

ABSTRACT
Aberrant mutational activation of FGFR2 is associated with endometrial cancers (ECs). AP24534 (ponatinib) currently undergoing clinical trials has been known to be an orally available multi-targeted tyrosine kinase inhibitor. Our biochemical kinase assay showed that AP24534 is potent against wild-type FGFR1-4 and 5 mutant FGFRs (V561M-FGFR1, N549H-FGFR2, K650E-FGFR3, G697C-FGFR3, N535K-FGFR4) and possesses the strongest kinase-inhibitory activity on N549H-FGFR2 (IC50 of 0.5 nM) among all FGFRs tested. We therefore investigated the effects of AP24534 on endometrial cancer cells harboring activating FGFR2 mutations and explored the underlying molecular mechanisms. AP24534 significantly inhibited the proliferation of endometrial cancer cells bearing activating FGFR2 mutations (N549K, K310R/N549K, S252W) and mainly induced G1/S cell cycle arrest leading to apoptosis. AP24534 also diminished the kinase activity of immunoprecipitated FGFR2 derived from MFE-296 and MFE-280 cells and reduced the phosphorylation of FGFR2 and FRS2 on MFE-296 and AN3CA cells. AP24534 caused substantial reductions in ERK phosphorylation, PLCγ signaling and STAT5 signal transduction on ECs bearing FGFR2 activating mutations. Akt signaling pathway was also deactivated by AP24534. AP24534 causes the chemotherapeutic effect through mainly the blockade of ERK, PLCγ and STAT5 signal transduction on ECs. Moreover, AP24534 inhibited migration and invasion of endometrial cancer cells with FGFR2 mutations. In addition, AP24534 significantly blocked anchorage-independent growth of endometrial cancer cells. We, for the first time, report the molecular mechanisms by which AP24534 exerts antitumor effects on ECs with FGFR2 activating mutations, which would provide mechanistic insight into ongoing clinical investigations of AP24534 for ECs.
KEYWORDS: Endometrial cancer, ERK, FGFR2 mutation, PLCγ, onatinib, STAT5
Introduction
According to Cancer Statistics 2015, endometrial cancer was estimated the fourth most common cancer cases among women in the United States with the ranks seventh for cancer-related deaths.1 Endometrial cancer is clinicopathologically subtyped into 2 variants by presence of aberrant estrogen signaling, estrogen-related type I endometrioid endometrial cancer, and estrogen-independent type II non-endometrioid serous endometrial cancer.2 Even though type II endometrial cancer accounts for less than 10% of endometrial cancer diagnosis, it is associated with poor prognosis and refractory to current therapy for type I endometrioid endometrial cancers. Surgery and adjuvant radiation therapy result in successful cure in patients with early stage type I endometrioid carcinomas, whereas type II non-endometrioid serous carcinomas, even though in the similar stage, is clinically more aggressive and is at high risk of metastasis and relapse. New therapeutic targets/agents for the treatment of metastatic and recurrent endometrial cancer have been required.3
Type I tumors has been characterized to bear frequent mutation in FGFR2, ARID1A, PTEN, KRAS, PIK3CA, and CTNNB-1. In addition, microsatellite instability (MSI) is predominant in 30% of type I tumors.4 Unlike Type I, Type II has unique genomic and transcriptomic profile of mutations in p53, p16, E-cadherin, and HER2 overexpression along with low level of oestorgen receptor/progesterone receptor.4 Recently, it has also been reported that loss of PTEN function and/or aberrant activation of upstream tyrosine kinase growth factor receptors caused PI3K/AKT signaling pathway to drive tumorigenesis.2
Mutation of fibroblast growth factor receptor 2 (FGFR2) was identified in about 12~16 % cases of primary uterine tumors5 and endometrial tumors.6 In addition, Pollock et al. reported that activating mutations of FGFR2 are present in both type I and type II endometrial cancer.5 Byron and Pollock highlighted that FGFR2 could be a molecularly therapeutic target in endometrial cancers.7
Specific FGF ligand binding in complex with heparin sulfate leads to homodimerization of FGFRs, subsequently inducing autophosphorylation in the cytoplasmic kinase domain.8 FGFR substrate 2 (FRS2), a key adaptor protein of FGFRs, can dock onto FGFRs, which leads to the activation of downstream signaling pathway such as PI3K/Akt and Ras/ERK kinases.8 Aberrant activities of FGFRs are implicated in various pathological disorders including congenital skeletal disorder, and cancers. Historically, FGFR1 amplification and overexpression was found in 10% of breast cancer9 and 21% of lung squamous cell carcinomas (SCC), while FGFR3 was mutated in 30% of urothelial cancers.10 FGFR2 germ line mutations have also been observed in Pfeiffer syndrome, Apert syndrome and Crouzon syndrome, and FGFR2 somatic mutation identical to those germ line mutations have been detected in various cancers including endometrial cancer. Recently, somatic mutations in FGFR2 and FGFR3 as well as recurrent FGFR3-TACC3 fusion were identified as oncogenic alterations in lung SCC. Nowadays, FGFR4 is newly being spotlighted as a molecular target in a range of tumor types in prostate, breast, pancreatic, and liver tissues, with previously established high frequency in rhabdomyosarcoma (RMS), as well as recent discovery of FGFR4 as a mediator of drug resistance in colorectal cancer. Several types of FGFR inhibitors have been developed including ATP-competitive and irreversible inhibitor (FIIN-1) as well as ATP-competitive and reversible inhibitors (PD173074, BGJ398, dovitinib, AZD4547, LY2874455, ponatinib). It has been reported that PD173074, known as a selective pan-FGFR inhibitor, induces selective growth inhibition and apoptosis of gastric (KatoIII, Snu16, and OCUM-2M)11 and endometrial (MFE-296, MFE-280, and AN3CA) cancer cells.6,12 More recently, brivanib, a dual kinase inhibitor of VEGFR and FGFR, was evaluated to show significant response rate (18%) and progression free survival (30.2%) in phase II clinical trial for patients with recurrent endometrial carcinomas.
AP24534 (ponatinib, Fig. 1A) currently undergoing clinical trials is an orally available multi-targeted tyrosine kinase inhibitor. AP24534 displays highly potent activities against native Bcr-Abl as well as mt-Bcr-Abls including T315I gate-keeper mutant.13 AP24534 belongs to the type II ATP-competitive kinase inhibitor class and the piperazine-trifluoromethylbenzamide moiety of AP24534 binds to the additional hydrophobic pocket induced by the DFG-out (inactive) conformation located adjacent to ATP binding site of Abl kinase domain.9,10 It has also been reported that AP24534 also has the inhibitory activity in hematologic malignancies, including FLT3, Kit, FGFR1, and PDGFRα.14 In addition, kinase profiling revealed that AP24534 also possesses strong kinase inhibitory activities against FGFR1-4.13 Consistent with its in vitro kinase inhibitory activities on FGFRs, it has also been reported that AP24534 suppresses growth of stem cell leukemia/lymphoma (SCLL) by targeting FGFR1.15 Rivera and coworkers demonstrated that AP24534 reduces the tumor growth in FGFR2-amplified or mutated endometrial and gastric tumor xenograft model.16 It was also reported that ponatinib is capable of targeting wild-type and mutant FGFR4 in RMS.17 Ponatinib has also been shown to effectively suppress proliferation of Ba/F3 cells harboring dovitinib-resistant FGFR2 mutants, and exhibited in vivo efficacy on FGFR2-deregulated endometrial cancer xenograft model.18 Clinical trial of ponatinib is currently being conducted for patients with FGFR2 mutation-positive recurrent or persistent endometrial cancers (NCT01888562).
Figure 1.
