

Abstract
Chemical biologists commonly seek out correlations between the physicochemical properties of molecules and their behavior in biological systems. However, a new paradigm is emerging for peptides in which conformation is recognized as the primary determinant of bioactivity and bioavailability. This review highlights an emerging body of work that directly addresses how a peptide’s conformation controls its biological effects, cell penetration, and intestinal absorption. Based on this work, the dream of mimicking the potency and bioavailability of natural product peptides is getting closer to reality.
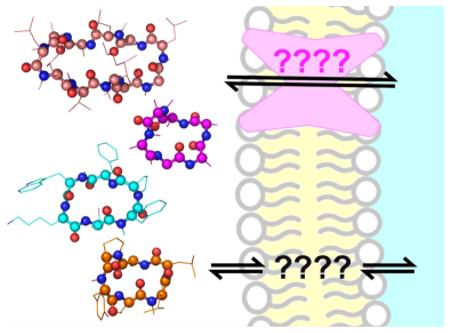
Discovering new probes and lead compounds for specific biological targets is a complex endeavor – so many molecules, so little time! To reduce this complexity and to promote bioactivity and bioavailability, chemical space can be filtered using parameters such as size, hydrophobicity, and hydrogen-bonding propensity.1,2 Applying these guidelines to small molecules can be informative, but there are no comparable guidelines for understanding the suitability of peptides as drugs. However, a growing body of work is revealing that, for peptides and their analogues, conformation plays a predominant role in determining their biological effects. Increasingly, previous “exceptions” to physicochemical rules are being evaluated as general scaffolds for highly bioactive and bioavailable molecules. In this Review, we highlight recent findings from a variety of fields that are converging on a new understanding of how conformation controls peptide bio-activity and bioavailability. This research is galvanizing academic and industrial chemists alike and driving a new wave of research into modified peptides as probes and therapeutics.
Bioactivity is the ability of a molecule to have a specific biological effect when introduced to a living system such as cultured cells or a whole organism. The bioactivity of most compounds is dictated by their specific targets and binding modes, but overarching trends can be observed for specific “privileged” compound classes such as benzodiazepines and certain natural product macrocycles.3–5 As a class, peptides can be bioactive against diverse targets because they can assume a variety of conformations. However, this flexibility can be a drawback, leaving peptides vulnerable to proteolytic degradation. Bioavailability is the extent to which a molecule, once introduced to a living organism, reaches its physiological target and persists long enough to have an effect. The major components of bioavailability through oral administration are breakdown in the gastrointestinal tract, gut absorption, distribution into circulation and physiological compartments, metabolism, and elimination. For peptides, each of these is a formidable obstacle.
Controlling biology with peptides is a century-old field,6,7 and great strides have been made in the fields of peptidomimetics and peptide drug development.8,9 Though we have come far, we have not yet matched the activities of orally bioavailable natural products. Only recently have studies directly addressed the factors that govern peptide bioactivity and bioavailability, and the emerging results indicate that modern tools for controlling conformation may finally unlock the full potential of peptides as biological agents.
TWO LESSONS FROM NATURAL PRODUCTS
Medicinal chemistry can be seen as the short-term effort of a few chemists to imitate the long-term efforts of an army of microbes; this is certainly true for the field of peptide design. α-amanitin, for example, is an astonishingly potent, bioavailable (and poisonous) natural product (Figure 1).10,11 α-amanitin acts by potently inhibiting RNA polymerase II, and its striking bioavailability (oral LD50 is less than 0.1 mg/kg; see Table 1) has prompted detailed investigations into its pharmacokinetics.12 Its mode of gut absorption is unknown, but once absorbed it concentrates in the liver because of facilitated diffusion by bile acid transporters.13 Structure–activity relationships reffect the known structure of the amanitin/RNA polymerase complex, solved by Kornberg in 1970,14 but also hint at specific conformational effects on bioactivity and bioavailability.12 Solution and crystal structures indicate that amanitin’s bicyclic scaffold is relatively rigid, and large perturbations to its rigidified structure, such as opening one of the rings, leads to complete loss of bioactivity and bioavailability. More subtle structural alterations, such as reduction, oxidation, or changes within the cross-link, have more subtle effects on overall toxicity that are not easily attributed to changes in target binding. Thus, α-amanitin’s locked conformation appears to promote not only potent, selective RNA polymerase binding but also gut absorption and facilitated transport into hepatocytes.
Figure 1.

“To be steady as a rock and always trembling.”144 α-Amanitin and cyclosporine A provide contrasting lessons from natural products. Both are head-to-tail cyclic peptides, and deviations from the 20 proteinogenic amino acids are shown in red. α-Amanitin is locked into a single conformation by virtue of a sulfone-indole intramolecular cross-link. This protects it from proteolytic degradation despite having a largely unmodified peptide backbone and appears to promote gut absorption and transport into liver cells by organic anion transport proteins (OATPs).12 Cyclosporine, by contrast, survives digestive proteases by virtue of its highly N-methylated backbone. It can change conformations in order to form intramolecular hydrogen bonds in nonpolar environments.23 This is hypothesized to promote passive diffusion through plasma membranes.
Table 1.
Bioactivity and Bioavailability of Selected Constrained Peptides and Controlsa
| name | no. of amino acids |
constraint | in vitro activity | cell penetration | activity/bioavailability | refs |
|---|---|---|---|---|---|---|
| α-amanitin | 8 | backbone cyclic, indole-sulfone cross-link |
Ki = 2 × 10−8 M for RNA polymerase II | ND | LD50 ≈ 0.1 mg/kg p.o. | 10, 12 |
| cyclosporine A | 11 | backbone cyclic | NA | 1.1 × 106 cm/s in RRCK cells | oral bioavailability 27% in rat | 24 |
| cyclo(-Leu-NMeDLeu-NMe- Leu-Leu-Pro-NMeTyr-) |
6 | backbone cyclic | NA | 4.9 × 106 cm/s in RRCK cells | oral bioavailability 28% in rat | 24 |
| Veber–Hirschmann peptide cyclo(-Pro-Phe-DTrp-Lys- Thr-Phe-) |
6 | backbone cyclic | Kd = 9.8 nM (hsst2), 15.1 nM (hsst5) | 5 × 105 cm/s in Caco-2 cells | oral bioavailability in rat negligible | 36 |
| cyclo(-Pro-Phe-NMeDTrp- NMeLys-Thr-NMeDPhe-) |
6 | backbone cyclic | Kd = 61.7 nM (hsst2), 60.3 nM (hsst5) | 4 × 106 cm/s in Caco-2 cells | oral bioavailability 9.9% in rat | 36 |
| cyclo(-NMeDAla-Ala-Ala- Ala-NMeAla-Ala-) |
6 | backbone cyclic | NA | ~2–3 × 105 cm/s in Caco-2 cells and in ex vivo rat intestine | ND | 134 |
| α-conotoxin Vc1.1 | 16 | two disulfides | IC50 = 1.7 nM for inhibition of Ca2+ channel currents in rat neurons |
ND | some activity in 0.36 and 3.6 μg i.m. boluses in rat models of neuropathic pain |
145 |
| cyclic Vc1.1 | 22 | two disulfides, backbone cyclic |
IC50 = 0.3 nM for inhibition of Ca2+ channel currents in rat neurons |
ND | 1.3 mg/kg p.o. as effective as 30 mg/kg gabapentin in rat model of neuropathic pain |
56 |
| des-Arg10-[Leu10]-kallidin | 9 | none | ND | ND | no effect p.o., 38% inhibition i.p. (1 mg/kg) in a rat model of visceral pain |
63 |
| Ckb-kal | 31 | four disulfides, backbone cyclic |
IC50 ≈ 0.2–0.4 μM for competition at the bradykinin receptor |
ND | 49% inhibition p.o., 42% inhibition i.p. (1 mg/kg) in a rat model of visceral pain |
63 |
| gp41-derived variant T649V626-662 |
37 | none | IC50 = 2.9 nM and >3000 nM for in vitro
HIV infectivity of HXBc2 and YU2 strains, respectively |
NA | None detected in plasma after 10 or 20 mg/kg p.o. |
102 |
| “stapled” gp41 variant T649V626-662 |
37 | two side chain olefin cross-links |
IC50 = 2.5 nM and 87 nM for in vitro HIV infectivity of HXBc2 and YU2 strains, respectively |
NA | 1.5 μg/mL and 2.3 μg/mL detected in plasma after 10 or 20 mg/kg p.o., respectively |
102 |
| Bid BH3 helix | 24 | none | Kd = 269 nM for Bcl-XL binding in vitro | no cell penetration or effects on leukemia cells in vitro | ND | 95 |
| “stapled” Bid BH3 helix | 24 | one side chain olefin cross-link |
Kd = 39 nM for Bcl-XL binding in vitro | cell penetration observed with dye-labeled analogues; IC50 = 1.6–10.2 μM for killing leukemia cells in vitro |
median survival increased from 5 to 11 days in mouse leukemia model, i.v. 10 mg/kg |
95 |
| HBS3 | 16 | Kd = 28 μM for nucleotide-free Ras | cell penetration observed with dye-labeled analogues; 75 μM peptide reduced Ras activation 5-fold in HeLa cells |
ND | 109 |
Abbreviations: p.o. (peroral administration), i.v. (intravenous injection), i.m. (intramuscular injection), i.p. (intraperitoneal injection). NA = not applicable. ND = not determined. hsst2 and hsst5 refer to different human somatostatin receptors.
