

Abstract
Hsp90 is a molecular chaperone implicated in many diseases including cancer and neurodegenerative disease. Most inhibitors target the ATPase site in Hsp90’s N-terminal domain, with relatively few inhibitors of other domains reported to date. Here, we show that peptides derived from a short helix at the C-terminus of Hsp90 show micromolar activity as Hsp90 inhibitors in vitro. These inhibitors do not block the N-terminal domain’s ATP-binding site, and thus are likely to bind at the C-terminal domain. Substitutions and helix stapling were applied to demonstrate structure–activity relationships and improve activity. These helical peptides will help guide the design of a new class of inhibitors of Hsp90’s C-terminal domain.
Keywords: Peptides, Stapled helices, Protein–protein interactions, Hsp90, Molecular chaperones
1. Introduction
Heat-shock protein 90 (Hsp90) is a ubiquitous cytosolic chaperone that assists in the folding and regulation of many signaling proteins.1 Since Hsp90 is known to interact with proteins involved in all the hallmarks of cancer, it has received considerable attention as a target for new anti-cancer therapies.2 Most classes of inhibitors reported to date, including purine analogues, resorcinols, and various derivatives of the natural product geldanamycin, target the nucleotide-binding pocket in the protein’s N-terminal domain. Geldanamycin derivatives have been the subject of numerous clinical trials.3 Recently, an alternate approach to Hsp90 inhibition has emerged, focused on targeting the C-terminal domain.4,5 Neckers and co-workers reported that the aminocoumarin natural product novobiocin inhibits the C-terminal domain of Hsp90.6 Subsequent work by Blagg and co-workers has discovered more potent derivatives of novobiocin’s coumarin core that inhibit Hsp90 without inducing the heat-shock response, a major shortcoming of N-terminal-domain inhibitors.4,7–9 Based on this empirical data and the complex mechanisms of Hsp90 and its cofactors, it is anticipated that additional C-terminal inhibitors will show similarly unique effects.
In a complementary approach, a number of groups have developed peptide-based inhibitors as a means of disrupting Hsp90 chaperone activity by preventing specific protein–protein interactions. Shepherdin is a nonapeptide derived from the Hsp90 client protein survivin, and this peptide inhibits Hsp90 by binding in the N-terminal domain’s ATP-binding site.10 PIER1, a 37-mer helical peptide from Hsp90 paralog gp96, inhibits Hsp90 by binding residues near the N-terminal ATP-binding site.11 Another helical peptide, TPR2A(301–312) from co-chaperone Hop, inhibits Hsp90 by binding to the MEEVD motif at Hsp90’s absolute C-terminus.12,13 Finally, the cyclic pentapeptide sansalvamide A-amide binds to the N-terminal and middle domains of Hsp90 and, in doing so, prevents binding of some client proteins known to bind Hsp90 via the C-terminal domain.14–16
In this work, we extend this set of designed peptide inhibitors of Hsp90. Other than the PIER1 peptide, we were unable to find reports of peptides derived from Hsp90 itself being tested as Hsp90 inhibitors. Since Hsp90 functions as a homodimer with independent dimerization interfaces between N-terminal domains and C-terminal domains,17 we hypothesized that Hsp90 inhibitors could be designed based on helices at the N-and C-terminal dimer interfaces. Since the C-terminal dimer interface becomes exposed upon nucleotide binding, peptides binding this portion of the interface would be expected to interrupt the Hsp90 chaperone cycle in a manner unique from existing inhibitors.