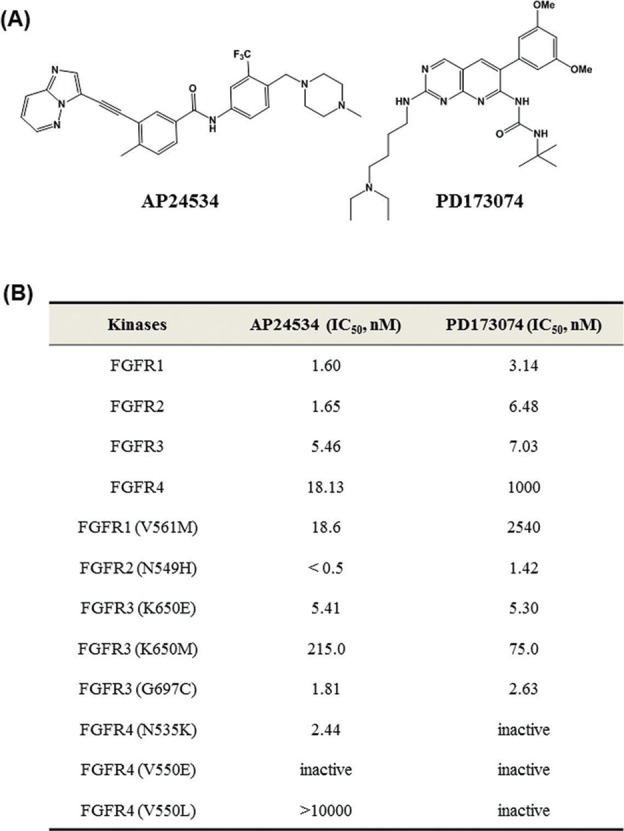
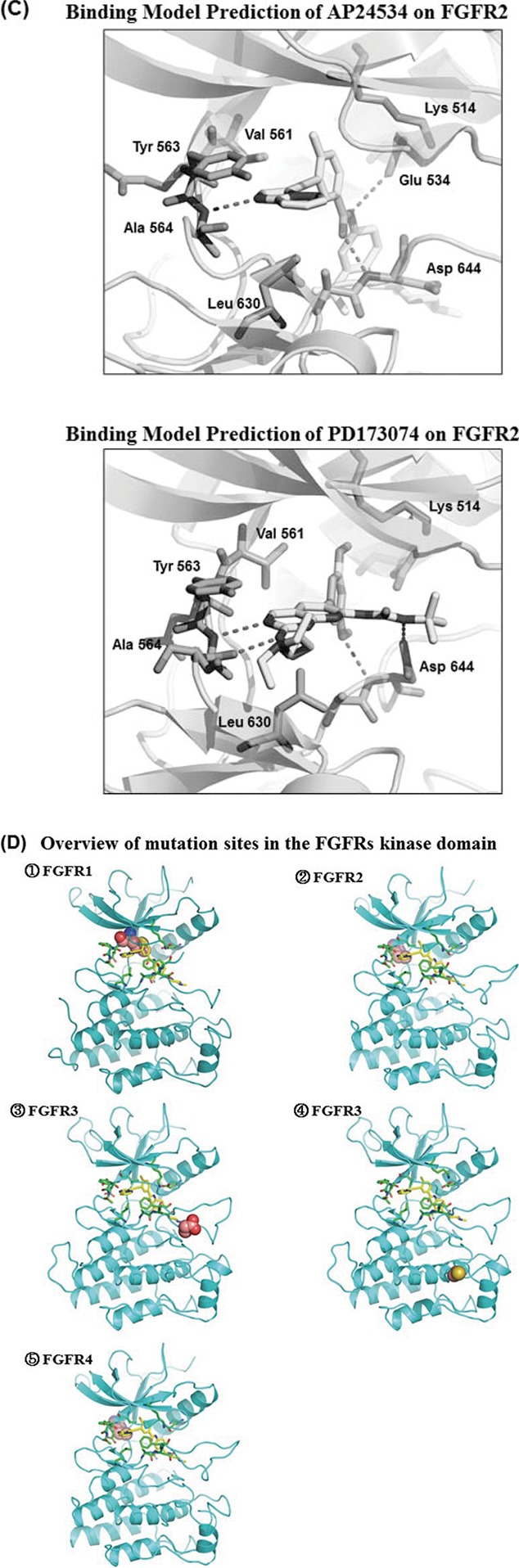
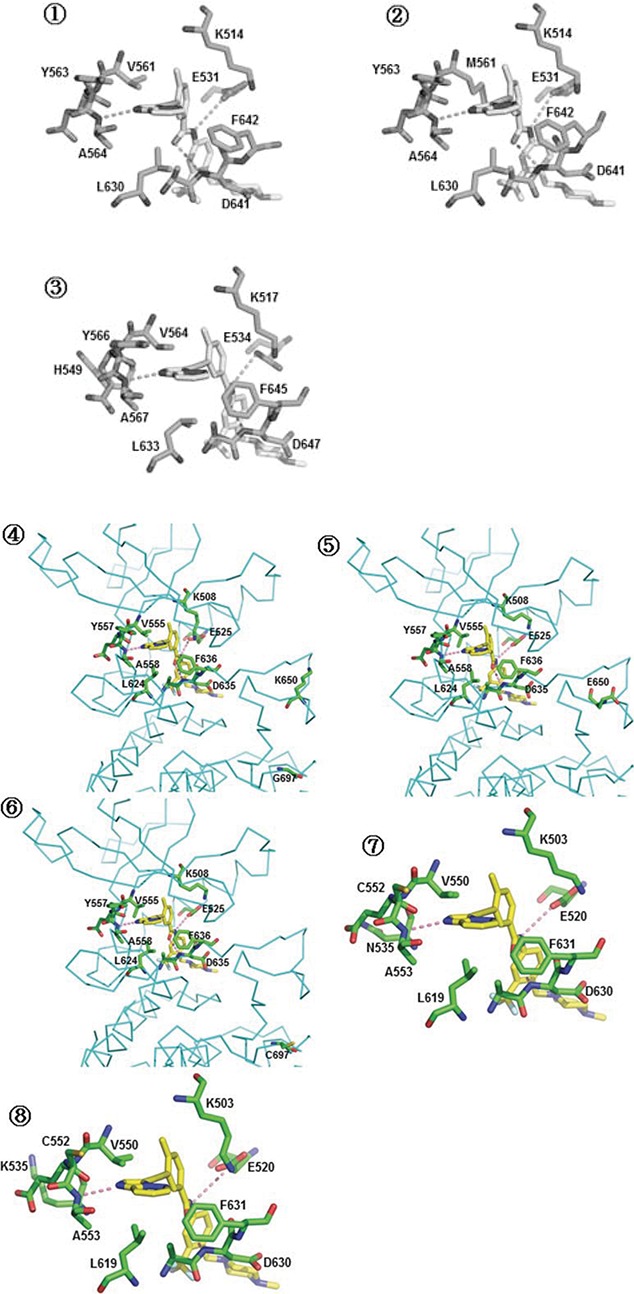
AP24534 inhibits in vitro kinase activity of FGFRs and the proliferation of endometrial cancer cells. (A) The chemical structure of AP24534 and PD173074. (B) IC50 values of AP24534 and PD173074 on wild-type FGFR1-4 and 8 mutant FGFRs kinase. In vitro kinase assays of FGFR were performed at Reaction Biology Corp. (C) Binding model prediction of AP24534 and PD173074 on FGFR2. (D) Overview of mutation sites in the FGFRs kinase domain. ① FGFR1 ② FGFR2 ③,④ FGFR3 ⑤ FGFR4 (E) Binding model prediction of AP24534 on wild-type (wt) and mutant (mt) FGFRs. ① wt-FGFR1 complex with AP24534 (pdb code: 4v04) ② Docking model of AP24534 on FGFR1 V561M mutant ③ Docking model of AP24534 on FGFR2 N549H mutant ④ wt-FGFR3 complex with AP24534 (Homology model) ⑤ Docking model of AP24534 on FGFR3 K650E mutant ⑥ Docking model of AP24534 on FGFR3 G697C mutant ⑦ wt-FGFR4 complex with AP24534 (pdb code; 4uxq) ⑧ Docking model of AP24534 on FGFR4 N535K mutant.
Figure 1.
(Continued).
Figure 1.
(Continued).
In this report, we for the first time present molecular mechanisms on how AP24534 strongly suppresses the proliferation of endometrial cancer cells harboring activating FGFR2 mutations.
