of 20 uL. One μg cDNA was applied in a 10 μL PCR mix using a QuantiTest SYBR Green PCR kit (QIAgen). Amplification of triplicate cDNA template samples was then performed with denaturation for 15 min at 95°C, followed by 40 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s and extension at 72°C for 30s. Values were normalized to the relative amount of 18S mRNA, which were obtained from a similar standard curve. The changes in fluorescence of SYBR green dye in every cycle were monitored by the ABI 7900 system software and threshold cycle (ΔΔCt) for each reaction was calculated. The relative amount of PCR products generated from each primer set was determined based on the Ct value. The following primers were used in the detection of SPHK1: human SPHK1 sense, 5′-ATGCACGAGGTGGTGAAC-3′, antisense 5′-TAGCCAGCATAATGGTTCAAG-3′.
Western blot analysis
Protein expression levels were determined as previously described.20 Briefly, whole cell lysates were harvested using 1X RIPA buffer. Lysates were centrifuged for 15 minutes at 14400 g at 4°C to pellet cell debris. Protein concentrations were determined using a BCA assay. Equal amounts of protein were loaded onto Bolt 4–12% Bis-Tris gels, separated by electrophoresis and transferred to PVDF membranes. Membranes were blocked in 5% BSA/TBS-T and incubated overnight at 4°C with primary antibody (1:1000). Immunodetection was performed with IgG HRP-linked antibodies (1:3000) and ECL reagent on a Bio-Rad ChemiDOC XP or ChemiDOC MP.
Reverse phase protein microarray construction and analysis
Pathway activation mapping was performed by reverse phase protein microarray (RPMA) as previously described.57-59 Briefly, cells were subjected to lysis with 2.5% solution of 2-mercaptoethanol in Tissue Protein Extraction Reagent (t-PER™ Pierce) /2X SDS Tris-Glycine 2X SDS buffe. The lysates were printed on glass-backed nitrocellulose array slides using an Aushon 2470 arrayer (Aushon BioSystems, Burlington, MA USA) equipped with 185 µm pins. Each sample was printed in triplicate. Arrays were blocked (I-Block) for 1h and subsequently probed with phosphorylated, cleaved or total protein antibodies. Detection was performed using a fluorescence-based tyramide signal amplification strategy using Streptavidin-conjugated IRDye680 (LI-COR Biosciences) was used as detection reagent. All antibodies were validated for single band specificity as well as for ligand-induction (for phospho-specific antibodies) by Western Blotting prior to use on the arrays as described previously.57-59 Each array was scanned using a TECAN LS laser scanner. After scanning, spot intensity was analyzed, data were normalized to total protein and a standardized, single data value was generated for each sample on the array by MicroVigene software V2.999 (VigeneTech, North Billerica, MA USA) as previously described.57
Apoptosis and cell viability assays
Apoptosis induced by SKI-II or SKI-178 in NKL, RNK16, normal NKs from health donors or NK cells from LGL patients was detected by flow cytometry with Annexin V -PE (BD PharMingen) and 7-amino-actinomycin D (7-AAD) staining using 5 × 105 cells per sample in triplicate. The percentage specific apoptosis was calculated using the follow formula: Apoptosis (%) = [(% Annexin V-PE positive cells) in treatment wells] - [(% Annexin V-PE positive cells) in control wells] × 100/ [(% Annexin V-PE positive cells) in control wells]. Cell viability was determined using CellTiter 96 Aqueous One Solution assay kit (Promega). The relative viable cell number was determined using a Cytation HT Multi-Detection Microplate Reader (Bio-TEK).
Cell cycle analysis
DNA content was analyzed using propidium iodide (PI) staining and flow cytometry. Briefly, cells were treated for the indicated time intervals with SKI-178 (5 μM) or DMSO and fixed in cold 70% EtOH and incubated for 3 hours at −20°C. Cells were then resuspended in 1X PBS with 100 μg/mL PI and 200 μg/mL Ribonuclease A (Abcam) for 30 minutes and analyzed with a FACS Calibur analyzer. Data was processed and analyzed with FlowJo V10 software.