Cyclosporine is another defining example of a peptide natural product with surprising bioactivity and bioavailability (Figure 1).15 After 40 years of research, we still lack definitive understanding of the mechanisms of gut uptake and cell penetration by cyclosporine. However, there is a great deal of indirect evidence that indicates a unique model for these processes. In aqueous solvent, cyclosporine assumes a conformation similar to that observed in the co-crystal structure of the cyclosporine/cyclophilin complex.16–21 In organic solvent, however, it inverts to form internal backbone hydrogen bonds, with its N-methylated amino acids pointing outward.22 This ability to assume a different conformation in hydrophobic environments is hypothesized to promote passive diffusion across plasma membranes.23 Practicable strategies for mimicking this “shapeshifting” capability have lagged due to the synthetic challenges of peptide cyclization and N-methylation, and the need to apply rigorous models of cell penetration and tissue absorption. Only recently have these obstacles been overcome, allowing for design and screening for model peptides that appear to diffuse through membranes in a cyclosporine-like manner (see below).24,25
Our current understanding of α-amanitin and cyclosporine provide contrasting examples of how conformation controls peptide bioactivity and bioavailability (Figure 1). In the case of cyclosporine, a flexible structure allows for a large, hydrophilic binding interface that can be conformationally “hidden” in order to promote passive diffusion. In α-amanitin’s case, nature has evolved a single rigid structure that avoids metabolic modification and degradation, takes advantage of existing transporters, and binds its target with high affinity. As we describe below, chemical biologists have taken both approaches to developing peptide probes, and either approach may be used to maximize bioactivity and bioavailability.
PEPTIDE ENGINEERING WITH SMALL EPITOPES
Given the critical role of conformation in molecular recognition, peptide chemists have long sought to improve natural peptides using conformational constraints. Throughout the late 1970s and 1980s, pioneers such as Hirschmann, Hruby, and Kessler used conformational constraints to recapitulate the active conformations of potent peptide hormones.26–31 In doing so, they revealed critical information on the binding modes of oxytocin, enkephalin, somatostatin, melanocortin, and other hormones and on the mechanisms of their associated receptors. Constrained analogues of these hormones became important probes for understanding endocrine signaling pathways and in some cases were developed as pharmaceuticals. A particularly illustrative example is the development of somatostatin analogues, recently reviewed by Ovadia et al.32 Somatostatin, discovered in 1972, is a hormone that counteracts the effects of human growth hormone.33 Subsequent analysis of the structural and conformational requirements for its activity by Veber, Hirschmann, and colleagues led to the design of potent constrained peptide analogues.27,34,35 This paved the way for the development of somatostatin analogues
A particularly illustrative example is the development of somatostatin analogues, recently reviewed by Ovadia et al.32 Somatostatin, discovered in 1972, is a hormone that counteracts the effects of human growth hormone.33 Subsequent analysis of the structural and conformational requirements for its activity by Veber, Hirschmann, and colleagues led to the design of potent constrained peptide analogues.27,34,35 This paved the way for the development of somatostatin analogues as treatments for acromegaly and other conditions that arise from an excess of growth hormone activity.33 These drugs are minimized forms of somatostatin that contain a disulfide-mediated macrocycle and artificial amino acids including a critical, turn-promoting D-tryptophan. These modifications promote a bioactive conformation and prolong the half-life in the blood, which allows for intravenous administration. Recent work has continued this progress, with a structure-guided N-methyl scan of a backbone-cyclic analogue resulting in a triply N-methylated version with substantial oral bioavailability (Figure 2a and Table 1).32,36 While the ability of the N-methylated peptide to undergo conformational change in hydrophobic environments was not addressed, the authors noted that the specific N-methylations that promoted bioavailability also appeared to stabilize the conformation responsible for potent bioactivity. Thus, evidence to date implies that, like α-amanitin, stabilized somatostatin analogues derive both target binding and bioavailability from a single, rigidified conformation.
Figure 2.

Engineered peptides with high bioactivity and/or bioavailability. These constrained peptides vary greatly in size and hydrophobicity and employ different chemical cross-links, cyclizations, and folding topologies. (a) Somatostatin mimic with ~60 nM binding affinity to human somatostatin receptors sst2 and sst5. This compound permeates Caco-2 monolayers and has 7% oral bioavailability in rats.36 (b) Caco-2-penetrant cyclic peptide scaffold found by Kessler, Hoffman, and co-workers.135 (c) Cyclic peptide scaffold found by Lokey and co-workers to have 28% oral bioavailability in mice.24 (d) Cyclized α-conotoxin that targets GABAB receptors and acts as an analgesic. Head-to-tail cyclization resulted in oral bioavailablility as judged by effects on pain-related phenotypes in rats.56 (e) Grafted analogue of kalata B that acts as an orally bioavailable analgesic that targets bradykinin receptors.63 (f) Stapled helix of Bid that is able to slow proliferation of leukemia xenografts.95 (g) Hydrogen-bond-surrogate helix that can penetrate cells and inhibit Ras.109
Similar optimization methodologies have been applied to other peptide hormones and small binding epitopes. Peptide ligands for melanocortin receptors were developed by cyclizing fragments of natural hormones to improve bioactivity, then N-methylating to enforce selectivity for specific receptor 37–40 One cyclic peptide that mimics α-melanocyte-stimulating hormone was shown to be orally bioavailable and effective as an antiobesity drug in rats.41 Another important example is the development of cyclic peptides that incorporate the RGD sequence from fibronectin as potent and selective integrin ligands.42,43 Several series of analogues were explored that first varied ring size and the placement of a D-amino acid and then later exhaustively searched for N-methylated analogues with improved selectivity and bioavailability.44–47 RGD-derived cyclic peptides are now standard tools for imaging, targeting, and manipulating integrins, and the RGD peptide Cilengitide is currently being tested as an antiangiogenic agent for the treatment of glioblastoma.48,49
All together, these studies share a long-term methodology: first cyclize the backbone to stabilize a biologically relevant conformation, then make rational substitutions with natural and unnatural amino acids to introduce distinct conformational preferences that promote target affinity and desired physical properties. Historically, N-methylation was applied in later stages, perhaps due to the difficulty of the chemistry involved, though thanks to the work of Kessler and others, synthesis of N-methylated peptides is more accessible than ever before.26,50–52 The effects of cyclization, substitution and N-methylation are typically non-additive and highly cooperative, making it difficult to predict or rationalize the results of even simple series of analogues. Even so, these “scanning” methodologies have established that conformational control of peptide bioavailability is possible, and indeed may be possible in most cases in which a small, contiguous epitope is found to mediate bioactivity.