Results
AP24534 strongly inhibits in vitro kinase activity of FGFR 1-4
Kinase inhibitory activities of AP24534 on wild-type FGFR 1-4 and 8 FGFR mutants (V561M-FGFR1, N549H-FGFR2, K650E-FGFR3, K650M-FGFR3, G697C-FGFR3, N535K-FGFR4, V550E-FGFR4, V550L-FGFR4) were assessed in biochemical kinase assay compared with those of PD173074, a potent and selective FGFR inhibitor. As shown in Fig. 1B, AP24534 is more potent on wild-type as well as mutant FGFRs enzymes than PD173074. Interestingly, among wild-type FGFR1-4, kinase inhibitory activity against FGFR4 of AP24534 was higher about 55-fold than PD173074. In addition, AP24534 also showed a remarkable kinase inhibitory activity (IC50 of 18.6 nM) against FGFR1-V561M gatekeeper mutant and this potency is higher over 100-fold compared with that of PD173074. In particular, both AP24534 and PD173074 showed the highest kinase-inhibitory activity on FGFR2-N549H mutant among all the wild-type and mutant FGFR1-4s tested. Mutations (K650E and K650M) at the lysine 650 residue of FGFR3 located in kinase domain result in the constitutive activation of the kinase19 and these mutants are associated with severe dwarfisms, TDII (thanatophoric dysplasia type II) and SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans).20,21 AP24534 displayed lower kinase inhibitory effect (IC50 of 215 nM) on FGFR3-K650M mutant than on other FGFR kinases. On the other hand, these 2 inhibitors exhibited great kinase-inhibitory activities against FGFR3-G697C mutant. The replacement of a glycine 697 residue by cysteine in the cytoplasmic kinase domain resulted in enhanced autophosphorylation activity, which may be caused by the ligand-independent dimerization of FGFR3.22 Most importantly, AP24534 displayed an IC50 value of 2.44 nM against FGFR4-N535K mutant whereas PD173074 was inactive on this mutant. It is of note that neither AP24534 nor PD173074 is capable of inhibiting kinase activity of FGFR4-V550 mutants (V550E and V550L). The point mutations occurred at Asn535 and Val550 within the hinge region of FGFR4 kinase domain are found in human rhabdomyosarcoma and exhibit an increased autophosphorylation through constitutive receptor activation.23 We performed molecular docking studies to predict binding modes of AP24534 and PD173074 on 9 wt- and mt-FGFR1-4s to explain its kinase-inhibitory activities.
Binding model prediction of AP24534 and PD173074 on FGFR2
The binding modes of AP24534 and PD173074 on FGFR2 based on docking studies are predicted in Fig. 1C. As shown in these predicted binding modes, AP24534 binds to FGFR2 in Type II binding fashion as AP24534 binds to Abl kinase.13 N1 on imidazo[1,2-b]pyridazine scaffold of AP24534 forms single hydrogen bond with Ala564 in the hinge region. The amide group of AP24534 makes a pair of hydrogen bonds with Asp644 backbone/Glu534 side chain of FGFR2. As for the predicted binding mode of PD173074 on FGFR2, N3 and 2-NH on pyrido[2,3-d]pyrimidine scaffold make 2 hydrogen bonds with Ala564 in the hinge segment. One of methoxy group of PD173074 interacts with the backbone NH of Asp644 while NH of urea group attached to tert-butyl group forms a hydrogen bond with the side chain carboxylate on Asp644. Our binding model analysis of AP24534 and PD173074 on FGFR2 explains how AP24534 and PD173074 tightly interact with the APT site of FGFR2, which leads to high potency of these 2 compounds against FGFR2. We also carried out docking studies on complexes between AP24534 and wt-FGFR1, V561M-FGFR1, N549H-FGFR2, wt-FGFR3, K650E-FGFR3, G697C-FGFR3, wt-FGFR4 and N535K-FGFR4. The locations of 5 point-mutations (V561M-FGFR1, N549H-FGFR2, K650E-FGFR3, G697C-FGFR3 and N535K-FGFR4) of FGFR1-4 are depicted in Fig. 1D. Analysis of the binding modes (Fig. 1E) predicted using these docking studies shows that AP24534 makes single hydrogen bond contact with the backbone NH of Ala564, A558 and A553 in the hinge region of FGFR1, FGFR3 and FGFR4, respectively. Also, the amide group of AP24534 forms a pair of hydrogen bonds with Asp641 backbone/Glu531 side chain, Asp635 backbone/Glu525 side chain and Asp630 backbone/Glu520 side chain in the kinase domain of FGFR1, FGFR3 and FGFR4, respectively. N549-FGFR2, making 3 internal critical hydrogen bonds with the backbone atoms of αC-β4 loop, is the molecular brake at the hinge region of FGFR2 and N549H mutation should loosen this molecular brake and cause gain of function in FGFR2.24 N535-FGFR4 is structurally corresponding to N549-FGFR2. Our docking models (Fig. 1E 3 and 8) reveal that AP24534 is capable of binding tightly to N549H-FGFR2 and N535K-FGFR4. Based on docking model of Fig. 1E (2), it is worthwhile to note that V561M, a gatekeeper mutant of FGFR1 would not encounter a steric collision with AP24534 but might make hydrophobic interaction with phenyl acetylene moiety of AP24534, which is consistent with the fact that AP24534 is potent (IC50 = 18.6 nM) on V561M-FGFR1 mutant in biochemical kinase assay. Taken together, analysis of our docking models (Fig. 1C and 1E) of AP24534 on 9 wt- and mt-FGFR1-4 explains how AP24534 could tightly interact with the kinase domain of these FGFRs.
Figure 3.
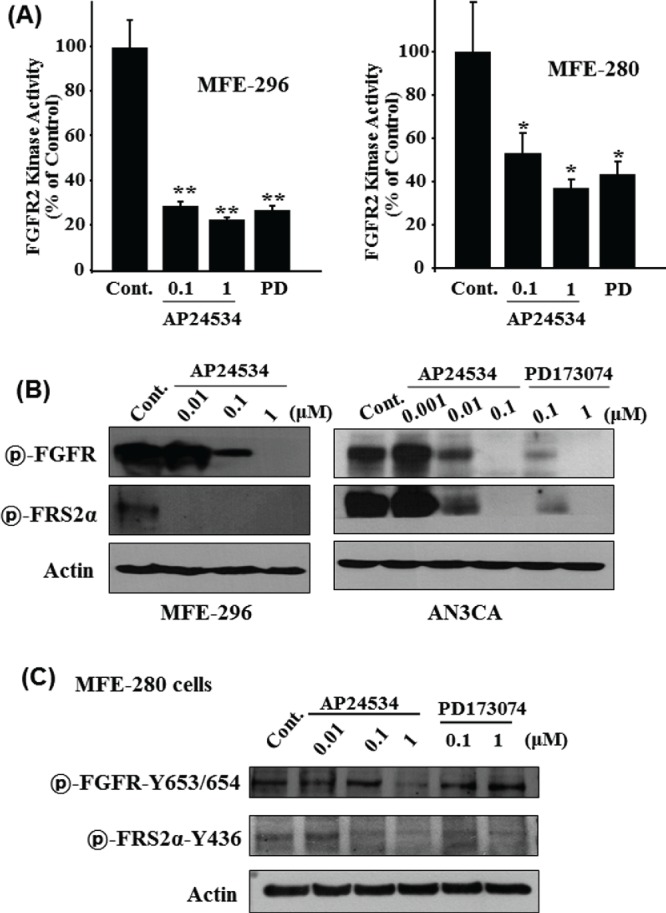
(A) MFE-296 cells were treated with the indicated concentrations of AP24534 (0.1 and 1 μM) or PD173074 (1 μM). FGFR2 was immunoprecipitated with FGFR2 antibody and FGFR2 kinase assay was performed as describe in Materials & Methods section. *, p < 0.01; **, p< 0.001; significantly different from vehicle control. (B, C) Cells were treated with indicated concentration of AP24534 or PD173074 for 48 h, and the phosphorylation of FGFR (Tyr653/654) and FRS2α (Tyr436) were assessed by Western blot analysis. Actin was measured for the confirmation of equal amounts of protein loaded.