2. Results and discussion
2.1. Design of peptides
The wealth of structural data of Hsp90 in complex with clients and co-chaperones provides numerous opportunities for the design of peptide-based inhibitors.18,19 Inspection of the dimer interfaces revealed the N-terminal helix (F22-Y38) and C-terminal helix (D680-G697) of Hsp90 as particularly attractive opportunities. Both of these helices feature multiple hydrophobic residues on consecutive turns buried in the opposite protomer. We performed computational alanine scanning of available structures of full-length yeast Hsp90 (PDB 2CG9) and N-domain-truncated human Hsp90 (PDB 3Q6M) using Rosetta,20,21 and found that a few highly conserved residues—I25, L28, and L31 in the N-helix and I688, Y689, I692, and L696 in the C-helix—are predicted to be hot spots that contribute substantially to the stability of the closed conformation of Hsp90 (Fig. 1A and B).
Figure 1.

Design of helical peptide inhibitors of Hsp90. (A) Sequences of peptides designed as potential inhibitors of Hsp90. Predicted hotspots in native sequences are shown in orange, substitutions are shown in red and residues involved in cyclization (peptides 9, 11–13, 16, 17) are underlined. *denotes (S)-α-methyl-pentenylalanine. (B) Two views of the Hsp90 C-helix D680-G697 (yellow ribbon, sequence identical to peptide 2) with predicted hotspots shown as orange sticks and the opposite protomer of Hsp90 shown as a gray surface. Structure shown is that of the middle and C-terminal domains of human Hsp90 (PDB 3Q6M).19 (C) Hsp90 inhibition at 250 μM peptide concentration. Hsp90 inhibition was measured in a luciferase renaturation assay as described.22 Heat-denatured luciferase was refolded in an Hsp90-dependent manner by rabbit reticulocyte lysate. Relative emission is measured as a percent of vehicle (2.5% DMSO). Error bars show standard deviation of three independent trials. (D) Detail of predicted hydrophobic pocket (green mesh) proximal to residue G695 (purple sticks).
Based on these predictions, N-helix and C-helix peptides were synthesized and tested for Hsp90 inhibition in a luciferase renaturation assay using rabbit reticulocyte lysate.22 At 250 lM, the N-helix (peptide 1) was inactive, while the C-helix (peptide 2) showed similar activity to known C-terminal inhibitor novobiocin (Fig. 1C). We designed a series of six point mutants of peptide 2 based on the structure of the C-helix and its binding site (PDB 3Q6M)19 and sequence tolerance prediction using Rosetta (Table S1). These were synthesized and assayed for inhibition of luciferase refolding at 250 μM (Figure 1). Four of these peptides (3, 4, 6, and 8) showed little improvement in activity, but substitution Y689W (peptide 5) showed mild improvement and G695L (peptide 7) showed strong improvement. At 250 μM, peptide 7 reduced Hsp90-mediated renaturation of luciferase almost to background levels. Computational solvent mapping using FTSite23 identified a putative hydrophobic binding pocket proximal to G695 (Fig. 1D). This provided a rationale for the increased activity of the G695L mutation, and for exploring additional substitutions at G695. Further, the isolated, 18-residue C-helix was only 20% helical when analyzed by circular dichroism (CD) spectroscopy (Table 1). Thus, we pursued further optimization of peptide 2 by substituting Y689 and G695 and by introducing conformational constraint via side-chain-to-side-chain lactamization and ringclosing metathesis.24–26
Table 1.