HIGHLY CONSTRAINED NATURAL PRODUCT PEPTIDES: LOCKED AND LOADED
Recent developments using highly constrained natural products have provided a striking counterpoint to the previous work on smaller cyclic peptides.53–55 In a startling report in 2010, Craik and co-workers reported that the α-conotoxin Vc1.1, a venom peptide produced by predatory cone snails, can be engineered to be orally active in rats using backbone macrocyclization.56 Vc1.1 is a 16-residue peptide with a short internal α-helix, an amidated C-terminus, and two intramolecular disulfide bonds. It is a potent analgesic that acts through GABAB receptors, though its specific mode of action is unclear. In designing the cyclic conotoxin, Craik and co-workers used previously published work on protein and peptide cyclization to derive a linear relationship between the distance between termini and the number of residues in a successful linker.56 On the basis of this empirical correlation, they tested cyclic Vc1.1 analogues with simple five-residue and six-residue linkers composed of glycine and alanine and found the six-residue linker afforded a molecule with optimal activity (Figure 2d). This cyclic Vc1.1 was more potent than linear Vc1.1 as a GABAB-mediated calcium channel blocker and was also more selective for this effect over inhibition of nicotinic acetylcholine receptors. Most importantly, cyclic Vc1.1 showed dose-dependent relief of neuropathic pain in rats when administered orally. Its activity at 1.3 mg/kg (see Table 1) was similar to the activity of gabapentin, a commonly prescribed oral analgesic, at 30 mg/kg, highlighting the potency and bioavailability of the cyclic conotoxin.
While the most advanced applications of natural product peptides are targeting their native receptors, significant efforts are being made to adapt these peptides as designer scaffolds for inhibition of other targets. Example scaffolds include highly disulfide-bonded scaffolds such as animal defensins and plant cyclotides,53–61 as well as naturally knotted “lasso” peptides.62 In a striking example, Tam and co-workers recently described an orally active bradykinin receptor antagonist based on the highly constrained peptide kalata B1.63 Kalata B1 is a member of the cyclotide family of natural products, which are plantderived head-to-tail cyclic peptides with multiple disulfides in a “cystine knot” topology.55,64 Kalata B1 is itself orally bioavailable; it is the active ingredient responsible for the oxytocin-like activity of an herbal tea prepared by native Congolese for inducing labor.65 Key sequences from established bradykinin receptor antagonist peptides were “grafted” within a loop of the 29-residue kalata B1. The most successful analogue (Figure 2e) showed pain relief in mice when orally administered at 1 mg/kg (Table 1). Equal effects were observed for intraperitoneal injection and oral administration, demonstrating that digestive tract stability and intestinal absorption were not the primary limiting factors for the observed pain relief. Taken together with studies on α-conotoxin work and other venom peptides and cyclotides, it is clear that larger, highly constrained cyclic peptides can be as potent and bioavailable as smaller cyclic peptides.
There are a few clues to explain the exceptional properties of these highly constrained peptides. Backbone cyclization is clearly important, but the effects of masking and restricting the termini cannot be understood solely through estimates of hydrophobicity or hydrogen-bonding potential. These cyclic peptides appear to be genuine “privileged structures,” because the structure of the cyclic Vc1.1 peptide was revealed to be nearly identical to that of the linear form,56 and NMR chemical shift data indicate that the grafted kalata analogue has a fold similar to that of Kalata B1.63 In fact, greater overall rigidity of the bioactive peptide epitope seems to affect binding in a receptor-specific way, allowing for selectivity tuning based on conformational restriction.56 It is also likely that, even for highly constrained peptides, further rigidification can reduce dynamic “breathing” of the structure, resulting in a longer overall halflife. This was observed directly for cyclic conotoxins, which had reduced inactivation due to disulfide shuffing in simulated intestinal fluid.56,58,66,67 Further investigations on these and related scaffolds will reveal how unique these privileged structures are and how well they can be used to target various extracellular proteins.
PLEASE RETURN YOUR HELICES TO AN UPRIGHT, LOCKED POSITION
The field of α-helix stabilization is well-understood with respect to biophysics and design, and so this field has also recently converged on understanding how conformational restriction affects activity in biological systems.68,69 This aspect has always been part of the study of constrained helices. Some of the earliest work in this area was Lerner and colleagues’ demonstration in 1988 that a simple peptide derived from malarial sporozoites could be “chemically shaped” to a more structured, immunogenic form.70 By the early 1990s, stabilization of α-helix structure was generalized by several groups through the formation of side chain lactams, disulfides, metal ion coordination complexes, and a variety of other linkages.71–75 Several reviews have described the biophysics and structure–activity relationships of these stabilized α-helical peptides.76–78 Because of their synthetic accessibility, helices stabilized by (i,i+4) lactam constraints have been extensively analyzed for bioactivity and bioavailability. For example, empirically optimized lactam cross-links increased the potency of N-terminal fragments of parathyroid hormone related protein (PTHrP) in vitro and in cell-based assays and were shown to alter transit times following subcutaneous administration.79–82 Analgesics derived from the peptide hormone nociceptin were successfully stabilized using two lactam bridges, resulting in a very potent antagonist for the associated opioid receptor whose effects lasted up to 1 h after injection.83,84 These and other examples have shown that, even without demonstration of oral bioavailability, lactam bridges clearly alter the distribution and degradation of helical peptides in whole organisms.
Despite steady progress using small peptide macrocycles and helices, there was one aspect of natural product peptides that still seemed like it might remain out of reach: cell penetration. While some highly constrained natural product peptides appear to enter cells,85 most work on cell-penetrating peptides was focused on endocytosis stimulated by polycationic peptides and proteins.86–88 Addition of arginines to cyclic peptides and protein surfaces has been shown to promote cell penetration,88–90 and recent work has demonstrated a key effect of peptide conformation on arginine-mediated internalization and endocytic escape.91 While these works have opened up one front on the problem of cell penetration, it has been more challenging to access additional mechanisms, especially those employed by amanitin, cyclosporine, and other potent natural products.
The idea that peptide conformation can control cell penetration was recently rekindled with reports that helices stabilized through hydrocarbon cross-links can indeed enter cells. Such helices were first described by Blackwell and Grubbs and later refined by Schafmeister and Verdine.92–94 They incorporate amino acids with olefinic side chains in order to enable an intramolecular metathesis reaction to form the “stapled” helix. A watershed report in 2004 by Walensky et al. demonstrated that these stapled helices can possess some striking biological properties.95 In this work, the BH3 helix of Bid, a commonly used tool for inducing apoptosis in vitro, was modified with an all-hydrocarbon “staple”. Stapled analogues possessed enhanced helicity in aqueous solution, improved target affinity, and superior resistance to degradation, mirroring results from other systems that used lactam bridges and other cross-link chemistries. However, the stapled Bid-BH3 helices were also able to penetrate living cells via an energy-dependent endocytosis mechanism and could trigger apoptosis via interactions with BCL-2 family members present in the cytosol and at the mitochondrial surface. The most potent stapled helix, shown in Figure 2f, increased median survival from 5 days to 11 days in leukemia xenograft mice (Table 1), demonstrating potential as an anticancer therapeutic.95 This strategy was subsequently applied to other protein–protein interactions relevant to cancer, including additional BCL-2 family members, p53, Notch, and estrogen receptor.96–101 One report described how addition of two hydrocarbon staples within the known HIV-fusion inhibitor enfuvirtide (Fuzeon) conferred substantial protease resistance and oral bioavailability.102 Taken together, these exciting results demonstrate that peptides as large as 36 amino acids can penetrate cells, and even be made orally bioavailable by controlling conformation. The effects of hydrocarbon stapling are, to date, best understood as a combination of favorable physicochemical properties (increased hydrophobicity and amphipathic patterning) and favorable conformational properties.103,104 However, stapled peptides are not universally endowed with these favorable properties, and there are intensive ongoing efforts to figure out how conformation controls the behavior of stapled helices in biological systems.
EMERGING STRATEGIES FOR CONFORMATIONAL CONTROL
Additional work has hinted that other methodologies for controlling peptide conformation may similarly boost bio-activity and bioavailability. Arora and co-workers have developed a complementary strategy for covalently constraining helical peptides that involves replacing the N-terminal (i,i+4) hydrogen bond with an isosteric covalent bond, an approach termed “hydrogen-bond surrogate” (HBS).105,106 This approach uses the same ring-closing metathesis chemistry as helix stapling but involves appending the olefins directly to the backbone rather than incorporating them within side chains (Figure 2g). This approach has been applied to a number of targets, producing helical inhibitors of BCL-XL, p53, gp41, Hif-1, and Ras.107–111 Several HBS-stabilized helices were reported with improved metabolic stability compared to that of non-stabilized controls, reduced cytotoxicity, effective cell penetration, and/or potent cellular activity consistent with selective inhibition of their intended targets.