AP24534 inhibits the proliferation of endometrial cancer cells
Treatment of human endometrial cancer cells (MFE-296, AN3CA, MFE-280 and Ishikawa) with AP24534 caused the decreased cell viability. AP24534 suppressed more strongly the growth of these endometrial cancer cell lines than PD173074 (Fig. 2A). As shown in the Fig. 2A, endometrial cancer cell lines (MFE-296, AN3CA, and MFE-280) harboring activating FGFR2 mutations are more sensitive to AP24534 as well as PD173074 rather than Ishikawa cell line having wild-type FGFR2. In particular, Ishikawa cells were very moderately affected by PD173074, a selective FGFR inhibitor. The survival of endometrial cancer cell lines harboring activating FGFR2 mutations might be more dependent on FGFR2 signaling compared with that of Ishikawa cell line expressing wild-type FGFR2.6 In contrast to endometrial cancer cell lines, normal human embryonic kidney (HEK293) cells were much less affected by AP24534 and PD173074. It is worthwhile to note that AP24534 and PD173074 displayed differential anti-growth effects on 3 endometrial cancer cell lines (MFE-296, AN3CA, and MFE-280) over HEK293, non-cancerous cell line. Both AP24534 and PD173074 exhibited higher antiproliferative effects on MFE-296 and AN3CA cell lines bearing FGFR2 kinase domain mutations than against MFE-280 cell line harboring S252W mutation in the extracellular domain of FGFR2. The anti-proliferative effect of AP24534 on the cell cycle distribution in endometrial cancer cells was assessed by FACS. Treatment with 0.1 μM AP24534 for 48 h caused an accumulation of MFE-296 and AN3CA cells predominantly in the G1 phase, with a concomitant decrease in the cell cycle progression through the S phase (Fig. 2B). In addition, AP24534 slightly induced the sub-G1 fraction, suggesting that it mainly induces G1/S cell cycle arrest, leading to apoptosis of MFE-296 and AN3CA cells.
Figure 2.
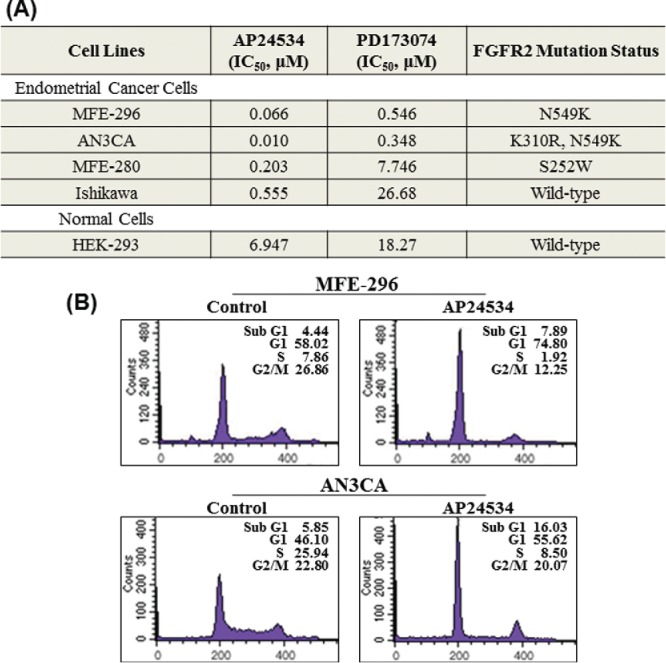
AP24534 inhibits the cell proliferation and FGFR2 kinase activity in endometrial cancer cells with activating FGFR2 mutations. (A) Cells were treated with indicated concentration of AP24534 for 48 h, and the cell viability was determined by the MTT reduction assay. We determined the value of IC50 of each compound. The concentration of chemicals producing 50% inhibition of cell growth (IC50) was estimated from semilog concentration-inhibition curves constructed using Prism software (GraphPad Software Inc..) (B) MFE-296 and AN3CA cells were treated with 0.1 μM of AP24534 for 48 h. The DNA content (propidium iodide) and cell cycle distribution was analyzed by flow cytometry as described in Material and Methods.
AP24534 inhibits the kinase activity of FGFR2 and its phosphorylation on FGFR2-activated endometrial cancer cells
We then investigated whether the anti-growth effect of AP24534 is attributed to the inhibition of FGFR2 in endometrial cancer cells bearing the FGFR2 mutations. As shown in Fig. 3A, AP24534 inhibited the kinase activity of immunoprecipitated FGFR2 derived from MFE-296 harboring N549K-FGFR2 mutation in a concentration-dependent manner and the kinase-inhibitory activity of AP24534 on FGFR2 is superior to that of PD173074 (1 μM). Compared with MFE-296, the proliferation of MFE-280 cells harboring S252W-FGFR2 mutation is less suppressed by AP24534 as well as PD173074. As expected, we found that the kinase-inhibitory activities of AP24534 and PD173074 on immunoprecipitated FGFR2 derived from MFE-280 cells are lower than those of AP24534 and PD173074 on immunoprecipitated FGFR2 derived from MFE-296 cells. These results suggest that the lower anti-proliferative activities of AP24534 and PD173074 on MFE-280 than against MFE-296 might arise from the differential kinase-inhibitory activities. Activated FGFR phosphorylates several tyrosine residues of FRS2, which is an adaptor protein that links FGFR activation to the MAP kinase signal transduction.8 In order to examine the effect of AP24534 on activation/phosphorylation of FGFR and FRS2α, we performed the western blot analysis. As a result, AP24534 exhibited a significant inhibitory effect on the phosphorylation of FGFR and FRS2α in MFE-296 and AN3CA cells in a concentration-dependent fashion (Fig. 3B). To a less extent, the level of phospho-FGFR and phospho-FRS2α is downregulated in MFE-280 by increasing concentration of AP24534 (Fig. 3C). In accordance with higher anti-proliferative activity of AP24534 on MFE-296, AN3CA and MFE280 as compared to that of PD173074, AP24534 inhibits more strongly the phosphorylation of FGFR and FRS2α in these 3 endometrial cancer cells rather than PD173074.
AP24534 inhibits the proliferation of endometrial cancer cells through ERK, Akt, PLCγ/PKC and STAT5 signaling pathway
Transphosphorylation of FGFRs has been known to cause the activation of 4 downstream pathways: RAS-RAF-MAPK, PI3K-Akt, signal transducer and activator of transcription (STAT) and phospholipase Cγ (PLCγ).8 To further elucidate the molecular basis for the AP24534-induced cell cycle arrest and apoptosis, we measured the expression as well as phosphorylation of ERK and Akt in endometrial cancer cells after AP24534 treatment of indicated concentration for 48 h. As shown in Fig. 4, ERK activation through phosphorylation was attenuated significantly by the treatment of AP24534 in MFE-296 (Fig. 4A), AN3CA cells (Fig. 4B) and MFE280 (Fig. 4C), while expression of total-ERK was not changed. PD173074 inhibited to a lesser extent phosphorylation of ERK than AP24534 on endometrial cancer cells. In addition, AP24534 treatment caused a modest reduction in phosphorylated Akt at serine 473 in MFE-296, AN3CA, and MFE280 cells while PD173074 exhibited no effect on Akt phosphorylation. The expression of PTEN, an inactivator of Akt/PKB, was not altered by AP24534 treatment in MFE-296 cells and MFE-280 (Figs. 4A, C). Furthermore, we examined whether AP24534 could modulate the activation of PLCγ/PKC and STAT signaling in endometrial cancer cells having FGFR2-N549K mutation. As shown in Fig. 4D, AP24534 significantly inhibited the phosphorylation of PLCγ, PKCα, and STAT5 in MFE-296 and AN3CA cells. On the other hand, AP24534 and PD173074 moderately inhibited the phosphorylation of PKCα in MFE280 cells (Fig. 4E).
Figure 4.