Mitochondrial depolarization assay
JC-1 is a cationic dye that permeates mitochondrial membranes and fluoresces red when it aggregates in healthy, high potential mitochondria, but fluoresces green when there are damaged, low potential mitochondria. JC-1 can therefore be used to indicate mitochondrial matrix potential (ΔΨM). Briefly, we treated NKL cells for 16h with SKI-178. Cells were incubated with 2 μg/mL JC-1 dye for 30 minutes at 37°C and immediately analyzed on a BD FACS-Calibur at 488 nM excitation. Data were collected at 529 nm for green fluorescence and 590 nm for red fluorescence. Results are expressed as percent of JC-1 fluorescence.
Lipid quantification by mass spectroscopy
Briefly, 5 × 106 of cells were homogenized by sonication in a 10mM Tris/HCl(pH 7.2). These samples then were subjected to lipid extraction in chloroform/methanol/water as originally described by Bligh and Dyer60 after internal standards had been added (C17-sphingosine-1-phosphate, C17-sphingosine, C17-sphinganine, C17-sphinganine-1-phosphate, C12-ceramide). Sphingoid bases and 1-phosphates was separated by HPLC (Agilent 1100) with a Luna 2.1mm × 50mm C18-reverse phase column (Phenomenex) and ceramide was separated on a LC-NH2-amino column (Sigma-Aldrich) before detection via multiple reaction monitoring (MRM) and quantification with an 4000QTrap mass spectrometer (a hybrid quadrupole-ion trap mass spectrometer, Sciex, Redwood City, CA USA). The peak areas for the sphingolipid subspecies were quantified according to the internal standards and normalized to total protein content as determined by BCA assay.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Alexander Wendling for technical support with flow cytometry. We thank Dr. Kevin Lynch and Dr. Yugesh Kharel for their advice (University of Virginia) and lipidomics work.
Authorship
FRL participated in research design, conducted experiments, performed data analysis and wrote the manuscript. XL participated in research design and conducted experiments. JH participated in research design and contributed to the writing of the manuscript. TF conducted experiments and contributing to the writing of the manuscript. JY contributed to the writing of the manuscript and provided vital new reagents. DJF participated in research design and contributed to the writing of the manuscript. TPL participated in research design and contributed to the writing of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA098472 (to T.P.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev 2014; 28:87-94; PMID:24679833; http://dx.doi.org/ 10.1016/j.blre.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poullot E, Zambello R, Leblanc F, Bareau B, De March E, Roussel M, Boulland ML, Houot R, Renault A, Fest T, et al.. Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann Oncol 2014; 25:2030-5; PMID:25096606; http://dx.doi.org/ 10.1093/annonc/mdu369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood 2011; 117:2764-74; PMID:21190991; http://dx.doi.org/ 10.1182/blood-2010-07-296962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leblanc F, Zhang D, Liu X, Loughran TP. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol 2012; 8:787-801; PMID:22830400; http://dx.doi.org/ 10.2217/fon.12.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang JM, Yang-Yen HF, Karras J, et al.. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 2001; 107:351-62; PMID:11160159; http://dx.doi.org/ 10.1172/JCI9940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I, et al.. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012; 120:3048-57; PMID:22859607; http://dx.doi.org/ 10.1182/blood-2012-06-435297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, Lagström S, Clemente MJ, Olson T, Jalkanen SE, et al.. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012; 366:1905-13; PMID:22591296; http://dx.doi.org/ 10.1056/NEJMoa1114885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SPHK1 as a new strategy against cancer. Curr Drug Targets 2008; 9:662-73; PMID:18691013; http://dx.doi.org/ 10.2174/138945008785132402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 2008; 111:4365-74; PMID:18270325; http://dx.doi.org/ 10.1182/blood-2007-08-106336 [DOI] [PubMed] [Google Scholar]