Data from dose–response experiments using the luciferase renaturation assay, circular dichroism spectroscopy experiments, a cell-based Hsp90 inhibition assay, and cytotoxicity measurements using a human epithelial cell line
| Peptide | Luciferase renaturation IC50a (μM) |
CD spectroscopy % helicityb |
MDA-kb2 cell culture |
|
|---|---|---|---|---|
| IC50c (μM) | LC50d (μM) | |||
| Novobiocin | 414 ± 28 | 231 ± 42 | 690±188 | |
| Coumermycin A1 |
28.1 ± 2.5 | 15.3 ± 4.1 | 19.1 ± 9.1 | |
| Geldanamycin | 0.282 ± 0.058 | <0.01e | >2f | |
| (2) | 233 ± 9.2 | 20 ± 1 | >160f | >160f |
| (7) | 94.5 ± 3.6 | 43 ± 8 | 18.6 ± 0.3 | 22.9 ± 3.4 |
| (9) | 37.9 ± 4.0 | 64 ± 4 | 18.8 ± 4.5 | 26.6 ± 4.4 |
| (10) | 14.4 ± 2.0 | 50 ± 3 | 15.8 ± 4.3 | 19.1 ± 2.0 |
| (11) | 168 ± 9.0 | 11 ± 1 | >120 | >120 |
| (12) | 137 ± 19 | 29 ± 4 | >67f | >67f |
| (13) | 37.0 ± 3.1 | 37 ± 4 | 121 ± 14 | 97 ± 20 |
| (14) | 38.0 ± 2.2 | 31 ± 5 | 10.4 ± 1.2 | 10.6 ± 3.4 |
| (15) | 32.0 ± 5.7 | 58 ± 4 | 16.8 ± 1.5 | 18.9 ± 1.7 |
| (16) | 8.43 ± 0.09 | 80 ± 2 | >27f | >27f |
| (17) | 4.11 ± 0.19 | 90 ± 2 | 0.73 ± 0.06 | 1.86 ± 0.11 |
From luciferase renaturation assay. IC50 values are ± standard deviation from three independent trials, each performed in triplicate. Values observed for novo-biocin, coumermycin A1, and geldanamycin are in accordance with previously reported values.15
Percent helicity as measured by circular dichroism at 222 nm using 100 μM peptide in 10 mM phosphate buffer at 23 °C.
Dose-dependent inhibition of glucocorticoid-receptor-dependent luciferase expression.
Concentration at which cell viability was reduced by 50% as determined using Promega Cell Titer Glo kit.
Complete suppression of luciferase activity at lowest concentration tested.
No effect observed at the compound’s solubility limit in cell culture media. While all compounds were soluble to 100 μM in phosphate buffer, some compounds had lower solubility limits in cell culture media.
2.2. Effects of stapling and tryptophan substitutions on helicity and in vitro potency
Stapling side chains using all-hydrocarbon cross-links has been shown to improve potency, alter selectivity, and promote protease resistance and cell penetration for a variety of helical peptides.27 We introduced all-hydrocarbon staples using α-methyl-(S)-pente-nylalanine residues in place of M691 and G695. Simply introducing these α-methylated, hydrophobic residues at these positions greatly improved helicity (50% helical for peptide 10 versus 20% for 2) and potency in the luciferase renaturation assay (14.4 μM IC50 for peptide 10 compared to 233 μM for 2). On-resin ring-closing metathesis was used to produce the all-hydrocarbon stapled analog 9. Peptide 9 had even more helical character (64%) but was a less potent Hsp90 inhibitor than the unstapled analog (IC50 = 40 μM), perhaps due to an inability of the stapled side chain to fully access the putative hydrophobic pocket near G695. Additional peptides incorporating α-methyl-(S)-pentenylalanine and all-hydrocarbon staples proved challenging to produce, and so a different chemistry for mediating side-chain stapling was implemented for further investigations.
Side-chain-to-side-chain lactam staples were introduced using Lys and Asp residues at (i, i + 4) positions. This configuration has been shown to promote helical conformations in a variety of short peptides.28 Since optimal stapling position cannot be determined a priori,27 we installed lactam staples at several positions predicted to be solvent-exposed by the structural model of C-helix binding (peptides 11–13). These peptides did not have high helical content as judged by CD spectroscopy, and peptide 12, the lactam-stapled analog of hydrocarbon-stapled peptide 9, showed only modest potency (137 μM) and helicity (29%). In fact, all three lactam-stapled peptides had modest increases in potency compared to the native C-helix sequence (2, IC50 = 233 ± 9 μM), with potency highest for peptide 13 (IC50 = 37 ± 3 μM, Fig. 2). Peptide 13 had a lactam staple at the C-terminal end, and had nearly double the helical character of the unmodified C-helix (Fig. 2). Thus, for these lactam-stapled analogs of the C-helix, we find that stapling along the presumed solvent-exposed face increases potency, and that the staple between positions 693 and 697 is optimal for both structure and Hsp90 inhibitory potency.