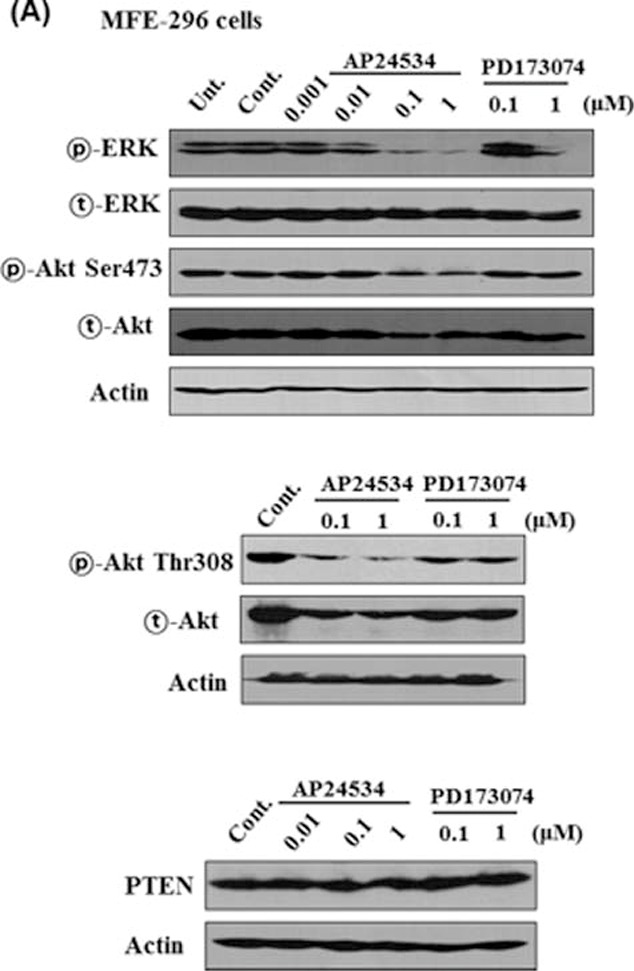
AP24534 decreases the phosphorylation of ERK and/or Akt in endometrial cancer cells. (A, C) MFE-296 cells and MFE280 cells were treated with indicated concentration of AP24534 and PD17307 for 48 h. Expression levels of both phosphorylated and total Akt and ERK1/2 were measured by Western blot analysis. PTEN expression was determined under the same experimental condition. (B) AN3CA cells were treated with AP24534 and PD173074 of the indicated concentrations for 48 h, and then activation/expression of Akt and ERK1/2 were examined by performing western blotting. (D) MFE-296 and AN3CA cells were treated with AP24534 of the indicated concentrations for 48 h, and then phosphorylation of PLCγ, PKCα, and STAT5 were assessed by immunoblot analysis. (E) In addition, phospho-level of PLCγ, and PKCα was analyzed in MFE280 under the same experimental condition.
Figure 4.
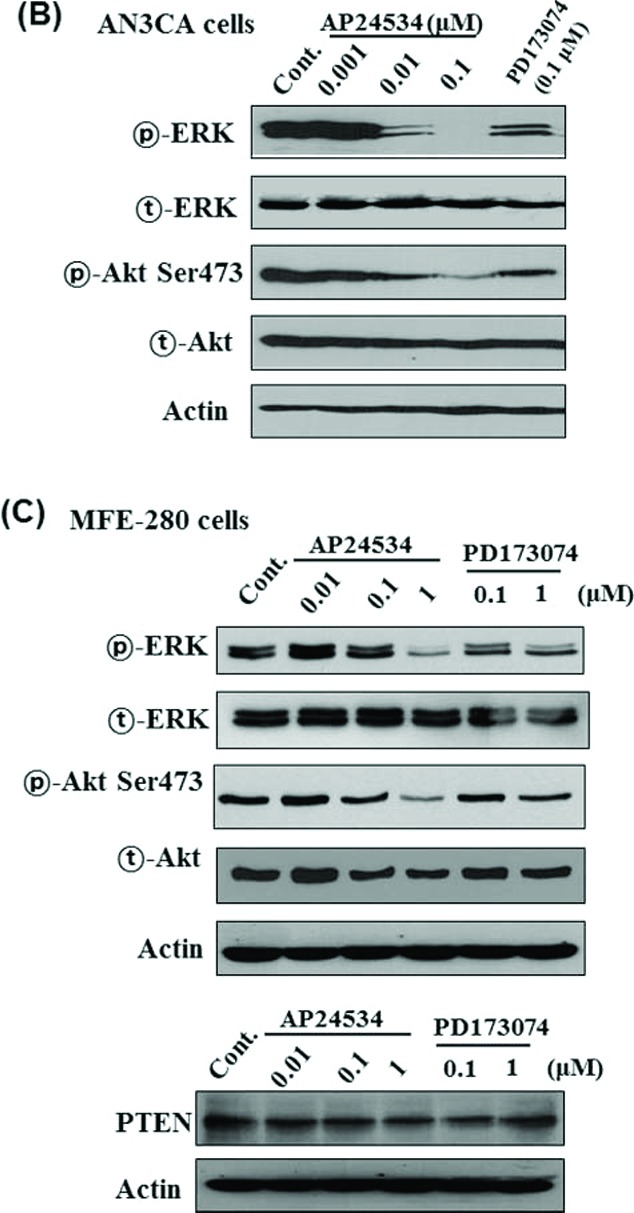
(Continued).
AP24534 inhibits the migration and invasion of endometrial cancer cells
PKCα has been known to be one of the important PKC isoforms for the progression of malignancies in cancer cell lines.25 Recently, Yang et al. reported that FGF2/STAT5 signaling cascade promotes angiogenesis in human brain endothelial cells. Thus, we examined whether AP24534 could block migration and invasion of endometrial cancer cells. We evaluated the inhibitory effect of AP24534 on migration and invasion of endometrial cancer cells using the wound-healing migration assay and Transwell invasion assay, respectively. As shown Fig. 5A, the migration capability of both MFE-296 and AN3CA cells was attenuated by AP24534 treatment. In addition, we further observed that AP24534 slightly reduces the invasion of AN3CA cells (Fig. 5B).
Figure 5.
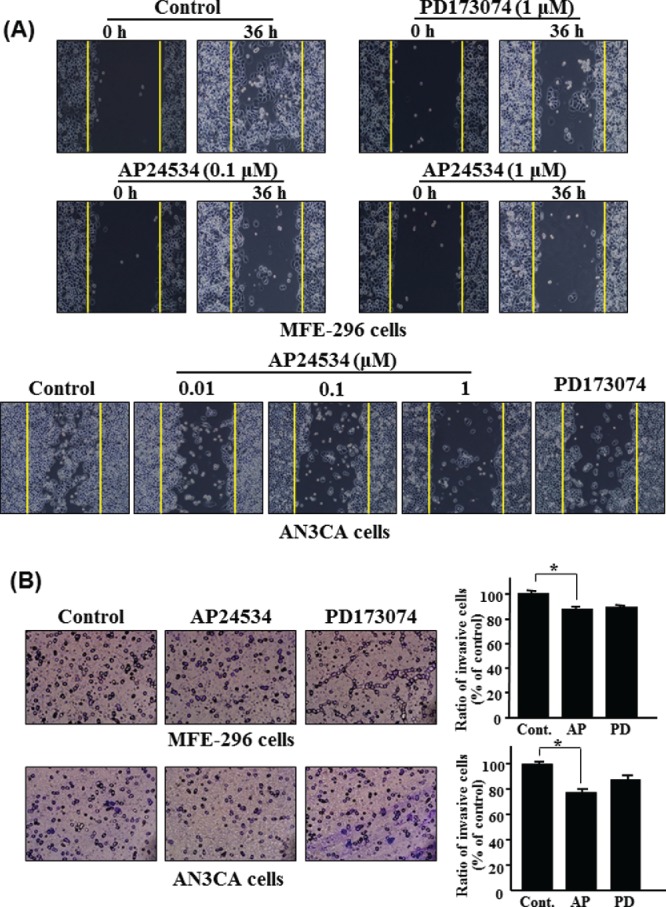
AP24534 inhibits the migration and invasion of endometrial cancer cells. (A) MFE-296 and AN3CA cells were treated with indicated concentration of AP24534 and PD173074. Cell migration was measured by using the Culture-Inserts, and wound closure was monitored by photography at 36 h following treatment with each compound. (B) After treatment of MFE-296 and AN3CA cells with AP24534 (1 μM) and PD173074 (1 μM) for 48 h, an in vitro invasion assay was performed using a 24-well Transwell unit with polycarbonate filters having a diameter of 6.5 mm and a pore size of 8.0 μm, and the number of invading cells per field was counted under light microscopy. *, significantly different between the groups compared (p < 0.01)
AP24534 abrogates the cell transformation of endometrial cancer cells
To clarify the potential therapeutic effect of AP24534 on endometrial cancer, we examined the inhibitory capability of AP24534 on anchorage-independent growth of MFE-296 and AN3CA cells. As illustrated in Fig. 6, MFE-296 cells and AN3CA cells exhibited significantly increased anchorage-independent growth for 3 weeks in 0.4% agar. In the presence of nanomolar concentration of AP24534, the colony formation was remarkably abolished, which is revealed by the reduced number of colonies (Fig. 6). Downregulation of FGFR2 signaling by AP24534 substantially inhibited anchorage-independent growth of MFE-296 and AN3CA cells.
Figure 6.