- 10.Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem 2014; 57:5509-24; PMID:24471412; http://dx.doi.org/ 10.1021/jm4011687 [DOI] [PubMed] [Google Scholar]
- 11.Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer 2013; 13:51-65; PMID:23235911; http://dx.doi.org/ 10.1038/nrc3398 [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Wang Y, Wan Z, Liu S, Cao Y, Zeng Z. Sphingosine kinase 1 and cancer: a systematic review and meta-analysis. PLoS One 2014; 9:e90362; PMID:24587339; http://dx.doi.org/ 10.1371/journal.pone.0090362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, et al.. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 2008; 112:1382-91; PMID:18511810; http://dx.doi.org/ 10.1182/blood-2008-02-138958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C, Delisle MB, Cuvillier O, Susini C, Bousquet C. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol Cancer Ther 2009; 8:809-20; PMID:19372554; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-1096 [DOI] [PubMed] [Google Scholar]
- 15.Sinha UK, Schorn VJ, Hochstim C, Chinn SB, Zhu S, Masood R. Increased radiation sensitivity of head and neck squamous cell carcinoma with sphingosine kinase 1 inhibition. Head Neck 2011; 33:178-88; PMID:20848438; http://dx.doi.org/ 10.1002/hed.21418 [DOI] [PubMed] [Google Scholar]
- 16.Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, Spiegel S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett 2005; 579:5313-7; PMID:16194537; http://dx.doi.org/ 10.1016/j.febslet.2005.08.055 [DOI] [PubMed] [Google Scholar]
- 17.Hengst JA, Wang X, Sk UH, Sharma AK, Amin S, Yun JK. Development of a sphingosine kinase 1 specific small-molecule inhibitor. Bioorg Med Chem Lett 2010; 20:7498-502; PMID:21050755; http://dx.doi.org/ 10.1016/j.bmcl.2010.10.005 [DOI] [PubMed] [Google Scholar]
- 18.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 2003; 63:5962-9; PMID:14522923 [PubMed] [Google Scholar]
- 19.Kennedy AJ, Mathews TP, Kharel Y, Field SD, Moyer ML, East JE, Houck JD, Lynch KR, Macdonald TL. Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J Med Chem 2011; 54:3524-48; PMID:21495716; http://dx.doi.org/ 10.1021/jm2001053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watters RJ, Fox TE, Tan SF, Shanmugavelandy S, Choby JE, Broeg K, Liao J, Kester M, Cabot MC, Loughran TP, et al.. Targeting glucosylceramide synthase synergizes with C6-ceramide nanoliposomes to induce apoptosis in natural killer cell leukemia. Leuk Lymphoma 2013; 54:1288-96; PMID:23181473; http://dx.doi.org/ 10.3109/10428194.2012.752485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP Jr. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A 2008; 105:16308-13; PMID:18852469; http://dx.doi.org/ 10.1073/pnas.0806447105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol 1996; 24:406-15; PMID:8599969 [PubMed] [Google Scholar]
- 23.Ward JM, Reynolds CW. Large granular lymphocyte leukemia. A heterogeneous lymphocytic leukemia in F344 rats. Am J Pathol 1983; 111:1-10; PMID:6837719 [PMC free article] [PubMed] [Google Scholar]
- 24.Jayadev S, Liu B, Bielawska AE, Lee JY, Nazaire F, Pushkareva MYu, Obeid LM, Hannun YA. Role for ceramide in cell cycle arrest. J Biol Chem 1995; 270:2047-52; PMID:7836432; http://dx.doi.org/ 10.1074/jbc.270.5.2047 [DOI] [PubMed] [Google Scholar]
- 25.Lee JY, Leonhardt LG, Obeid LM. Cell-cycle-dependent changes in ceramide levels preceding retinoblastoma protein dephosphorylation in G2/M. Biochem J 1998; 334(Pt 2):457-61; PMID:9716505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takai N, Ueda T, Kawano Y, Nishida M, Nasu K, Narahara H. C2-ceramide exhibits antiproliferative activity and potently induces apoptosis in endometrial carcinoma. Oncol Rep 2005; 14:1287-91; PMID:16211298 [PubMed] [Google Scholar]