Figure 2.

(A) Dose–response behavior of C-helix peptide 2, analogs 13, 16, and 17, C-terminal-domain inhibitors novobiocin and coumermycin A1, and N-terminal-domain inhibitor geldanamycin using an in vitro luciferase renaturation assay.22 Error bars show standard deviations from three independent trials, and curve fits reflect IC50 values shown in Table 1. (B) Circular dichroism spectra for C-helix peptide 2 and analogs 13–17.
From the initial modeling and the first substitution series (peptides 1–8), it was clear that substitutions for Y689 and G695 could produce more potent analogs of the C-helix. We incorporated Trp residues for G695 (14) or both Y689 and G695 (15) and assessed α-helicity and inhibitory potency. The single Trp substitution mildly improved helical structure, but incorporating both Trp substitutions (15) increased helicity to 58%. Despite this difference, peptides 14 and 15 both had similar inhibitory potencies. The IC50 values for 14 and 15 are similar to those of the most potent lactam-stapled and all-hydrocarbon-stapled analogs (13 and 9), providing indirect evidence of the relevance of the putative hydrophobic pocket near G695. Combining lactam staples with the singleand double-tryptophan substitutions resulted in peptides with very high degrees of helical structure (80% and 90% helical for 16 and 17, respectively) and single-digit-micromolar inhibitory potency in the luciferase refolding assay (8.4 and 4.1 μM for 16 and 17, respectively). The most potent peptide tested, peptide 17, represents a fifty-fold improvement in activity over the native C-helix peptide. 4.1 μM for 16 and 17, respectively). The most potent peptide tested, peptide 17, represents a fifty-fold improvement in activity over the native C-helix peptide.
2.3. Testing peptides in a cell culture assay of Hsp90 inhibition
The most potent peptides were then examined for Hsp90 inhibition with the MDA-kb2 cell line.29 This cell line, derived from breast cancer cell line MDA-MB-453, contains a stably transfected firefly luciferase reporter under the control of a mouse mammary tumor virus promoter that contains response elements for both the androgen receptor (AR) and glucocorticoid receptor (GR). Both receptors are activated in an Hsp90-dependent manner, and the AR response in the MDA-kb2 cell line was previously used to test the effects of inhibitors of the Hsp90 cochaperone FKBP52 on the AR-Hsp90-FKBP52 complex.30,31 We surmised that this assay would be straightforward and convenient for assessing relative activities of Hsp90 inhibitors in cell culture. To our knowledge, this work represents the first report of this assay for the direct testing of Hsp90 inhibitors.
Cell-based assays were used to broadly assess the cell penetration, cytotoxicity, and cellular effects of each of our Hsp90 inhibitors. Specifically, we measured the dose dependence of each inhibitor’s effects on luciferase expression following activation of GR with dexamethasone, and LC50 values were also measured under identical conditions to quantitate cytotoxicity. As positive controls, we first tested the C-terminal inhibitors coumermycin A1 and novobiocin and the N-terminal inhibitor geldanamycin. All of these compounds suppressed the production of the Hsp90-dependent luciferase reporter, with some accompanying toxicity from novobiocin and coumermycin (Table 1). After validating with these two classes of Hsp90 inhibitors, we examined the effects of the C-helix (peptide 2) and derivatives 9–17. Several peptides (2, 11, 12, 16) showed neither activity nor cytotoxicity up to their solubility limits. Most peptides containing tryptophans (14, 15, and 17, but not 16) were cytotoxic without an appreciable effect on luciferase induction at sub-toxic concentrations. Visual inspection of cells treated with 14, 15, and 17 indicated a rapid and complete disruption of the plasma membrane, suggesting that these peptides are membrane-lytic and that toxicity is unrelated to Hsp90 inhibition. Within these peptides, a helical structure would position the tryptophan-substituted positions adjacent to the cationic face established by R687, R690, and K693 (Fig. 3). This patterning of aromatic and cationic side chains is known to cause helical peptides to be membrane-lytic. For example, in a series of amphipathic helices studied by Rekdal et al., this relative orientation of tryptophans and cationic residues correlated with increased cytotoxicity against a number of mammalian cell lines.32 Interestingly, peptide 7, which differs from 14 in that a leucine is substituted for G695 instead of a tryptophan, is less potent and less cytotoxic than 14. A notable exception is peptide 16, which exhibited no measurable toxicity up to its solubility limit (LC50 >27 μM). The reason why 16 was not lytic, despite the potent toxicity of non-stapled analog 14 and Y689W-substituted analog 17, is likely due at least in part to its constrained conformation; this remains a topic for future investigations.