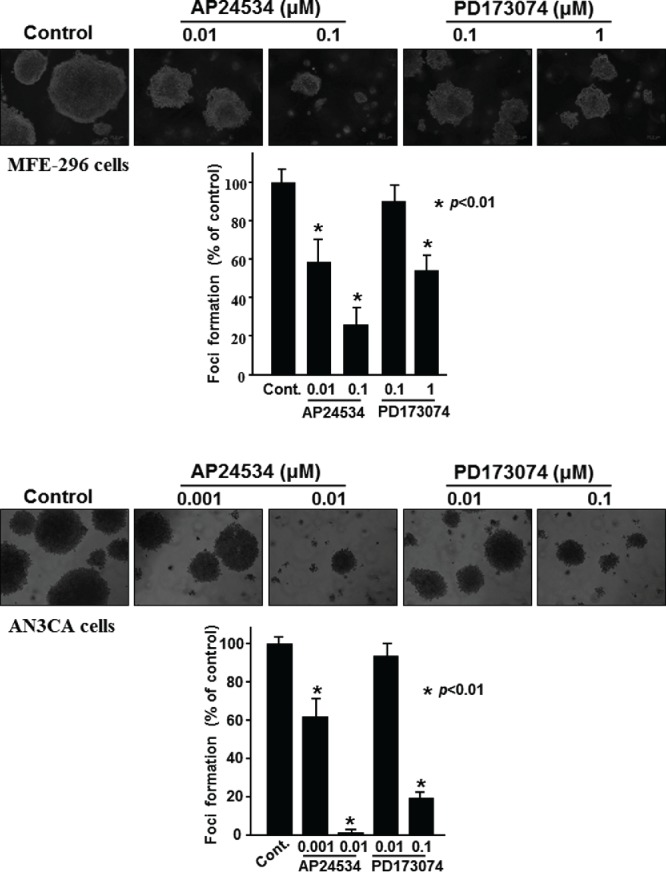
AP24534 inhibits the anchorage-independent growth of endometrial cancer cells. (A) MFE-296 cells were treated with AP24534 (0.01 and 0.1 μM) and PD 173074 (0.1 and 1 μM) twice a week for 3 wks in the soft-agar colony formation assay. (B) AN3CA cells were treated with AP24534 (0.001 and 0.01 μM) and PD173074 (0.01 and 0.1 μM) as described in Material and Methods. Colonies were counted using a digital microscope (Olympus DP71, USA) Colony counts from different treatment groups were subjected to statistical analysis. *, p < 0.01, significantly different from vehicle control.
Discussion
A various human tumors such as multiple myeloma, breast, prostate, colon, and endometrial cancers have been implicated the activating mutation and overexpression of FGFRs.5,6,8,26-28 Particularly, activating mutations of FGFR2 have been known to be associated with ~16% of endometrial cancers.6,7 Recently, Gatius et al. reported that endometrioid endometrial carcinomas show higher expression of FGFR2 than those of nonendometrioid.29 Authors suggested that FGFR2 has a dual function in endometrium. In normal endometrium, FGFR2 can inhibit cell proliferation during menstrual cycle, however, it is regarding as an oncogene in endometrial carcinoma.29 Knockdown of FGFR2 using shRNA vector and small molecule pan-FGFR inhibitor such as PD173074 resulted in cell cycle arrest and induction of cell death in endometrial cancer cells.7 Clinical evaluations targeting to EGFR, VEGFR and AKT/mTOR signaling pathways have been performed for endometrial cancers.30-32 However, the effect of these targeted therapies is not notable and ultimate so far. Activating FGFR2 mutation could be an important therapeutic target for endometrial cancers. Several other studies have attempted to elucidate the anti-tumor effects of AP24534 in vitro and in vivo. For example, Byron et al. reported that ponatinib inhibits the proliferation of Ba/F3 cells harboring dovitinib-resistant FGFR2 mutations.18 In addition, subcutaneous xenograft model of SNU16 (gastric cancer), UMUC14 (renal cancer), and AN3CA cells were demonstrated to inhibit tumor growth through downregulation of FGFR and FRS2 phosphorylation.16 Moreover, inhibition of FGFR2 and mTOR activity by combined oral treatment of both AP24534 and ridaforolimus in nude mice exerted to synergistic antitumor effect in endometrial cancer xenograft model.33
AP24534 currently undergoing clinical trials is a potent multi-targeted kinase inhibitor. In this study, we investigated antitumor effects and mechanisms of AP24534 on endometrial cancer cells harboring activating FGFR2 mutations. Our biochemical kinase assay data showed that AP24534 is potent against wild-type FGFR1-4 and 5 mutant FGFRs (V561M-FGFR1, N549H-FGFR2, K650E-FGFR3, G697C-FGFR3, N535K-FGFR4) and displays the strongest kinase-inhibitory activity on N549H-FGFR2 (IC50 of 0.5 nM) among all FGFRs tested. FGFR1-V561M that is gatekeeper mutant corresponding to Abl-T315I and EGFR-T766M mutants has been reported to confer the resistance to FGFR inhibitors including PD173074.34,35 N549H mutation in FGFR2 is associated with Crouzon Syndrome36 and this mutation activates the kinase in a ligand-independent manner by loosening the molecular brake at the kinase hinge region.24 Also worthy to note is that AP24534 exhibited an IC50 value of 2.44 nM against FGFR4-N535K mutant whereas PD173074 was inactive on this mutant. We then investigated cellular effects of AP24534 and PD173074 on MFE-296 (N549K), AN3CA (K310R/N549K), and MFE-280 (S252W) endometrial cancer cells bearing FGFR2-activating mutations. As presented in the Fig. 2A, MFE-296, AN3CA, and MFE-280 cells are more sensitive to AP24534 as well as PD173074 rather than Ishikawa cell line having wild-type FGFR2. AP24534 suppressed more strongly the growth of these endometrial cancer cell lines than PD173074. FACS analysis showed that AP24534 mainly induces G1/S cell cycle arrest, leading to apoptosis of MFE-296 and AN3CA cells.
As shown in Fig. 3, low concentration (0.1 μM) of AP24534 significantly inhibited the FGFR2 kinase activity as well as phosphorylation of FGFR and FRS2α in endometrial cancer cells harboring FGFR2-activating mutations. In addition, AP24534 distinctly decreased the phosphorylation of both ERK and Akt (Figs. 4A, B) in MFE-296 and AN3CA cells. By contrast, PD173074 did not affect the activation of Akt in MFE-296 and AN3CA cell lines, which is consistent with the report of Byron et al.12 AN3CA cells harboring FGFR2 double mutations (K310R & N549K) were the most sensitive to AP24534 among FGFR2-deregulated endometrial cancer cells tested (Fig. 2A). AN3CA cells were also sensitive to PD173074, which is consistent to the previous report12 even though AN3CA cells were less sensitive to PD173074 than to AP24534. We showed that AP24534 possesses the inhibitory effect on activation of PLCγ-PKCα as well as STAT5 phosphorylation in endometrial cancer cells harboring activating FGFR2 mutations. Attenuation of downstream signaling of FGFR2 by AP24534 accounts for its inhibitory migration effect on endometrial cancer cells.
FGFRs have been reported to be involved in various cellular processes such as apoptosis, proliferation, migration and angiogenesis.37 Inhibition of FGFR1 and FGFR2 activity in glioma C6 cells is associated with reduced tumor vascularization as well as expression of VEGF.38 We observed that AP24534 is capable of suppressing the migration and invasion of MFE-296 and AN3CA cells. These findings suggest that FGFR2 would be a valid target in inhibition of the migration and/or invasion capability of endometrial cancer cells harboring FGFR2-activating mutations. Taken together, this is undoubtedly that AP24534 possesses excellent inhibitory ability against FGFR2-deregulated endometrial cancer cells. In addition, antitumor effect of AP24534 against endometrial cancer cells with activating FGFR2 mutations might be mainly associated with the blockade of ERK, PLCγ and STAT5 signal transduction.
We, for the first time, present the molecular mechanisms by which AP24534 exerts antitumor effects on ECs having activating FGFR2 mutations, which would provide mechanistic insight into ongoing clinical trials of AP24534 for ECs.