Encouraged by the successes of helix stapling and HBS strategies, a number of other strategies for constraining peptides are emerging. Many of these have yet to be evaluated in vivo but show promise in model systems and cultured cells. Existing strategies for conformational constraint, such as (i,i+4) lactamization, are being pushed to new limits. Fairlie and co-workers have recently reported a variety of peptides with multiple (i,i+4) lactam bridges, all with impressive aqueous helicity and serum stability despite a variety of primary sequences and binding partners.112 Copper-catalyzed azide– alkyne 1,3-dipolar Huisgen cycloaddition has been used to constrain (i,i+4) side chains within peptides derived from PTHrP and from the transcriptional coactivator BCL9.113–115 Tetrazole-enone photocycloaddition led to cell permeability and modest cellular activity for a peptide inhibitor of Mdm2 and MdmX, and these were further optimized by linking (i,i+7) cysteine residues with a hydrophobic bisarylmethylene group.116,117 Introducing a novel metal coordination chemistry for (i,i+4) side chain linkages, Ball and co-workers reported the stabilization of helical structure using interactions between acid side chains and rhodium cations to yield biocompatible protein ligands.118,119 New covalent constraints are also being applied to non-helical peptides. Heinis and Winter reported a strategy for the covalent macrobicyclization of peptides using the reaction between symmetrical tris-bromomethylbenzene and three cysteine side chains.120,121 Smaller peptide bicycles were recently reported by Quartararo et al. that take advantage of lactam bridges across a peptide macrocycle to generate peptide bicycles that act as potent, selective protein ligands.122 These and similar strategies are reaching a critical stage where their bioactivities and bioavailabilities are being evaluated. Within a decade, there will likely be multiple synthetic platforms for the preparation of constrained peptides suitable for biological probes and pharmaceutical lead compounds.
SHAPE SORTERS: SYSTEMATIC STUDIES USING MODEL SYSTEMS
There are now many diverse examples of peptides whose bioactivity and bioavailability are controlled by conformation. However, understanding the underlying mechanisms was historically limited by the need for meaningful in vitro models of bioavailability. Throughout the late 1980s and 1990s, groundbreaking work by Borchardt promoted in vitro models for investigating bioavailability, most notably the Caco-2 gut epithelial cell line for the measurement of intestinal permeability.123,124 Following extensive applications of peptides and proteins to these systems, these authors concluded that desolvation of amide bonds was the primary barrier to passive transport of peptides through biological membranes (transcellular permeability) and that electrostatic charge and molecule size limits transport of peptides through junctions between cells (paracellular permeability).125–129 To understand specific effects of conformation on peptide uptake in gut epithelium, explicit comparisons were made among sets of cyclic peptides, turn peptides, and N-methylated peptides using Caco-2 cells.126,127,130 These studies showed that conformation can powerfully alter permeability in ways that cannot be fully attributed to physicochemical factors. Later development of artificial membranes, including the parallel artificial membrane permeability assay (PAMPA),131 has helped to further deconvolute transcellular, paracellular, and active transport mechanisms and better explain peptide uptake. Throughout these works, investigators have often noted that conformation plays a fundamental role in peptide permeability but often explained this by correlating increased permeability to hydrophobicity, size, or other physical parameters.127,129,132,133
In the past few years, two distinct efforts have used model systems to go beyond physiochemical correlations and address specific conformations that control cell penetration, gut absorption, and peptide degradation. Both involve systematic screening of model peptides, followed by detailed structural analysis of unusually bioavailable compounds. As part of the first effort, Hoffman, Gilon, and co-workers examined a series based on the model hexapepetide Phe-(Gly)4-Phe to determine the effects of several backbone modifications on various aspects of bioavailability. The authors independently addressed the effects of cyclization, ring size, N-methylation, and C-methylation (Gly to Ala substitutions) within this relatively hydrophilic model peptide using a diverse panel of assays including ex vivo penetration of rat intestine, Caco-2 permeability, liposome bilayer penetration, PAMPA, and degradation in brush border membrane vesicles.130 They found that the absorption of these hydrophilic peptides is paracellular and remains so despite all backbone modifications tested. They also found that cyclization was the only backbone modification that improved paracellular transport. Using data from high-resolution size exclusion chromatography and NMR, they argued that cyclization reduced overall size and stabilized a single conformation, leading to the observed tissue penetration.
Continuing this effort, recent work by the Kessler and Hoffman groups examined the effects of N-methylation on intestinal permeability using the model hexapeptide cyclo-[D-Ala-(Ala)5].134 Fifty-four peptides comprising all possible N-methylation patterns were analyzed using an extensive panel of permeability assays. Some highly penetrant peptides were also conformationally homogeneous in aqueous solution, and these were selected for structural analysis by NMR. This led to the identification of two specific structural templates for highly penetrant cyclic, N-methylated peptides (one is shown in Figure 2c and Figure 3c; data given in Table 1).135 One of their template structures overlaid well with a turn motif from cyclosporine, a turn motif also shared by the orally bioavailable analogue of somatostatin (Figure 3).36 In contrast to cyclosporine, however, transcellular penetration was judged unlikely for the model cyclic hexapeptide due to poor penetration observed with model membranes. Instead, the authors argued that a receptor-mediated mechanism was likely and would also explain the dependence on a specific conformation. This would match the example of α-amanitin in which a single, locked conformation mediates absorption and bioactivity. However, several of the highly penetrant peptides discovered in these works were conformationally heterogeneous, and specific alternate modes of cell penetration were not exclusively ruled out. Further work with this set of peptides, particularly on variants substituted with diverse side chains, will clarify whether “bottom-up” design using these cyclic peptide templates will promote bioactivity and bioavailability in the resulting inhibitors.
Figure 3.

New exceptions or new rules? Four head-to-tail cyclic peptides with significant oral bioavailability. Peptide backbones are shown in ball-and- stick, with side chains shown as thin lines; oxygens are in red, nitrogens are in blue, and hydrogens are omitted for clarity. All four peptides share similar turn conformations, despite having been developed via different strategies. (a) Three-dimensional structure of cyclosporine A in chloroform, thought to be representative of its structure when passing through lipid membranes.22,23,136 (b) NMR structure of the optimized, orally bioavailable somatostatin mimic shown in Figure 2a.36 (c) NMR structure of an orally bioavailable scaffold found by Hoffman, Kessler, and co-workers, similar to the peptide shown in Figure 2b.135 (d) NMR structure of an orally bioavailable scaffold found by Lokey and co-workers, shown in Figure 2c.24
In a second major approach to this problem, the Lokey group has examined cyclic peptide permeability in a slightly different manner. Their approach was similar to that of Kessler and Hoffman, but their initial focus was solely on transcellular penetration as judged by PAMPA, and significant allowances were made to accommodate a conformation-switching model as has been proposed for cyclosporine. Initial studies of cyclic hexapeptide diastereomers revealed that PAMPA permeability correlated with the capacity for the peptide to form intramolecular backbone hydrogen bonds.136 A computational algorithm was developed in collaboration with the Jacobsen group that could rationalize this result by finding a low-energy conformation for the cyclic peptide in a low-dielectric environment and then calculating the energy difference that would occur when this conformation was transferred to a high-dielectric environment.25 This energy difference was used to predict whether a given cyclic peptide could rapidly partition between aqueous and nonpolar environments, like cyclosporine does, thus promoting passive cell penetration. Ongoing efforts are rapidly improving these and other computational models of membrane penetration by small molecules and peptides.137–141 These models, especially ones based on predictive physical simulation rather than empirical correlations,142 will be critical for understanding whether the effects of peptide conformation can be understood using traditional physicochemical descriptors such as partition coefficients, or whether new descriptors will need to be developed to accurately describe these effects.
More recently, the Lokey group showed that mild N-methylation chemistry, applied to solid-phase peptide synthesis, does not methylate amides that are internally hydrogen-bonded in organic solvent. This was used to screen a library of cyclic peptide diastereomers for those that produced N-methylation patterns indicative of internal structure.24 The predicted backbone–backbone hydrogen bonds were confirmed using hydrogen–deuterium exchange and NMR solution structures. According to their previous work, the selectively N-methylated peptides were predicted to more readily penetrate membranes by passive diffusion. A selectively N-methylated scaffold (Figure 3d) was indeed shown to possess superior passive membrane permeability, metabolic stability, and oral bioavailability (Table 1), lending further credence to this “cyclosporine-like” approach.