Figure 3.
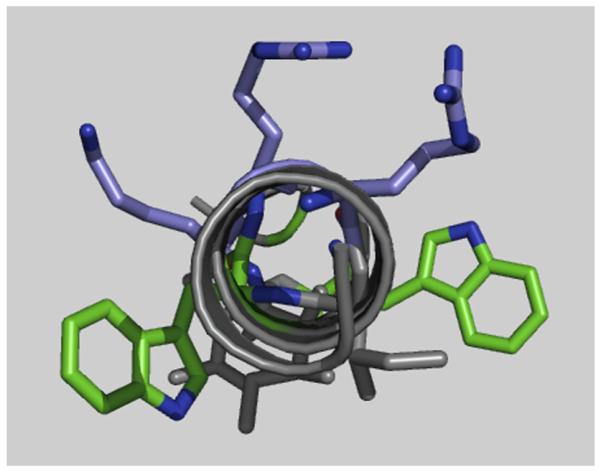
Relative orientation of tryptophans (green sticks) with respect to the cationic face established by two arginines and a lysine (purple sticks) within an idealized helical structure of peptide 10 (gray cartoon).
2.4. Fluorescence polarization competition assays provide evidence that C-helix peptides bind outside the N-terminal domain
Based on the design of these peptides, it is anticipated that they bind the C-terminal domain of Hsp90. Most demonstrations of C-terminal-domain binding have been cumbersome or indirect, using pull-downs with truncation mutants of Hsp90 or photoaffinity labeling.5,6 However, a fluorescence polarization assay using fluoresceinated geldanamycin has provided a more direct means of measuring competitive binding at the N-terminal domain’s ATP-binding site.33 We demonstrated displacement of fluorescein-geldanamycin by unlabeled geldanamycin and by another N-terminal domain inhibitor, radicicol. Under similar conditions, a thousand-fold excess of peptides 2, 9, 10, or 17 failed to displace fluorescein-geldanamycin (Fig. 4). Given the relative potencies measured in the luciferase renaturation assay, competition for N-terminal binding would be expected in this concentration range if these peptides inhibited Hsp90 via effects on the N-terminal domain’s ATP-binding site. This provides evidence against a mode of action involving the N-terminal domain of Hsp90. A dye-labeled analog of peptide 15 was tested by fluorescence polarization with several other proteins including cochaperone protein Aha1 to rule out nonspecific binding. Other than binding to bovine serum albumin at 1–10 μM, binding to other proteins was not observed up to 5 μM (see Supplementary Information). We synthesized and purified a fluorescein-labeled version of 17 to try to measure direct binding to full-length Hsp90, but no significant increase in polarization was observed at the maximum concentration of Hsp90 possible for this assay (see Supplementary Information). While the present evidence argues against binding or allosteric effects at the N-terminal domain’s ATP-binding site, direct binding to the C-terminal domain cannot be definitively established at this time.