Materials and Methods
Inhibitors
AP24534, 3(imidazo[1,2b]pyridazin 3 ylethynyl) 4 methyl N (4 ((4 methylpiperazin 1 yl)methyl) 3 (trifluoromethyl)phenyl)banzamide, and PD173074, N-[2-[[4-(diethylamino)butyl]amino]-6-(3,5,-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl]-N´-(1,1,-dimethylethyl)-urea, were synthesized in over 97% purity. All inhibitors were prepared as 10 mM DMSO stock solutions and stored at 20°C. Serial dilution of 10 mM DMSO stock solutions was carried out just prior to use in each experiment.
Materials
DMEM, RPMI1640, and fetal bovine serum (FBS) were purchased from WelGENE (Korea). Primary antibodies for phospho-Akt (S473), phospho-ERK (T202/Y204), Akt, phospho-FRS (Y436), phospho-FGFR (Y653/654), phospho-PLCγ (Y783), phospho-PKCα (S660), PTEN, and Actin were obtained from Cell Signaling Technology. The FGFR (Bek) antibody and secondary horseradish peroxidase-conjugated anti-rabbit/mouse antibodies were procured from Santa Cruz Biotechnology.
Biochemical in vitro kinase assay
The biochemical kinase-inhibitory activities of AP24534 and PD173074 on human FGFR1-4 protein kinases were performed at Reaction Biology Corp. (Malvern, PA, USA) using Kinase profiler assay.
Construction of FGFR2 homology model and docking study
The crystal structures of the FGFR2 tyrosine kinase domain reported to date are all in the DFG-in active conformation. In order to construction of FGFR2 kinase domain structure with the DFG-out conformation, we adopted the crystal structure of FGFR4 kinase domain bound with a DFG-out inhibitor AP24534 (pdb code: 4UXQ) as the most appropriate template. The FGFR2 kinase is composed of 289 amino acids. The Discovery Studio v4.5 (2015) was used for homology model construction (Biovia, San Diego, USA). Sequence analysis identified homologs for FGFR2 kinase sequences by searching the BLAST search (comparison matrix, BLOSUM 62; E-threshold, 10) using NCBI server. Finally, the modeled structure was refined by energy minimization for molecular docking.
For the binding model prediction of AP24534 and PD173074 on FGFR2 kinase domain, AP24534 and PD173074 were built using Maestro build panel and energy minimized using the Impact module in the Schrödinger software package. The starting coordinate of the FGFR2 kinase domain was further modified for docking calculation. The protein structure was minimized using the Protein Preparation Wizard by applying an OPLS force field. The prepared protein and the ligand were employed to build energy grids using the default value of protein atom scaling (1.0 Å) within a cubic box, defined as the centroid of the ATP binding pocket. After Grid generation, the ligand was docked with the protein by using Glide 6.0 module (Glide, Version 6.0, 2013) in extra precision mode (XP). The best-docked poses were selected as the lowest Glide score. The molecular graphics for the refined docking model of AP24534 and PD173074 with FGFR2 were generated using PyMol package (http://www.pymol.org).
MTT assays
Endometrial cancer (MFE-296) cells were maintained in 40% RPMI 1640 + 40% DMEM medium + 20% FBS. AN3CA and Ishikawa cells were cultured in DMEM enriched with 10% FBS, each containing 100 units penicillin and streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. Human cell lines were taken from culture substrate with 0.05% trypsin-0.02% EDTA and plated at a density of 2 × 103 cells/well in 96 well plates and then incubated at 37 °C for 24 hours in a humidified atmosphere with 5% CO2 prior to treatment of various concentration (10-fold serial dilution, 6 points) of test compounds. The cell viability was assessed by the conventional 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. MTT assays were carried out with CellTiter 96® (Promega, Madison, WI, US) according to the manufacturer's instructions. The absorbance at 590 nm was recorded using EnVision2103 (PerkinElmer; Boston, MA, US). The IC50 was calculated using GraphPad Prism 4.0 software.
Flow cytometry
Treated cells were trypsinized and washed with ice-cold PBS once, fixed by gentle addition of ice-cold 70% ethanol and incubated overnight at 4°C. The fixed cells were then washed with PBS and stained with 20 μg/ml of propidium iodide containing 10 μg/ml RNaseA. The stained cells were incubated at room temperature for 30 min in the dark. The DNA contents of the cells (1×104 cells/experimental group) were analyzed by a FACSCalibur flow cytometry using the CellQuest analysis program (Becton Dickinson Immunocytometry Systems, CA, USA).
Immunoprecipition and kinase assay
FGFR2 kinase activity was quantified by using a Universal Tyrosine Kinase Assay Kit (Takara) according to manufacturer's instructions. FGFR2 proteins were collected from the MFE-296 cell lysates by overnight immunoprecipitation with an anti-FGFR2 antibody (Bek C-17; Santacruze Biotechnology). The FGFR2 immune complexes were washed 3 times with PBS and diluted with kinase reaction buffer including 2-mercaptoethanol. Immobilized tyrosine kinase substrate (poly[Glu-Tyr] was incubated for 30 min at 37 °C with each sample in the presence of kinase-reacting solution and ATP. Addition of ATP solution starts phosphorylation of tyrosine. Samples were washed 4 times with washing buffer, blocked with blocking solution, and incubated for 30 min at 37 °C with anti-phosphotyrosine antibody (PY20) conjugated to horseradish peroxidase (HRP). The absorbance of the phosphorylated substrate was measured at 450 nm with plate reader.
Western blot analysis
Endometrial cancer cells were incubated with AP24534 or PD173074 for 48 h. Cells were lysed on ice for 30 min in cell lysis buffer [20 nmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L sodium vanadate, 1 μg/ml leupeptin, and 1 mmol/L phenylmethylsulfonyl fluoride] and centrifuged at 13,000 × g for 15 min. The protein concentration of the supernatant was measured by using the BCA reagents (Pierce, Rockfold, IL, USA). Protein was separated by running through 8-10% SDS-PAGE gel and transferred to the PVDF membrane (Gelman Laboratory, Ann Arbor, MI, USA). The blots were blocked with 5% non-fat dry milk-TBST buffer [TBS containing 0.1% Tween-20] for 1 h at room temperature. The membranes were incubated for 2 h at room temperature with 1:1000 dilution of each antibody. Equal lane loading was assured using actin (Chemicon, Millipore, CA). The blots were rinsed 3 times with TBST buffer for 10 min each. Washed blots were treated with 1:5000 dilution of the horseradish peroxidase conjugated-secondary antibody for 1 h and washed again 3 times with TBST buffer. The transferred proteins were visualized with an enhanced chemiluminescence detection kit (iNtRON Biotechnology, Seoul, Korea).
Wound migration assay
The migration of MFE-296 and AN3CA cells was measured by using the Culture-Inserts (Ibidi). Cells were seeded at a density of 3 × 105 cells/ml in each well of Culture-Inserts. After 24-h incubation, the Culture-Inserts were removed, and 500 μm cell-free gap were created. The dish containing these cells were then treated with the medium containing inhibitor and incubated at 37 °C. Phase contrast images of the closed gap were captured 0 h (control) and 36 h of incubation using an inverted microscope (magnification, 10x).
Invasion assay
The invasion of MFE-296 and AN3CA cells was measured by using the Transwell chambers (Chemicon, Millipore, CA) according to the manufacturer's protocol. Briefly, the cells were seeded onto the membrane of the upper chamber of the transwell at a concentration of 2 × 105/ml in 500 ul of serum free medium. The medium at the lower chamber contained inhibitors with 10% Fetal Bovine Serum as a source of chemoattractants. Cells that passed through the Matrigel coated membrane were stained with Cell Stain Solution containing crystal violet supplied in the Transwell Invasion assay (Chemicon, Millipore, CA) and photographed after 48 hours of incubation. The relative invasion was determined by counting the number of cells in 13 fields that invaded the Matrigel matrix relative to the number of cells that migrated through the control insert.
Anchorage-independent growth assay
MFE-296 and AN3CA cells suspended in 0.4% agarose solution were poured over hard-bottomed agar previously solidified in each 60 mm dish. Cells were exposed to different concentrations of AP24534 and PD173074 twice a week for 3 weeks and incubated at 37 °C in 95% air/5% CO2. After 3 weeks of incubation, anchorage-independent growth was scored using a light microscope. The experiments were replicated 3 times, and a representative set of data is photographed for presentation.