The Kessler/Hoffman/Gilon and Lokey/Jacobsen approaches each provide a long-awaited glimpse into how conformation controls bioavailability, and both use systematically varied collections of cyclic hexamers to uncover scaffolds with surprising oral bioavailability. Both have provided scaffolds for future development, and both remarkably “re-confirmed” that the same orally active somatostatin analogue represents a uniquely bioavailable scaffold among variants of N-methylated, cyclic hexapeptides.24,36,135 However, the differences between these lines of inquiry serve to highlight their different conclusions. The Kessler/Hoffman groups used alanine-based cyclic peptides and focused on cell-based penetration assays, leading them to isolate single structures that promote paracellular or receptor-mediated transport (as hypothesized for α-amanitin). The Lokey group used more hydrophobic, leucine-rich cyclic hexapeptides and focused on passive membrane diffusion, explicitly looking for a cyclosporine-like mode of transport. This clearly makes a difference, since alteration of even a single leucine to serine diminished, but did not abrogate, cell penetration within their best scaffold.24 Even so, whether looking for an amanitin-like or cyclosporine-like mechanism, both groups have found what they sought: highly bioavailable scaffolds based on conformationally constrained backbones. This fact bodes very well for the future of peptide drug design.
CONCLUSIONS
After decades of envy directed at natural products such as α-amanitin and cyclosporine, chemical biologists can now point to a critical mass of research demonstrating that peptide bioactivity and bioavailability can be controlled by conformation. These recent insights were catalyzed by advances in synthetic methodology and by systematic adoption of standard in vitro and organism-based assays for peptide bioavailability. Model systems have provided new frameworks and atomistic models for understanding and improving the metabolic stability, gut uptake, and cell penetration of constrained peptides. Meanwhile, researchers from diverse subfields of peptide engineering – hormone mimicry, natural products, helix stabilization, non-helical peptide scaffolds, computational design, and model systems – are all converging on the same question: how can peptides with intrinsically high bioactivity and bioavailability be rationally designed? In the next decade, researchers will answer this question by evaluating new structural scaffolds and expanding model systems to incorporate the interplay between backbone conformation and substituted side chains.143 The answers may be different depending on cellular target, tissue of interest, or transport mechanism. In all cases, it has become clear that control over peptide conformation is critical for the advancement of these exciting tools and therapeutics.
ACKNOWLEDGMENTS
The authors thank the Lokey and Kessler laboratories for supplying structure files and helpful comments. J.G. is supported by NIH K12GM074869.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- (2).Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- (3).Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery – an underexploited structural class. Nat. Rev. Drug Discovery. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- (4).Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- (5).Duarte CD, Barreiro EJ, Fraga CAM. Privileged structures: A useful concept for the rational design of new lead drug candidates. Mini-Rev. Med. Chem. 2007;7:1108–1119. doi: 10.2174/138955707782331722. [DOI] [PubMed] [Google Scholar]
- (6).Scott EL. On the influence of intravenous injections of an extract of the pancreas on experimental pancreatic diabetes. Am. J. Physiol. 1912;29:306–310. [Google Scholar]
- (7).Banting FG, Best CH, Collip JB, MacLeod JJR, Noble EC. The effect of pancreatic extract (insulin) on normal rabbits. Am. J. Physiol. 1922;62:162–176. [Google Scholar]
- (8).Adessi C, Soto C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- (9).Vagner J, Qu HC, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wieland T, Faulstich H. Amatoxins, phallotoxins, phallolysin, and antamanide – biologically-active components of poisonous amanita mushrooms. CRC Crit. Rev. Biochem. 1978;5:185–260. doi: 10.3109/10409237809149870. [DOI] [PubMed] [Google Scholar]
- (11).Hallen HE, Luo H, Scott-Craig JS, Walton JD. Gene family encoding the major toxins of lethal Amanita mushrooms. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19097–19101. doi: 10.1073/pnas.0707340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wieland T, Faulstich H. 50 years of amanitin. Experientia. 1991;47:1186–1193. doi: 10.1007/BF01918382. [DOI] [PubMed] [Google Scholar]
- (13).Letschert K, Faulstich H, Keller D, Keppler D. Molecular characterization and inhibition of amanitin uptake into human hepatocytes. Toxicol. Sci. 2006;91:140–149. doi: 10.1093/toxsci/kfj141. [DOI] [PubMed] [Google Scholar]
- (14).Lindell TJ, Weinberg F, Morris PW, Roeder RG, Rutter WJ. Specific inhibition of nuclear RNA polymerase II by alpha-amanitin. Science. 1970;170:447–449. doi: 10.1126/science.170.3956.447. [DOI] [PubMed] [Google Scholar]
- (15).Schreiber SL, Crabtree GR. The mechanism of action of cyclosporine-A and FK506. Immunol. Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- (16).Wenger RM, France J, Bovermann G, Walliser L, Widmer A, Widmer H. The 3D structure of a cyclosporine analog in water is nearly identical to the cyclophilin-bound cyclosporine conformation. FEBS Lett. 1994;340:255–259. doi: 10.1016/0014-5793(94)80149-5. [DOI] [PubMed] [Google Scholar]
- (17).Altschuh D, Vix O, Rees B, Thierry JC. A conformation of cyclosporine-A in aqueous environment revealed by the X-ray structure of a cyclosporine-FAB complex. Science. 1992;256:92–94. doi: 10.1126/science.1566062. [DOI] [PubMed] [Google Scholar]
- (18).Fesik SW, Gampe RT, Eaton HL, Gemmecker G, Olejniczak ET, Neri P, Holzman TF, Egan DA, Edalji R, Simmer R, Helfrich R, Hochlowski J, Jackson M. NMR studies of [U-C13]cyclosporin A bound to cyclophilin: bound conformation and portions of cyclosporine involved in binding. Biochemistry. 1991;30:6574–6583. doi: 10.1021/bi00240a030. [DOI] [PubMed] [Google Scholar]
- (19).Fesik SW, Neri P, Meadows R, Olejniczak ET, Gemmecker G. A model of the cyclophilin cyclosporine-A (CSA) complex from NMR and X-ray data suggests that CSA binds as a transition-state analog. J. Am. Chem. Soc. 1992;114:3165–3166. [Google Scholar]
- (20).Kallen J, Spitzfaden C, Zurini MGM, Wider G, Widmer H, Wuthrich K, Walkinshaw MD. Structure of human cyclophilin and its binding-site for cyclosporine-A determined by X-ray crystallography and NMR spectroscopy. Nature. 1991;353:276–279. doi: 10.1038/353276a0. [DOI] [PubMed] [Google Scholar]
- (21).Spitzfaden C, Weber HP, Braun W, Kallen J, Wider G, Widmer H, Walkinshaw MD, Wuthrich K. Cyclosporine-A - cyclophilin complex formation - a model based on X-ray and NMR data. FEBS Lett. 1992;300:291–300. doi: 10.1016/0014-5793(92)80866-f. [DOI] [PubMed] [Google Scholar]
- (22).Kessler H, Kock M, Wein T, Gehrke M. Reinvestigation of the conformation of cyclosporine-A in chloroform. Helv. Chim. Acta. 1990;73:1818–1832. [Google Scholar]
- (23).Navia MA, Chaturvedi PR. Design principles for orally bioavailable drugs. Drug Discovery Today. 1996;1:179–189. [Google Scholar]
- (24).White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SSF, Kalgutkar AS, Bauman JN, Zhang YZ, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: Successful in silico prediction of the relative permeabilities of cyclic peptides. J. Am. Chem. Soc. 2006;128:14073–14080. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- (26).Chatterjee J, Gilon C, Hoffman A, Kessler H. N-Methylation of peptides: a new perspective in medicinal chemistry. Acc. Chem. Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- (27).Veber DF, Freidinger RM, Perlow DS, Paleveda WJ, Holly FW, Strachan RG, Nutt RF, Arison BH, Homnick C, Randall WC, Glitzer MS, Saperstein R, Hirschmann R. A potent cyclic hexapeptide analog of somatostatin. Nature. 1981;292:55–58. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]