Figure 4.
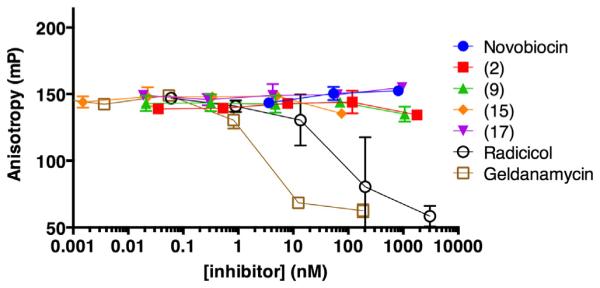
Displacement of fluoresceinated geldanamycin (1 nM) from human Hsp90 (90 nM) by known Hsp90 inhibitors and synthetic peptides.
3. Conclusion
Herein we report the design and in vitro characterization of peptides derived from the C-helix of Hsp90 as inhibitors of Hsp90’s C-terminal domain. These peptides inhibit Hsp90 in a manner distinct from the traditional paradigm of N-terminal domain inhibition. Within a set of C-helix peptides with substitutions and conformational constraints, we find that overall activity correlated with overall helical character. This supports the further design of constrained helices that target the C-terminal domain of Hsp90. Potency in an Hsp90-dependent luciferase renaturation assay was improved from 233 μM for the 18-residue C-helix alone to 4.1 μM for a lactam-stapled, Trp-substituted analog. These are relatively potent, considering that the most potent C-terminal domain inhibitors to date are synthetic novobiocin derivatives with EC50 values of 1–4.5 μM in the cell-based version of this assay.9 Cytotoxicity, likely via membrane lysis, was prominent for peptides with highly hydrophobic substitutions. This property is typical among early design iterations of stapled peptides, and future studies will optimize hydrophobic surface area, helical structure, and charge in order to minimize cytotoxicity and maximize cell penetration.27 Previous inhibitors of Hsp90’s C-terminal domain showed distinct differences in cellular effects compared to the more traditional N-terminal domain inhibitors.2 Therefore, it is anticipated that, once cytotoxicity is minimized, these peptides will be useful for further elucidating the unique consequences of inhibiting Hsp90’s C-terminal domain.
Supplementary Material
Acknowledgements
This work was supported by a Smith Family Foundation Award for Excellence in Biomedical Research to J.A.K. J.G. is supported by NIH/NIGMS IRACDA grant K12GM074869.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2014.06.006.
References and notes
- 1.Taipale M, Jarosz DF, Lindquist S. Nat. Rev. Mol. Cell Biol. 2010;11:515. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Carbonero R, Carnero A, Paz-Ares L. Lancet Oncol. 2013;14:e358. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 3.Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee W-C. J. Med. Chem. 2010;53:3. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]
- 4.Donnelly A, Blagg B. Curr. Med. Chem. 2008;15:2702. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BS. J. ACS Chem. Biol. 2011;6:800. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcu MG, Schulte TW, Neckers LJ. Natl Cancer Inst. 2000;92:242. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 7.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J. Org. Chem. 2008;73:2130. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 8.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. J. Org. Chem. 2008;73:8901. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang X, Hembruff S, Donnelly A, Rajewski RA, Blagg BS, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA. BMC Cancer. 2011;11:468. doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MG, Meli M, Dohi T, Fortugno P, Nefedova Y, Gabrilovich DI, Colombo G, Altieri DC. Cancer Cell. 2005;7:457. doi: 10.1016/j.ccr.