- 27.Waring P, Khan T, Sjaarda A. Apoptosis induced by gliotoxin is preceded by phosphorylation of histone H3 and enhanced sensitivity of chromatin to nuclease digestion. J Biol Chem 1997; 272:17929-36; PMID:9218417; http://dx.doi.org/ 10.1074/jbc.272.29.17929 [DOI] [PubMed] [Google Scholar]
- 28.Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 2003; 116:3677-85; PMID:12917355; http://dx.doi.org/ 10.1242/jcs.00735 [DOI] [PubMed] [Google Scholar]
- 29.Hans F, Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene 2001; 20:3021-7; PMID:11420717; http://dx.doi.org/ 10.1038/sj.onc.1204326 [DOI] [PubMed] [Google Scholar]
- 30.D'Amours D, Sallmann FR, Dixit VM, Poirier GG. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci 2001; 114:3771-8; PMID:11707529 [DOI] [PubMed] [Google Scholar]
- 31.Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 2002; 7:321-8; PMID:12101391; http://dx.doi.org/ 10.1023/A:1016119328968 [DOI] [PubMed] [Google Scholar]
- 32.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ 2000; 7:102-11; PMID:10713725; http://dx.doi.org/ 10.1038/sj.cdd.4400597 [DOI] [PubMed] [Google Scholar]
- 33.Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, Scorilas A. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Adv Hematol 2012; 2012:524308; PMID:21941553; http://dx.doi.org/ 10.1155/2012/524308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004; 23:2838-49; PMID:15077147; http://dx.doi.org/ 10.1038/sj.onc.1207556 [DOI] [PubMed] [Google Scholar]
- 35.Kim JA, Lee J, Margolis RL, Fotedar R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene 2010; 29:1702-16; PMID:20062077; http://dx.doi.org/ 10.1038/onc.2009.464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al.. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 2012; 19:140-54; PMID:22284361; http://dx.doi.org/ 10.1016/j.chembiol.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou L, Cai X, Han X, Xu N, Chang DC. CDK1 switches mitotic arrest to apoptosis by phosphorylating Bcl-2/Bax family proteins during treatment with microtubule interfering agents. Cell Biol Int 2014; 38:737-46; PMID:24677263; http://dx.doi.org/ 10.1002/cbin.10259 [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999; 19:8469-78; PMID:10567572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teramo A, Gattazzo C, Passeri F, Lico A, Tasca G, Cabrelle A, Martini V, Frezzato F, Trimarco V, Ave E, et al.. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood 2013; 121:3843-54, S3841; PMID:23515927; http://dx.doi.org/ 10.1182/blood-2012-07-441378 [DOI] [PubMed] [Google Scholar]
- 40.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer 2010; 10:489-503; PMID:20555359; http://dx.doi.org/ 10.1038/nrc2875 [DOI] [PubMed] [Google Scholar]
- 41.Loughran TP Jr, et al.. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia 2015; Apr;29(4):886-94; doi: 10.1038/leu.2014.298; Epub 2014 Sep 13; http://dx.doi.org/ 10.1038/leu.2014.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem 2009; 78:743-68; PMID:19231986; http://dx.doi.org/ 10.1146/annurev.biochem.78.072407.103733 [DOI] [PubMed] [Google Scholar]
- 43.Kothapalli R, Kusmartseva I, Loughran TP. Characterization of a human sphingosine-1-phosphate receptor gene (S1P5) and its differential expression in LGL leukemia. Biochim Biophys Acta 2002; 1579:117-23; PMID:12427546; http://dx.doi.org/ 10.1016/S0167-4781(02)00529-8 [DOI] [PubMed] [Google Scholar]
- 44.Sato K, Tomura H, Igarashi Y, Ui M, Okajima F. Possible involvement of cell surface receptors in sphingosine 1-phosphate-induced activation of extracellular signal-regulated kinase in C6 glioma cells. Mol Pharmacol 1999; 55:126-33; PMID:9882706 [DOI] [PubMed] [Google Scholar]