Statistical analysis
Values were expressed as the mean ±SE of at least 3 independent experiments. Statistical significance was determined by Student's t test and p < 0.05 was considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
The authors acknowledge to Dr. Heidi Greulich of Harvard Medical School, who provided MFE-296 cells and MFE-280 cells. In addition, we also thanks to Prof. Churl-Ki Min of Ajou University, who provided AN3CA cells.
Funding
This work was supported by Korea Institute of Science and Technology (KIST), and the Creative/Challenging Research Program (2011-0028676) of the National Research Foundation of Korea (NRF), a grant (D33400) of Korea Basic Science Institute, and Creative Fusion Research Program through the Creative Allied Project of the National Research Council of Science & Technology (CAP-12-1-KIST).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5-29; PMID:25559415; http://dx.doi.org/ 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol 2011; 8:261-71; PMID:21221135; http://dx.doi.org/ 10.1038/nrclinonc.2010.216 [DOI] [PubMed] [Google Scholar]
- 3.Bregar A, Robison K, Dizon DS. Update on the chemotherapeutic management of endometrial cancer. Clin Adv Heamatol Oncol 2014; 12:659-65; PMID:25658891 [PubMed] [Google Scholar]
- 4.O'Hara AJ, Bell DW. The genomics and genetics of endometrial cancer. Adv Genomics Genet. 2012; 2012:33-47; PMID:22888282; http://dx.doi.org/ 10.2147/AGG.S28953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007; 26:7158-62; PMID:17525745; http://dx.doi.org/ 10.1038/sj.onc.1210529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA 2008; 105:8713-7; PMID:18552176; http://dx.doi.org/ 10.1073/pnas.0803379105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byron SA, Pollock PM. FGFR2 as a molecular target in endometrial cancer. Future Oncol 2009; 5:27-3; PMID:19243295; http://dx.doi.org/ 10.2217/14796694.5.1.27 [DOI] [PubMed] [Google Scholar]
- 8.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010; 10:116-29; PMID:20094046; http://dx.doi.org/ 10.1038/nrc2780 [DOI] [PubMed] [Google Scholar]
- 9.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, Ellis IO, Reis-Filho JS. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res 2007; 9:R23; PMID:17397528; http://dx.doi.org/ 10.1186/bcr1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 1999; 23:18-20; PMID:10471491; http://dx.doi.org/ 10.1038/12615 [DOI] [PubMed] [Google Scholar]
- 11.Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, Elbi C, Lutterbach B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res 2008; 68:2340-8; PMID:18381441; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5229 [DOI] [PubMed] [Google Scholar]
- 12.Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA, Goodfellow PJ, Pollock PM. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res 2008; 68:6902-7; PMID:18757403; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0770 [DOI] [PubMed] [Google Scholar]
- 13.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009; 16:401-12; PMID:19878872; http://dx.doi.org/ 10.1016/j.ccr.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gozgit JM, Wong MJ, Wardwell SD, Tyner JW, Loriaux MM, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Druker BJ, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia (AML) and other hematologic malignancies. Mol Cancer Ther 2011; 10:1028-35; PMID:21482694; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia 2013; 27:32-40; PMID:22781593; http://dx.doi.org/ 10.1038/leu.2012.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 2012; 11:690-9; PMID:22238366; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0450 [DOI] [PubMed] [Google Scholar]
- 17.Li SQ, Cheuk AT, Shern JF, Song YK, Hurd L, Liao H, Wei JS, Khan J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS one 2013; 8:e76551; PMID:24124571; http://dx.doi.org/ 10.1371/journal.pone.0076551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, Blais SP, Neubert TA, Mohammadi M, Pollock PM. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 2013; 15:975-88; PMID:23908597; http://dx.doi.org/ 10.1593/neo.121106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webster MK, D'Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol Cell Biol 1996; 16:4081-7; PMID:8754806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwata T, Chen L, Li C, Ovchinnikov DA, Behringer RR, Francomano CA, Deng CX. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet 2000; 9:1603-13; PMID:10861287; http://dx.doi.org/ 10.1093/hmg/9.11.1603 [DOI] [PubMed] [Google Scholar]
- 21.Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet 1999; 8:35-44; PMID:9887329; http://dx.doi.org/ 10.1093/hmg/8.1.35 [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Hiraishi Y, Wang H, Sumi KS, Hayashido Y, Toratani S, Kan M, Sato JD, Okamoto T. Constitutive activating mutation of the FGFR3b in oral squamous cell carcinomas. Int J Cancer 2005; 117:166-8; PMID:15880580; http://dx.doi.org/ 10.1002/ijc.21145 [DOI] [PubMed] [Google Scholar]
- 23.Taylor JGT, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, Yu Y, Chen QR, Shah K, Youngblood V, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009; 119:3395-407; PMID:19809159; http://dx.doi.org/ 10.1172/JCI39703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell 2007; 27:717-30; PMID:17803937; http://dx.doi.org/ 10.1016/j.molcel.2007.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu TT, Hsieh YH, Hsieh YS, Liu JY. Reduction of PKC alpha decreases cell proliferation, migration, and invasion of human malignant hepatocellular carcinoma. J Cell Biochem 2008; 103:9-20; PMID:17486587; http://dx.doi.org/ 10.1002/jcb.21378 [DOI] [PubMed] [Google Scholar]
- 26.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 2007; 39:870-4; PMID:17529973; http://dx.doi.org/ 10.1038/ng2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res 2001; 61:3541-3; PMID:11325814 [PubMed] [Google Scholar]
- 28.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000; 7:165-97; PMID:11021964; http://dx.doi.org/ 10.1677/erc.0.0070165 [DOI] [PubMed] [Google Scholar]
- 29.Gatius S, Velasco A, Azueta A, Santacana M, Pallares J, Valls J, Dolcet X, Prat J, Matias-Guiu X. FGFR2 alterations in endometrial carcinoma. Mod Pathol 2011; 24:1500-10; PMID:21725289; http://dx.doi.org/ 10.1038/modpathol.2011.110 [DOI] [PubMed] [Google Scholar]
- 30.Oza AM, Eisenhauer EA, Elit L, Cutz JC, Sakurada A, Tsao MS, Hoskins PJ, Biagi J, Ghatage P, Mazurka J, et al. Phase II study of erlotinib in recurrent or metastatic endometrial cancer: NCIC IND-148. J Clin Oncol 2008; 26:4319-25; PMID:18591547; http://dx.doi.org/ 10.1200/JCO.2007.15.8808 [DOI] [PubMed] [Google Scholar]
- 31.Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, Walker C, Ramondetta LM, Burke TW, Gershenson DM, et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 2010; 116:5415-9; PMID:20681032; http://dx.doi.org/ 10.1002/cncr.25515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stefansson IM, Salvesen HB, Akslen LA. Vascular proliferation is important for clinical progress of endometrial cancer. Cancer Res 2006; 66:3303-9; PMID:16540684; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1163 [DOI] [PubMed] [Google Scholar]
- 33.Gozgit JM, Squillace RM, Wongchenko MJ, Miller D, Wardwell S, Mohemmad Q, Narasimhan NI, Wang F, Clackson T, Rivera VM. Combined targeting of FGFR2 and mTOR by ponatinib and ridaforolimus results in synergistic antitumor activity in FGFR2 mutant endometrial cancer models. Cancer Chemother Pharmacol 2013; 71:1315-23; PMID:23468082; http://dx.doi.org/ 10.1007/s00280-013-2131-z [DOI] [PubMed] [Google Scholar]
- 34.Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, Kéri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol 2004; 11:691-701; PMID:15157880; http://dx.doi.org/ 10.1016/j.chembiol.2004.02.029 [DOI] [PubMed] [Google Scholar]
- 35.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010; 2:62ra93; PMID:21160078; http://dx.doi.org/ 10.1126/scitranslmed.3001451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev 2005; 16:187-203; PMID:15863034; http://dx.doi.org/ 10.1016/j.cytogfr.2005.03.001 [DOI] [PubMed] [Google Scholar]
- 37.Acevedo VD, Ittmann M, Spencer DM. Paths of FGFR-driven tumorigenesis. Cell Cycle 2009; 8:580-8; PMID:19182515; http:dx.doi.org/ 10.4161/c1.6.4.7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auguste P, Gursel DB, Lemiere S, Reimers D, Cuevas P, Carceller F, Di Santo JP, Bikfalvi A. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res 2001; 61:1717-26; PMID:11245488 [PubMed] [Google Scholar]