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 11.Wu S, Dole K, Hong F, Noman ASM, Issacs J, Liu B, Li ZJ. Biol. Chem. 2012;287:19896. doi: 10.1074/jbc.M112.343848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horibe T, Kohno M, Haramoto M, Ohara K, Kawakami KJ. Transl. Med. 2011;9:8. doi: 10.1186/1479-5876-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horibe T, Kawamoto M, Kohno M, Kawakami KJ. Biosci. Bioeng. 2012;114:96. doi: 10.1016/j.jbiosc.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Vasko RC, Rodriguez RA, Cunningham CN, Ardi VC, Agard DA, McAlpine SR. ACS Med. Chem. Lett. 2010;1:4. doi: 10.1021/ml900003t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sellers RP, Alexander LD, Johnson VA, Lin C-C, Savage J, Corral R, Moss J, Slugocki TS, Singh EK, Davis MR, Ravula S, Spicer JE, Oelrich JL, Thornquist A, Pan C-M, McAlpine SR. Bioorg. Med. Chem. 2010;18:6822. doi: 10.1016/j.bmc.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramsey DM, McConnell JR, Alexander LD, Tanaka KW, Vera CM, McAlpine SR. Bioorg. Med. Chem. Lett. 2012;22:3287. doi: 10.1016/j.bmcl.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratzke C, Mickler M, Hellenkamp B, Buchner J, Hugel T. Proc. Natl. Acad. Sci. 2010;107:16101. doi: 10.1073/pnas.1000916107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee C-C, Lin T-W, Ko T-P, Wang AH-J. PLoS ONE. 2011;6:e19961. doi: 10.1371/journal.pone.0019961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kortemme T, Baker D. Proc. Natl. Acad. Sci. 2002;99:14116. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kortemme T, Kim DE, Baker D. Sci. Signal. 2004;2004:pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 22.Galam L, Hadden MK, Ma Z, Ye Q-Z, Yun B-G, Blagg BSJ, Matts RL. Bioorg. Med. Chem. 2007;15:1939. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngan C-H, Hall DR, Zerbe B, Grove LE, Kozakov D, Vajda S. Bioinformatics. 2012;28:286. doi: 10.1093/bioinformatics/btr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bock JE, Gavenonis J, Kritzer JA. ACS Chem. Biol. 2013;8:488. doi: 10.1021/cb300515u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chorev M, Roubini E, McKee RL, Gibbons SW, Goldman ME, Caulfield MP, Rosenblatt M. Biochemistry (Mosc.) 1991;30:5968. doi: 10.1021/bi00238a022. [DOI] [PubMed] [Google Scholar]
- 26.Blackwell HE, Grubbs RH. Angew. Chem., Int. 1998;37:3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 27.Walensky LD, Bird GHJ. Med. Chem. 2014:57. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gómez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Proc. Natl. Acad. Sci. 2010;107:11686. doi: 10.1073/pnas.1002498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson VS, Bobseine K, Lambright CR, Gray LE. Toxicol. Sci. 2002;66:69. doi: 10.1093/toxsci/66.1.69. [DOI] [PubMed] [Google Scholar]
- 30.Morishima Y, Murphy PJM, Li D-P, Sanchez ER, Pratt WBJ. Biol. Chem. 2000;275:18054. doi: 10.1074/jbc.M000434200. [DOI] [PubMed] [Google Scholar]
- 31.Leon JTD, Iwai A, Feau C, Garcia Y, Balsiger HA, Storer CL, Suro RM, Garza KM, Lee S, Kim YS, Chen Y, Ning Y-M, Riggs DL, Fletterick RJ, Guy RK, Trepel JB, Neckers LM, Cox MB. Proc. Natl. Acad. Sci. 2011;108:11878. doi: 10.1073/pnas.1105160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rekdal Ø, Haug BE, Kalaaji M, Hunter HN, Lindin I, Israelsson I, Solstad T, Yang N, Brandl M, Mantzilas D, Vogel HJJ. Biol. Chem. 2012;287:233. doi: 10.1074/jbc.M111.279281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du Y, Moulick K, Rodina A, Aguirre J, Felts S, Dingledine R, Fu H, Chiosis GJ. Biomol. Screen. 2007;12:915. doi: 10.1177/1087057107306067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.