- 45.Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J 2003; 22:5491-500; PMID:14532121; http://dx.doi.org/ 10.1093/emboj/cdg540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beverly LJ, Howell LA, Hernandez-Corbacho M, Casson L, Chipuk JE, Siskind LJ. BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochem J 2013; 452:111-9; PMID:23480852; http://dx.doi.org/ 10.1042/BJ20130147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Luo LF, Lu J, Li L, Liu YF, Wang J, Liu H, Song H, Jiang H, Chen SJ, et al.. FTY720 induces apoptosis of M2 subtype acute myeloid leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS One 2014; 9:e103033; PMID:25050888; http://dx.doi.org/ 10.1371/journal.pone.0103033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shakor AB, Atia M, Ismail IA, Alshehri A, El-Refaey H, Kwiatkowska K, Sobota A. Curcumin induces apoptosis of multidrug-resistant human leukemia HL60 cells by complex pathways leading to ceramide accumulation. Biochim Biophys Acta 2014; 1841(12):1672-82; PMID:25240837; http://dx.doi.org/ 10.1016/j.bbalip.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 49.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J 2008; 22:954-65; PMID:18039929; http://dx.doi.org/ 10.1096/fj.06-7859rev [DOI] [PubMed] [Google Scholar]
- 50.Taha TA, Kitatani K, El-Alwani M, Bielawski J, Hannun YA, Obeid LM. Loss of sphingosine kinase-1 activates the intrinsic pathway of programmed cell death: modulation of sphingolipid levels and the induction of apoptosis. FASEB J 2006; 20:482-4; PMID:16507765; http://dx.doi.org/ 10.1096/fj.05-4412fje [DOI] [PubMed] [Google Scholar]
- 51.Dick TE, Hengst JA, Fox TE, Colledge AL, Kale VP, Sung SS, Sharma A, Amin S, Loughran TP Jr, Kester M, et al.. The apoptotic mechanism of action of the sphingosine kinase 1 selective Inhibitor SKI-178 in human acute myeloid leukemia cell lines. J Pharmacol Exp Ther 2015; 352:494-508; PMID:25563902; http://dx.doi.org/ 10.1124/jpet.114.219659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang ST, Cidlowski JA. Phosphorylation status modulates Bcl-2 function during glucocorticoid-induced apoptosis in T lymphocytes. FASEB J 2002; 16:825-32; PMID:12039864; http://dx.doi.org/ 10.1096/fj.01-0852com [DOI] [PubMed] [Google Scholar]
- 53.Andersson EI, Rajala HL, Eldfors S, Ellonen P, Olson T, Jerez A, Clemente MJ, Kallioniemi O, Porkka K, Heckman C, et al.. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J 2013; 3:e168; PMID:24317090; http://dx.doi.org/ 10.1038/bcj.2013.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al.. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med 2010; 16:1421-8; PMID:21102457; http://dx.doi.org/ 10.1038/nm.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al.. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013; 23:107-20; PMID:23273921; http://dx.doi.org/ 10.1016/j.ccr.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lahm D, Lee LK, Bettelheim FA. Age dependence of freezable and nonfreezable water content of normal human lenses. Invest Ophthalmol Visual Sci 1985; 26:1162-5; PMID:4019108 [PubMed] [Google Scholar]
- 57.Paweletz CP, Charboneau L, Bichsel VE, Simone NL, Chen T, Gillespie JW, Emmert-Buck MR, Roth MJ, Petricoin EF III, Liotta LA. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 2001; 20:1981-9; PMID:11360182; http://dx.doi.org/ 10.1038/sj.onc.1204265 [DOI] [PubMed] [Google Scholar]
- 58.Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, Stratton SP, Liotta L, Longo C, Pellacani G, et al.. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res 2012; 5:403-13; PMID:22389437; http://dx.doi.org/ 10.1158/1940-6207.CAPR-11-0427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia W, Petricoin EF 3rd, Zhao S, Liu L, Osada T, Cheng Q, Wulfkuhle JD, Gwin WR, Yang X, Gallagher RI, et al.. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res 2013; 15:R85; PMID:24044505; http://dx.doi.org/ 10.1186/bcr3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959; 37:911-7; PMID:13671378; http://dx.doi.org/ 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.