

Abstract
Histone pre-mRNAs are cleaved at the 3′ end by a complex that contains U7 snRNP, the FLICE-Associated Huge protein (FLASH) and Histone pre-mRNA Cleavage Complex (HCC) consisting of several polyadenylation factors. Within the complex, the N-terminus of FLASH interacts with the N-terminus of the U7 snRNP protein Lsm11 and together they recruit the HCC. FLASH through its distant C-terminus independently interacts with the C-terminal SANT/Myb-like domain of Nuclear Protein, Ataxia-Telangiectasia locus (NPAT), a transcriptional co-activator required for expression of histone genes in S-phase. To gain structural information on these interactions, we used mass spectrometry to monitor hydrogen/deuterium (H/D) exchange in various regions of FLASH, Lsm11 and NPAT alone or in the presence of their respective binding partners. Our results indicate that the FLASH-interacting domain in Lsm11 is highly dynamic, while the more downstream region required for recruiting the HCC exchanges deuterium slowly and likely folds into a stable structure. In FLASH, a stable structure is adopted by the domain that interacts with Lsm11 and this domain is further stabilized by binding Lsm11. Notably, both H/D exchange experiments and in vitro binding assays demonstrate that Lsm11, in addition to interacting with the N-terminal region of FLASH, also contacts the C-terminal SANT/Myb-like domain of FLASH, the same region that binds NPAT. However, while NPAT stabilizes this domain, Lsm11 causes its partial relaxation. These competing reactions may play a role in regulating histone gene expression in vivo.
Keywords: U7 snRNP, FLASH, Lsm11, NPAT, Mass Spectrometry
Introduction
Animal replication-dependent histones mRNAs differ in many respects from all other eukaryotic mRNAs [1]. They lack a polyA tail and instead terminate in a conserved stem-loop that consists of a 6-base pair stem and a 4-nucleotide loop. The concentration of all classes of histone mRNAs is high in S-phase, ensuring timely and synchronous supply of all classes of histone proteins for de novo chromatin formation, and rapidly declines upon completion of DNA replication [1]. This tight cell cycle-dependent regulation of histone mRNA levels in animal cells is largely mediated by the unique stem-loop structure [1].
Generation of histone mRNAs during the S-phase results from simultaneous activation of transcription of histone genes and the unique 3′ end processing mechanism that converts histone pre-mRNAs into mature histone mRNAs. Transcription of all five classes of histone genes in S phase is coordinated by the transcriptional co-activator Nuclear Protein, Ataxia-Telangiectasia locus (NPAT) [2–5], which is extensively phosphorylated by cyclin E/Cdk2 within the C-terminal half at the G1-S phase transition. However, how this modification activates histone gene expression remains obscure [2,4]. Histone pre-mRNAs are cleaved 4–5 nucleotides after the stem-loop by a dedicated ribonucleoprotein processing complex that differs from the canonical cleavage and polyadenylation machinery [6]. At the core of this complex is U7 snRNP that consists of an approximately 60-nucleotiode U7 snRNA and a unique Sm ring, containing the U7-specific proteins Lsm10 and Lsm11 and five proteins shared with the spliceosomal snRNPs: SmB, D3, E, F and G [7,8]. The 5′ end of U7 snRNA is partially complementary to the histone downstream element (HDE), a 10–15 nucleotide sequence in histone pre-mRNA located 10–15 nucleotides after the stem-loop. The base pairing interaction between these two sequences brings the U7 snRNP to the vicinity of the cleavage site in the pre-mRNA substrate.
Lsm11 is significantly larger than any other Sm/Lsm protein and its extended N-terminal region interacts with the N-terminal region of the 220 kDa FLICE-Associated Huge Protein (FLASH) [9]. Together, Lsm11 and FLASH form a binding site for a subset of factors involved in cleavage and polyadenylation that we collectively refer to as the histone pre-mRNA cleavage complex (HCC) [10,11]. The HCC consist of symplekin, CstF64 and all five subunits of the cleavage and polyadenylation specificity factor (CPSF), including the CPSF73 endonuclease and its homologue, CPSF100. Of the multiple subunits of the HCC, only CPSF73, CPSF100, symplekin [11–14] and possibly CstF64 [15,16] are indispensable for 3′ end processing of histone pre-mRNAs.