- (28).Veber DF, Holly FW, Nutt RF, Bergstrand SJ, Brady SF, Hirschmann R, Glitzer MS, Saperstein R. Highly-active cyclic and bicyclic somatostatin analogs of reduced ring size. Nature. 1979;280:512–514. doi: 10.1038/280512a0. [DOI] [PubMed] [Google Scholar]
- (29).Hruby VJ, Balse PM. Conformational and topographical considerations in designing agonist peptidomimetics from peptide leads. Curr. Med. Chem. 2000;7:945–970. doi: 10.2174/0929867003374499. [DOI] [PubMed] [Google Scholar]
- (30).Hruby VJ, Alobeidi F, Kazmierski W. Emerging approaches in the molecular design of receptor-selective peptide ligands - conformational, topographical and dynamic considerations. Biochem. J. 1990;268:249–262. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kessler H. Peptide conformations 0.19. Conformation and biological-activity of cyclic-peptides. Angew. Chem., Int. Ed. Engl. 1982;21:512–523. [Google Scholar]
- (32).Ovadia O, Greenberg S, Laufer B, Gilon C, Hoffman A, Kessler H. Improvement of drug-like properties of peptides: the somatostatin paradigm. Expert Opin. Drug Discovery. 2010;5:655–671. doi: 10.1517/17460441.2010.493935. [DOI] [PubMed] [Google Scholar]
- (33).Vale W, Brazeau P, Grant G, Nussey A, Burgus R, Rivier J, Ling N, Guillemi R. Observations on action mode of somatostatin, hypothalamic factor inhibiting growth-hormone secretion. C. R. Hebd. Seances Acad. Sci., Ser. D. 1972;275:2913–2916. [PubMed] [Google Scholar]
- (34).Freidinger RM, Veber DF, Perlow DS, Brooks JR, Saperstein R. Bioactive conformation of luteinizing-hormone-releasing hormone - evidence from a conformationally constrained analog. Science. 1980;210:656–658. doi: 10.1126/science.7001627. [DOI] [PubMed] [Google Scholar]
- (35).Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzer MS, Saperstein R, Hirschmann R. Conformationally restricted bicyclic analogs of somatostatin. Proc. Natl. Acad. Sci. U.S.A. 1978;75:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Improving oral bioavailability of peptides by multiple N-methylation: Somatostatin analogues. Angew. Chem., Int. Ed. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- (37).Al-Obeidi F, Castrucci AM, Hadley ME, Hruby VJ. Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: design based on molecular dynamics. J. Med. Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- (38).Doedens L, Opperer F, Cai M, Beck JG, Dedek M, Palmer E, Hruby VJ, Kessler H. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. J. Am. Chem. Soc. 2010;132:8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Linde Y, Ovadia O, Safrai E, Xiang Z, Portillo FP, Shalev DE, Haskell-Luevano C, Hoffman A, Gilon C. Structure-activity relationship and metabolic stability studies of backbone cyclization and N-methylation of melanocortin peptides. Biopolymers. 2008;90:671–682. doi: 10.1002/bip.21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclic .alpha.- melanotropins based on quenched dynamic simulations. J. Am. Chem. Soc. 1989;111:3413–3416. [Google Scholar]
- (41).Hess S, Linde Y, Ovadia O, Safrai E, Shalev DE, Swed A, Halbfinger E, Lapidot T, Winkler I, Gabinet Y, Faier A, Yarden D, Xiang Z, Portillo FP, Haskell-Luevano C, Gilon C, Hoffman A. Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J. Med. Chem. 2008;51:1026–1034. doi: 10.1021/jm701093y. [DOI] [PubMed] [Google Scholar]
- (42).Pierschbacher MD, Hayman EG, Ruoslahti E. Location of the cell-attachment site in fibronectin with monoclonal antibodies and proteolytic fragments of the molecule. Cell. 1981;26:259–267. doi: 10.1016/0092-8674(81)90308-1. [DOI] [PubMed] [Google Scholar]
- (43).Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- (44).Aumailley M, Gurrath M, Muller G, Calvete J, Timpl R, Kessler H. Arg-Gly-Asp constrained within cyclic pentapeptides - strong and selective inhibitors of cell-adhesion to vitronectin and laminin fragment-P1. FEBS Lett. 1991;291:50–54. doi: 10.1016/0014-5793(91)81101-d. [DOI] [PubMed] [Google Scholar]
- (45).Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, Kessler H. N-methylated cyclic RGD peptides as highly active and selective alpha(v)beta(3) integrin antagonists. J. Med. Chem. 1999;42:3033–3040. doi: 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- (46).Goodman SL, Holzemann G, Sulyok GAG, Kessler H. Nanomolar small molecule inhibitors for αvβ6, αvβ5, and αvβ3 integrins. J. Med. Chem. 2002;45:1045–1051. doi: 10.1021/jm0102598. [DOI] [PubMed] [Google Scholar]
- (47).Dawson R, Muller L, Dehner A, Klein C, Kessler H, Buchner J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003;332:1131–1141. doi: 10.1016/j.jmb.2003.08.008. [DOI] [PubMed] [Google Scholar]
- (48).Mas-Moruno C, Rechenmacher F, Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010;10:753–768. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schottelius M, Laufer B, Kessler H, Wester H.-J. r. Ligands for mapping αvβ3-integrin expression in vivo. Acc. Chem. Res. 2009;42:969–980. doi: 10.1021/ar800243b. [DOI] [PubMed] [Google Scholar]
- (50).Chatterjee J, Laufer B, Kessler H. Synthesis of N-methylated cyclic peptides. Nat. Protoc. 2012;7:432–444. doi: 10.1038/nprot.2011.450. [DOI] [PubMed] [Google Scholar]
- (51).Wilson RM, Stockdill JL, Wu XY, Li XC, Vadola PA, Park PK, Wang P, Danishefsky SJ. A fascinating journey into history: exploration of the world of isonitriles en route to complex amides. Angew. Chem., Int. Ed. 2012;51:2834–2848. doi: 10.1002/anie.201106628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Biron E, Kessler H. Convenient synthesis of N-methylamino acids compatible with Fmoc solid-phase peptide synthesis. J. Org. Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- (53).King GF. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011;11:1469–1484. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- (54).Lewis RJ, Garcia ML. Therapeutic potential of venom peptides. Nat. Rev. Drug Discovery. 2003;2:790–802. doi: 10.1038/nrd1197. [DOI] [PubMed] [Google Scholar]
- (55).Craik DJ, Cemazar M, Daly NL. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr. Opin. Drug Discovery Dev. 2006;9:251–260. [PubMed] [Google Scholar]
- (56).Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem., Int. Ed. 2010;49:6545–6548. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- (57).Getz JA, Rice JJ, Daugherty PS. Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem. Biol. 2011;6:837–844. doi: 10.1021/cb200039s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Clark RJ, Akcan M, Kaas Q, Daly NL, Craik DJ. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon. 2012;59:446–455. doi: 10.1016/j.toxicon.2010.12.003. [DOI] [PubMed] [Google Scholar]
- (59).Tam JP, Lu YA, Yang JL, Chiu KW. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8913–8918. doi: 10.1073/pnas.96.16.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Garcia AE, Tai KP, Puttamadappa SS, Shekhtman A, Ouellette AJ, Camarero JA. Biosynthesis and antimicrobial evaluation of backbone-cyclized alpha-defensins. Biochemistry. 2011;50:10508–10519. doi: 10.1021/bi201430f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lehrer RI, Cole AM, Selsted ME. Theta-defensins: cyclic peptides with endless potential. J. Biol. Chem. 2012;287:27014–27019. doi: 10.1074/jbc.R112.346098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Maksimov MO, Pan SJ, Link AJ. Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 2012;29:996–1006. doi: 10.1039/c2np20070h. [DOI] [PubMed] [Google Scholar]
- (63).Wong CTT, Rowlands DK, Wong CH, Lo TWC, Nguyen GKT, Li HY, Tam JP. Orally active peptidic bradykinin B-1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem., Int. Ed. 2012;51:5620–5624. doi: 10.1002/anie.201200984. [DOI] [PubMed] [Google Scholar]
- (64).Craik DJ, Daly NL, Mulvenna J, Plan MR, Trabi M. Discovery, structure and biological activities of the cyclotides. Curr. Protein Pept. Sci. 2004;5:297–315. doi: 10.2174/1389203043379512. [DOI] [PubMed] [Google Scholar]
- (65).Gran L. Effect of a polypeptide isolated from kalata-kalata (Oldenlandia-affinis DC) on estrogen dominated uterus. Acta Pharmacol. Toxicol. (Copenhagen) 1973;33:400–408. doi: 10.1111/j.1600-0773.1973.tb01541.x. [DOI] [PubMed] [Google Scholar]