In addition to the U7 snRNP, FLASH and the HCC, 3′ end processing of histone pre-mRNA also requires the Stem-Loop Binding Protein (SLBP) that tightly interacts with the conserved stem-loop structure [17] and stabilizes binding of the U7 snRNP to the HDE [18]. SLBP remains associated with the stem-loop in the mature histone mRNA following processing and is detected as a component of the histone mRNP in the cytoplasm [19].
In both vertebrates and Drosophila, biosynthesis of histone mRNAs takes place at Histone Locus Bodies (HLBs), microscopically detectable nuclear structures that assemble at large clusters of multiple histone genes [20]. HLBs are highly enriched in NPAT, FLASH and components of the core U7 snRNP, including Lsm10, Lsm11 and U7 snRNA [21]. It is believed that the assembly of HLBs improves the efficiency and fidelity of histone gene expression by concentrating the low abundance transcription and processing factors [22]. The HLB may also serve as a maturation site for the U7 snRNP where the core U7 snRNP particle is loaded with FLASH and all necessary polyadenylation factors and hence converted into an active endonuclease [23].
Identifying and characterizing the complex network of interactions that occur within the HLB is critical to understand both the biogenesis of this nuclear sub-domain and the mechanisms that tightly restrict expression of histone genes to the S-phase. We recently demonstrated that the C-terminal domain of FLASH directly interacts with the last 25 amino acids of NPAT [24]. Interestingly, a homologous SANT/Myb-like domain capable of interacting with the same region of NPAT also exists at the end of the unrelated vertebrate protein Yin Yang 1-associated protein-related protein (YARP). YARP and its Drosophila orthologue Mute are enriched in HLBs [23–25] and may function in part as a repressor of histone gene transcription [25,26]. Unpublished NMR spectroscopic data demonstrate that the C-terminal SANT/Myb-like domain in FLASH and YARP folds into three α-helices. The structure of the C-terminal region of NPAT is unknown [24]. No structural information is also available for the N-terminal regions of FLASH and Lsm11 in the free and bound state, in part due to low solubility of the untagged proteins.
To gain initial insight into structural aspects of proteins involved in histone gene expression, we used hydrogen/deuterium (H/D) exchange coupled to mass spectrometry (MS). In this method, a protein of interested is incubated in deuterated buffer and exchange of the backbone amide hydrogens for deuteria is monitored by mass spectrometry (MS). In principal, amide hydrogens that are part of a hydrogen bond networking do not exchange with solvent unless they are temporarily destabilized. For this reason, H/D exchange coupled to mass spectrometry is a valuable method of probing dynamics and mapping potential structural elements in the studied protein in solution [27,28]. H/D exchange has also been successfully applied to characterize changes in protein dynamics that occur as a result of protein-protein interactions and formation of large multi-subunit complexes. H/D exchange requires low protein concentrations and may be applied to proteins that are unsuitable for X-ray crystallography or exceed sizes amenable for NMR.
Using H/D exchange coupled to MS, we identified stable and highly dynamic regions in FLASH and Lsm11 and precisely delineated the domains that bring these two proteins together. Based on our studies, we propose that region of FLASH that interacts with Lsm11 is not limited to the N-terminus but also includes the distant C-terminal SANT/Myb-like domain. This unexpected interaction at the FLASH C-terminus may play a regulatory role by preventing formation of a complex between FLASH with NPAT and committing FLASH to formation of the fully assembled and processing-competent U7 snRNP.
Results
H/D exchange in the N-terminal region of FLASH
The first 138 amino acids of the nearly 2000 amino acids of FLASH are sufficient to interact with the N-terminal region of Lsm11 [9]. Lsm11-binding site in FLASH contains amino acids 100-135 and FLASH-binding site in Lsm11, amino acids 20-65 [29]. The N-terminal regions of FLASH and Lsm11 recruit a specific subset of polyadenylation factors termed Histone pre-mRNA Cleavage Complex (HCC) to the U7 snRNP [10] (Fig. 1). We refer to this composite U7 snRNP as holo-U7 snRNP to discriminate it from the core particle solely consisting of the U7-specific ring and the U7 snRNA (Fig. 1). The interaction of the FLASH/Lsm11 complex with the HCC depends on the highly conserved LDLY motif in FLASH (amino acids 55-58) and a less precisely defined region in Lsm11 located between amino acids 65-130 [10]. These two regions likely contact one or more polyadenylation factors of the HCC, although the exact network of interactions that bring this complex to FLASH and Lsm11 remains unknown.
Fig. 1. Key proteins involved in histone gene expression.
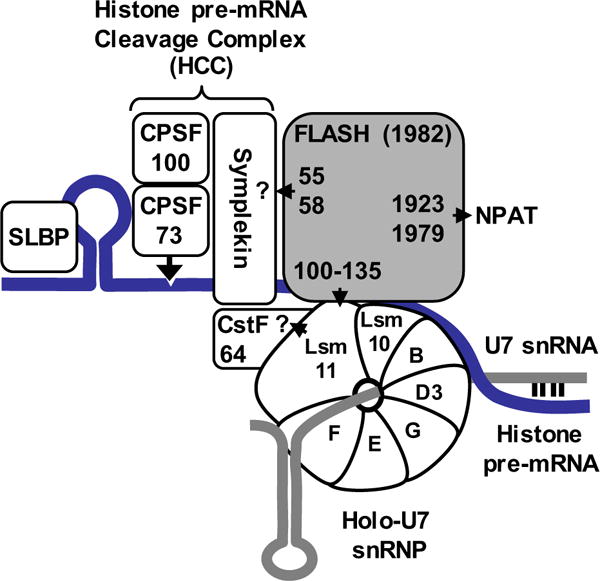
Known components of U7-dependent complex that cleaves histone pre-mRNA (blue line) at the 3′ end. 5′ end of U7 snRNA (grey line) base pairs with histone pre-mRNA. The U7 snRNA and the heptameric Sm ring together form the core U7 snRNP that upon interacting with FLASH recruits the HCC, giving rise to holo-U7 snRNP. Only four essential subunits of the HCC are shown: symplekin, CPSF100, CPSF73 and CstF64. Numbers in FLASH indicate amino acids that are required for the interaction of FLASH with the HCC, Lsm11 and NPAT. The arrangement of the four polyadenylation subunits in the HCC is hypothetical. The identity of the subunits that interact with FLASH and Lsm11 has not been determined, as indicated by question marks.
To identify potential structural elements in FLASH and better understand how FLASH may interact with its binding partners in processing, we monitored the fraction of exchange of the amide hydrogens with deuteria in three related FLASH variants: FLASH 159N, FLASH 138N and MiniFLASH. FLASH 159N contains the first 159 amino acids of FLASH, whereas FLASH 138N is its shorter version, containing amino acids 29-138 (Fig. 2A). MiniFLASH is a natural splice variant of full length FLASH in which the first 138 amino acids are directly linked to the last 52 amino acids, with the entire internal part being deleted via exon skipping (Fig. 2A) [24]. The C-terminal portion of MiniFLASH folds into a triple α-helical SANT/Myb-like domain and interacts with the C-terminal region of NPAT [24]. A homologous SANT/Myb-like domain with the ability to interact with NPAT is also present at the end of another HLB constituent of unknown role, YARP and its Drosophila orthologue, Mute [24,25]. As in our previous studies, the three FLASH variants were N-terminally fused to GST to facilitate their solubility and stability during bacterial expression. The GST fusion proteins retained the ability to interact with Lsm11 and fully supported processing of histone pre-mRNAs in vitro. These observations indicate that the GST tag had only minor, if any, effect on biological activity and structure of the expressed FLASH variants.
Fig. 2. Hydrogen/Deuterium (H/D) exchange in the N-terminal region of FLASH.