- (66).Halai R, Caaghan B, Daly NL, Clark RJ, Adams DJ, Craik DJ. Effects of cyclization on stability, structure, and activity of alpha-conotoxin RgIA at the alpha 9 alpha 10 nicotinic acetylcholine receptor and GABA(B) receptor. J. Med. Chem. 2011;54:6984–6992. doi: 10.1021/jm201060r. [DOI] [PubMed] [Google Scholar]
- (67).Carstens BB, Clark RJ, Daly NL, Harvey PJ, Kaas Q, Craik DJ. Engineering of conotoxins for the treatment of pain. Curr. Pharm. Des. 2011;17:4242–4253. doi: 10.2174/138161211798999401. [DOI] [PubMed] [Google Scholar]
- (68).Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr. Opin. Chem. Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets, in Methods in Enzymology: Protein Engineering for Therapeutics. In: Wittrup KD, Verdine GL, editors. Vol. 203. Elsevier Academic Press Inc.; San Diego: 2012. pp. 3–33. Pt B. [DOI] [PubMed] [Google Scholar]
- (70).Satterthwait AC, Arrhenius T, Hagopian RA, Zavala F, Nussenzweig V, Lerner RA. Conformational restriction of peptidyl immunogens with covalent replacements for the hydrogen bond. Vaccine. 1988;6:99–103. doi: 10.1016/s0264-410x(88)80007-0. [DOI] [PubMed] [Google Scholar]
- (71).Osapay G, Taylor JW. Multicyclic polypeptide model compounds 0.2. Synthesis and conformational properties of a highly alpha-helical uncosapeptide constrained by 3 side-chain to side-chain lactam bridges. J. Am. Chem. Soc. 1992;114:6966–6973. [Google Scholar]
- (72).Osapay G, Taylor JW. Multicyclic polypeptide model compounds 0.1. Synthesis of a tricyclic amphiphilic alpha-helical peptide using an oxime resin, segment-condensation approach. J. Am. Chem. Soc. 1990;112:6046–6051. [Google Scholar]
- (73).Felix AM, Heimer EP, Wang CT, Lambros TJ, Fournier A, Mowles TF, Maines S, Campbell RM, Wegrzynski BB, Toome V, Fry D, Madison VS. Synthesis, biological-activity and conformational-analysis of cyclic GRF analogs. Int. J. Pept. Protein Res. 1988;32:441–454. doi: 10.1111/j.1399-3011.1988.tb01375.x. [DOI] [PubMed] [Google Scholar]
- (74).Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the α-helical conformation. Curr. Opin. Chem. Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Taylor JW. The synthesis and study of side-chain lactam-bridged peptides. Biopolymers. 2002;66:49–75. doi: 10.1002/bip.10203. [DOI] [PubMed] [Google Scholar]
- (76).Ghadiri MR, Choi C. Secondary structure nucleation in peptides - transition-metal ion stabilized alpha-helices. J. Am. Chem. Soc. 1990;112:1630–1632. [Google Scholar]
- (77).Lieberman M, Sasaki T. Iron(II) organizes a synthetic peptide into 3-helix bundles. J. Am. Chem. Soc. 1991;113:1470–1471. [Google Scholar]
- (78).Estieu-Gionnet K, Guichard G. Stabilized helical peptides: overview of the technologies and therapeutic promises. Expert Opin. Drug Discovery. 2011;6:937–963. doi: 10.1517/17460441.2011.603723. [DOI] [PubMed] [Google Scholar]
- (79).Condon SM, Morize I, Darnbrough S, Burns CJ, Miller BE, Uhl J, Burke K, Jariwala N, Locke K, Krolikowski PH, Kumar NV, Labaudiniere RF. The bioactive conformation of human parathyroid hormone. Structural evidence for the extended helix postulate. J. Am. Chem. Soc. 2000;122:3007–3014. [Google Scholar]
- (80).Chorev M, Roubini E, McKee RL, Gibbons SW, Goldman ME, Caulfield MP, Rosenblatt M. Cyclic parathyroid hormone-related protein antagonists: lysine 13 to aspartic acid 17 [i to (i+ 4)] side chain to side chain lactamization. Biochemistry. 1991;30:5968–5974. doi: 10.1021/bi00238a022. [DOI] [PubMed] [Google Scholar]
- (81).Barbier JR, Neugebauer W, Morley P, Ross V, Soska M, Whitfield JF, Willick G. Bioactivities and secondary structures of constrained analogues of human parathyroid hormone: Cyclic lactams of the receptor binding region. J. Med. Chem. 1997;40:1373–1380. doi: 10.1021/jm960743o. [DOI] [PubMed] [Google Scholar]
- (82).Morley P, Whitfield JF, Willick GE, Ross V, MacLean S, Barbier JR, Isaacs RJ, Andreassen TT. The effect of monocyclic and bicyclic analogs of human parathyroid hormone (hPTH)-(1–31)NH2 on bone formation and mechanical strength in ovariectomized rats. Calcif. Tissue Int. 2001;68:95–101. doi: 10.1007/BF02678147. [DOI] [PubMed] [Google Scholar]
- (83).Charoenchai L, Wang H, Wang JB, Aldrich JV. High affinity conformationally constrained nociceptin/orphanin FQ(1–13) amide analogues. J. Med. Chem. 2008;51:4385–4387. doi: 10.1021/jm800394v. [DOI] [PubMed] [Google Scholar]
- (84).Ambo A, Hamazaki N, Yamada Y, Nakata E, Sasaki Y. structure–activity studies on nociceptin analogues: ORL1 receptor binding and biological activity of cyclic disulfide-containing analogues of nociceptin peptides. J. Med. Chem. 2001;44:4015–4018. doi: 10.1021/jm010092i. [DOI] [PubMed] [Google Scholar]
- (85).Cascales L, Henriques ST, Kerr MC, Huang Y-H, Sweet MJ, Daly NL, Craik DJ. Identification and characterization of a new family of cell-penetrating peptides. J. Biol. Chem. 2011;286:36932–36943. doi: 10.1074/jbc.M111.264424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm. Res. 2004;21:389–393. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- (87).Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Delivery Rev. 2008;60:452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Cronican JJ, Thompson DB, Beier KT, McNaughton BR, Cepko CL, Liu DR. Potent delivery of functional proteins into mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem. Biol. 2010;5:747–752. doi: 10.1021/cb1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Liu T, Liu Y, Kao HY, Pei DH. Membrane permeable cyclic peptidyl inhibitors against human peptidylprolyl isomerase Pin1. J. Med. Chem. 2010;53:2494–2501. doi: 10.1021/jm901778v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Appelbaum JS, LaRochelle JR, Smith BA, Balkin DM, Holub JM, Schepartz A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem. Biol. 2012;19:819–830. doi: 10.1016/j.chembiol.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Blackwell HE, Grubbs RH. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem., Int. Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- (93).Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000;122:5891–5892. [Google Scholar]
- (94).Miller SJ, Blackwell HE, Grubbs RH. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. J. Am. Chem. Soc. 1996;118:9606–9614. [Google Scholar]
- (95).Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of Apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Phillips C, Roberts LR, Schade M, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH, Read CM, Scott A, Brown DG, Xu B, Irving SL. Design and structure of stapled peptides binding to estrogen receptors. J. Am. Chem. Soc. 2011;133:9696–9699. doi: 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]
- (98).Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–U157. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007;129:2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- (102).Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He XY, Gavathiotis E, Sodroski JG, Walensky LD. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14093–14098. doi: 10.1073/pnas.1002713107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Guo ZJ, Mohanty U, Noehre J, Sawyer TK, Sherman W, Krilov G. Probing the alpha-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010;75:348–359. doi: 10.1111/j.1747-0285.2010.00951.x. [DOI] [PubMed] [Google Scholar]
- (104).Kutchukian PS, Yang JS, Verdine GL, Shakhnovich EI. All-atom model for stabilization of alpha-helical structure in peptides by hydrocarbon staples. J. Am. Chem. Soc. 2009;131:4622–4627. doi: 10.1021/ja805037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Wang D, Chen K, Kulp JL, Arora PS. Evaluation of biologically relevant short α-helices stabilized by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc. 2006;128:9248–9256. doi: 10.1021/ja062710w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Chapman RN, Dimartino G, Arora PS. A highly stable short α-helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc. 2004;126:12252–12253. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- (107).Bao J, Dong XY, Zhang JZH, Arora PS. Dynamical binding of hydrogen-bond surrogate derived Bak helices to antiapoptotic protein Bcl-x L. J. Phys. Chem. B. 2009;113:3565–3571. doi: 10.1021/jp809810z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Wang D, Lu M, Arora PS. Inhibition of HIV-1 fusion by hydrogen-bond-surrogate-based alpha helices. Angew. Chem., Int. Ed. 2008;47:1879–1882. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]