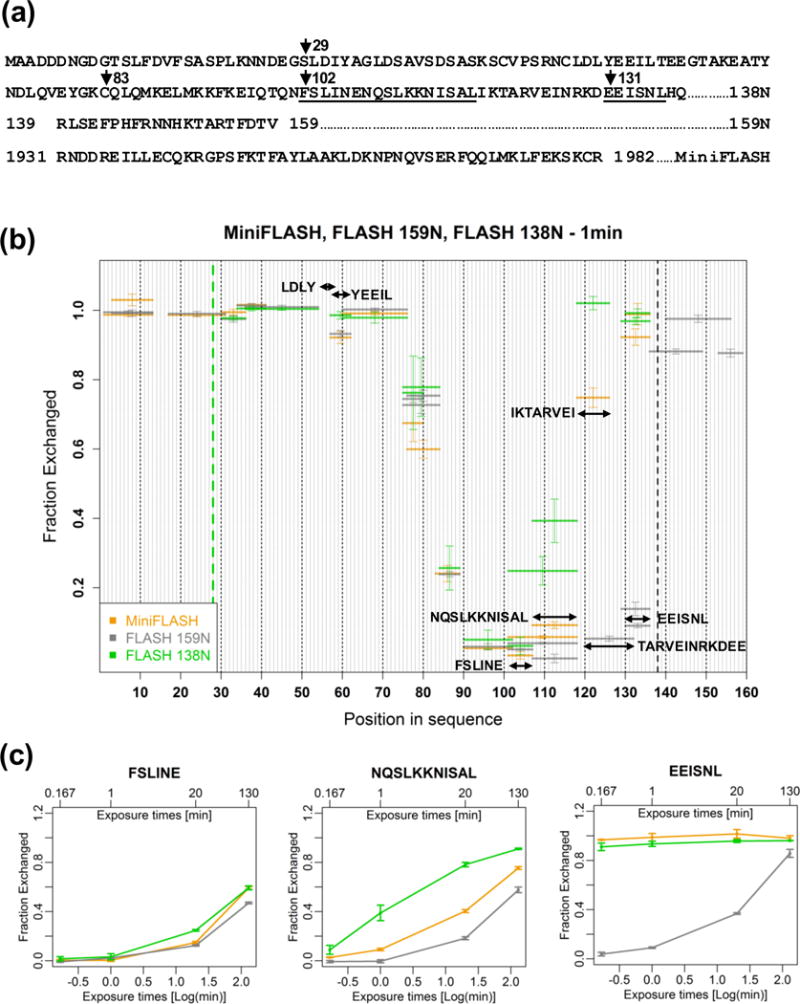
A. Sequences of the three FLASH variants, each containing a single GST tag at the N-terminus. FLASH 138N has amino acids 1–28 deleted. With the exception of cysteine 83 being mutated to alanine in FLASH 138N and FLASH 159N, the three proteins are identical in the region 29–138. In MiniFLASH, the first 138 amino acids are fused to the C-terminal region of FLASH (amino acids 1931-1982) as a result of alternative splicing. Peptides analyzed in panel C are underlined. B. H/D exchange patterns for FLASH 138N (green), FLASH 159N (grey) and MiniFLASH (orange) after 1 min incubation in D2O buffer. The horizontal lines indicate individual peptides and the error bars are standard deviations from at least three independent experiments. The green and black dashed lines delimit the N- and C-terminal regions, respectively, that differ among the three FLASH variants. The C-terminal domain of MiniFLASH is not shown in the plot. C. Time-dependent H/D exchange for three selected peptides (102FSLINE107, 108NQSLKKNISAL118 and 131EEISNL136) from FLASH 138N (green), FLASH 159N (grey) and MiniFLASH (orange). H/D exchange was analyzed at four time points plotted on a logarithmic scale. Time in minutes (0.167 min, 1 min, 20 min, 130 min) is shown at the top of the graph. Error bars represent standard deviations calculated from at least three independent experiments.
H/D exchange in FLASH 159N, FLASH 138N and MiniFLASH was monitored at different time points following the transfer of each protein to deuterated (D2O-containing) buffer, with each experiment being carried out at least three independent times. Student’s t-test was used to calculate the p-value for the same (or in few instances nearly identical) peptides identified in all three FLASH variants and results with the p-value ≤ 0.025 were considered as significantly different (Table S1).
In Fig. 2B, the fraction of exchange after 1 min was plotted for those peptides that were identified in all three FLASH variants, with the exception of the DSAVSDSASKSCVPSRNC peptide (amino acids 37-54) that was missing for MiniFLASH and the TARVEINRKDEE peptide (amino acids 121-132) that was identified only in FLASH 159N. In FLASH 138N and MiniFLASH this region is represented by the partially overlapping IKTARVEI peptide. Almost the entire N-terminal region (up to amino acid 75) in each FLASH variant displays rapid deuterium incorporation, with all amide protons being exchanged for deuteria within 1 min of incubation. The only notable exception in this region is the YEEIL peptide (amino acids 58-62) that contains the tyrosine (Y) from the highly conserved LDLY motif (amino acids 55-58). In FLASH 159N and MiniFLASH, this peptide compared to the surrounding sequences displays at 0.167 min (10 s) of incubation lower rate of deuterium uptake (Fig. S1). Interestingly, this slower exchange is not observed for the YEEIL peptide in FLASH 138N (Fig. S1). Overall, these observations may indicate that the YEEIL peptide and possibly the preceding LDL sequence that interacts with the HCC form a highly dynamic secondary structure and that the first 28 amino acids of FLASH are important for proper folding of this structure.