- (109).Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011;7:585–587. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Henchey LK, Porter JR, Ghosh I, Arora PS. High specificity in protein recognition by hydrogen-bond-surrogate alpha-helices: selective inhibition of the p53/MDM2 complex. ChemBioChem. 2010;11:2104–2107. doi: 10.1002/cbic.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of hypoxia inducible factor 1-transcription coactivator interaction by a hydrogen bond surrogate alpha-helix. J. Am. Chem. Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gomez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Downsizing human, bacterial, and viral proteins to short water-stable alpha helices that maintain biological potency. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11686–11691. doi: 10.1073/pnas.1002498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang SM. Design of triazole-stapled BCL9 alpha-helical peptides to target the beta-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 2012;55:1137–1146. doi: 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Scrima M, Le Chevalier-Isaad A, Rovero P, Papini AM, Chorev M, D’Ursi AM. Cu-I-catalyzed azide-alkyne intramolecular i-to-(i+4) side-chain-to-side-chain cyclization promotes the formation of helix-like secondary structures. Eur. J. Org. Chem. 2010:446–457. [Google Scholar]
- (115).Cantel S, Isaad ALC, Scrima M, Levy JJ, DiMarchi RD, Rovero P, Halperin JA, D’Ursi AM, Papini AM, Chorev M. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+4 intramolecular side-chain to side-chain azide - Alkyne 1,3-dipolar cycloaddition. J. Org. Chem. 2008;73:5663–5674. doi: 10.1021/jo800142s. [DOI] [PubMed] [Google Scholar]
- (116).Madden MM, Muppidi A, Li ZY, Li XL, Chen JD, Lin Q. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53-Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorg. Med. Chem. Lett. 2011;21:1472–1475. doi: 10.1016/j.bmcl.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Madden MM, Vera CIR, Song WJ, Lin Q. Facile synthesis of stapled, structurally reinforced peptide helices via a photoinduced intramolecular 1,3-dipolar cycloaddition reaction. Chem. Commun. 2009:5588–5590. doi: 10.1039/b912094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR, Ball ZT. Hybrid organic–inorganic inhibitors of a PDZ interaction that regulates the endocytic fate of CFTR. Angew. Chem., Int. Ed. 2012;51:7217–7220. doi: 10.1002/anie.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Zaykov AN, Ball ZT. A general synthesis of dirhodium metallopeptides as MDM2 ligands. Chem. Commun. 2011;47:10927–10927. doi: 10.1039/c1cc13169a. [DOI] [PubMed] [Google Scholar]
- (120).Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem. Biol. 2012;7:817–821. doi: 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- (121).Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- (122).Quartararo JS, Wu P, Kritzer JA. Peptide bicycles that inhibit the Grb2 SH2 domain. ChemBioChem. 2012;13:1490–1496. doi: 10.1002/cbic.201200175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human-colon carcinoma cell-line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- (124).Artursson P, Borchardt RT. Intestinal drug absorption and metabolism in cell cultures: Caco-2 and beyond. Pharm. Res. 1997;14:1655–1658. doi: 10.1023/a:1012155124489. [DOI] [PubMed] [Google Scholar]
- (125).Pauletti GM, Okumu FW, Borchardt RT. Effect of size and charge on the passive diffusion of peptides across Caco-2 cell monolayers via the paracellular pathway. Pharm. Res. 1997;14:164–168. doi: 10.1023/a:1012040425146. [DOI] [PubMed] [Google Scholar]
- (126).Okumu FW, Pauletti GM, VanderVelde DG, Siahaan TJ, Borchardt RT. Effect of restricted conformational flexibility on the permeation of model hexapeptides across Caco-2 cell monolayers. Pharm. Res. 1997;14:169–175. doi: 10.1023/a:1012092409216. [DOI] [PubMed] [Google Scholar]
- (127).Knipp GT, Vander Velde DG, Siahaan TJ, Borchardt RT. The effect of beta-turn structure on the passive diffusion of peptides across Caco-2 cell monolayers. Pharm. Res. 1997;14:1332–1340. doi: 10.1023/a:1012152117703. [DOI] [PubMed] [Google Scholar]
- (128).Burton PS, Conradi RA, Ho NFH, Hilgers AR, Borchardt RT. How structural features influence the biomembrane permeability of peptides. J. Pharm. Sci. 1996;85:1336–1340. doi: 10.1021/js960067d. [DOI] [PubMed] [Google Scholar]
- (129).Kim DC, Burton PS, Borchardt RT. A correlation between the permeability characteristics of a series of peptides using an in vitro cell culture model (Caco-2) and those using an in situ perfused rat ileum model of the intestinal mucosa. Pharm. Res. 1993;10:1710–1714. doi: 10.1023/a:1018961828510. [DOI] [PubMed] [Google Scholar]
- (130).Hess S, Ovadia O, Shalev DE, Senderovich H, Qadri B, Yehezkel T, Salitra Y, Sheynis T, Jelinek R, Gilon C, Hoffman A. Effect of structural and conformation modifications, including backbone cyclization, of hydrophilic hexapeptides on their intestinal permeability and enzymatic stability. J. Med. Chem. 2007;50:6201–6211. doi: 10.1021/jm070836d. [DOI] [PubMed] [Google Scholar]
- (131).Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998;41:1007–1010. doi: 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- (132).Knipp GT, Ho NF, Barsuhn CL, Borchardt RT. Paracellular diffusion in Caco-2 cell monolayers: effect of perturbation on the transport of hydrophilic compounds that vary in charge and size. J. Pharm. Sci. 1997;86:1105–1110. doi: 10.1021/js9700309. [DOI] [PubMed] [Google Scholar]
- (133).Pauletti GM, Jeffrey A, Siahaan TJ, Gangwar S, Borchardt RT. Improvement of oral peptide bioavailability: Peptidomimetics and prodrug strategies. Adv. Drug Delivery Rev. 1997;27:235–256. doi: 10.1016/s0169-409x(97)00045-8. [DOI] [PubMed] [Google Scholar]
- (134).Ovadia O, Greenberg S, Chatterjee J, Laufer B, Opperer F, Kessler H, Gilon C, Hoffman A. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol. Pharm. 2011;8:479–487. doi: 10.1021/mp1003306. [DOI] [PubMed] [Google Scholar]
- (135).Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S, Ovadia O, Greenberg S, Gilon C, Hoffman A, Kessler H. Intestinal permeability of cyclic peptides: common key backbone motifs identified. J. Am. Chem. Soc. 2012;134:12125–12133. doi: 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- (136).Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006;128:2510–2511. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- (137).Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- (138).Refsgaard HHF, Jensen BF, Brockhoff PB, Guldbrandt M, Christensen MS. In silico prediction of membrane permeability from calculated molecular parameters. J. Med. Chem. 2005;48:805–811. doi: 10.1021/jm049661n. [DOI] [PubMed] [Google Scholar]
- (139).Bemporad D, Essex JW, Luttmann C. Permeation of small molecules through a lipid bilayer: a computer simulation study. J. Phys. Chem. B. 2004;108:4875–4884. [Google Scholar]
- (140).Ekins S, Waller CL, Swaan PW, Cruciani G, Wrighton SA, Wikel JH. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods. 2000;44:251–272. doi: 10.1016/s1056-8719(00)00109-x. [DOI] [PubMed] [Google Scholar]
- (141).Marrink SJ, Berendsen HJC. Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J. Phys. Chem. 1996;100:16729–16738. [Google Scholar]
- (142).Leung SSF, Mijalkovic J, Borrelli K, Jacobson MP. Testing Physical models of passive membrane permeation. J. Chem. Inf. Model. 2012;52:1621–1636. doi: 10.1021/ci200583t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Rand AC, Leung SSF, Eng H, Rotter CJ, Sharma R, Kalgutkar AS, Zhang Y, Varma MV, Farley KA, Khunte B, Limberakis C, Price DA, Liras S, Mathiowetz AM, Jacobson MP, Lokey RS. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. MedChemComm. 2012;3:1282–1289. doi: 10.1039/C2MD20203D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Nemerov H. Trees, in Mirrors and Windows. University of Chicago Press; Chicago: 1958. [Google Scholar]
- (145).Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005;1059:149–158. doi: 10.1016/j.brainres.2005.08.009. [DOI] [PubMed] [Google Scholar]