A region of a clearly reduced exchange begins between FLASH amino acids 85-90, with the position of the C-terminal flank of this region differing among the three proteins (Fig. 2B). The protected region is the longest in FLASH 159N and extends to amino acid 136, whereas in FLASH 138N and MiniFLASH it ends near amino acid 115 and 125, respectively, with the differences observed in this region between the analyzed FLASH variants being statistically valid (Table S1). Note that FLASH 159N, FLASH 138N and MiniFLASH share the same sequence up to amino acid 138, so the shift in the position of this upstream stability boundary likely reflects the effect of the unique C-terminus in each protein.
To better illustrate the differences between FLASH 159N, FLASH 138N and MiniFLASH, we compared time-course of H/D exchange for three peptides located downstream of amino acid 100 (Fig. 2C and Table S2). The FSLINE peptide (amino acids 102-107) displays a very similar kinetics of exchange in all three proteins, with less than 45% of hydrogens being exchanged for deuteria after 130 min of incubation in D2O buffer. The different behavior of FLASH 159N, FLASH 138N and MiniFLASH within the C-terminal region is evident for the neighboring peptide NQSLKKNISAL (amino acids 108-118), which in FLASH 138N exchanges substantially faster, and is most clear for the EEISNL peptide (amino acids 131-136). In FLASH 138N and MiniFLASH, this peptide undergoes almost complete exchange during 1 min of incubation in D2O buffer, with a comparable level of exchange for this peptide in FLASH 159N requiring 130 min (Fig. 2C).
Based on the results of H/D exchange we conclude that the first 75 amino acids of FLASH, with the exception of a short segment in the vicinity of the conserved LDLY motif, are dynamic in solution and likely lack any stable structural elements. In contrast, the region located downstream of amino acid 90, previously shown to interact with Lsm11 [29], displays severely slower exchange of backbone amide protons and likely adopts a stable structure. This structure extends to amino acid ~135 in FLASH 159N but is partially destabilized beyond amino acid 120 in FLASH 138N and MiniFLASH. Thus, the region of FLASH between amino acids 139-159 that is present only in FLASH 159N, although likely disordered on its own right based on the rapid H/D exchange, provides an optimal sequence context for the most efficient and stable folding of the upstream Lsm11-interaction site.
H/D exchange in FLASH bound to Lsm11
To identify potential conformational changes in FLASH upon interaction with the N-terminal Lsm11, we compared the level of H/D exchange in FLASH 159N in the apo-state and in the presence of Lsm11 (amino acids 1–169). Lsm11 was N-terminally fused to Maltose Binding protein (MBP), which efficiently prevented proteolytic degradation of Lsm11 during bacterial expression and was essential to maintain FLASH/Lsm11 complex in solution. The first 75-amino acids of FLASH remain highly dynamic when Lsm11 is bound, with virtually all amide protons being exchanged for deuteria within 10 s (0.167 min) of incubation (Fig. S2). The region located between residues 90 and 136 that shows reduced exchange in the unbound state becomes significantly more protected in the complex with Lsm11 and this effect is most visible after 130 min incubation (Fig. 3, Table S3). The NQSLKKNISALIK peptide (amino acids 108-120) shows the highest stability in the complex, with only 10% of hydrogens being exchanged after 130 min incubation. This contrasts with more than 60% of hydrogens that are exchanged during the same time in the unbound protein (Fig. 3A).
Fig. 3. H/D exchange in FLASH in the apo-state and bound to Lsm11.

A. FLASH 159N and an equimolar mixture of FLASH 159N and Lsm11 (amino acids 1–169) were incubated in D2O for 130 min and the extent of H/D exchange for the same peptides in the unbound (red) and Lsm11-bound (blue) state was determined by mass spectrometry. Results shown for each peptide are an average of at least three independent experiments, with the error bars representing standard deviations. The 108NQSLKKNISALIK120 peptide is indicated with an arrow and amino acid 138 is indicated with a black dashed line. B. Differential plot of H/D exchange presented in panel A. Δ Fraction Exchanged was calculated by subtracting Fraction Exchanged values for FLASH 159N in the presence of Lsm 169N (bound state, blue) from the values obtained in the apo-state (red). Peptides with p-values below 0.025 in the t-test are labeled in bright purple.
We also monitored H/D exchange in MiniFLASH and FLASH 138N bound to the N-terminal Lsm11 and obtained similar results. Again, the first 75 amino acids of FLASH remain highly dynamic in the complex and exchange virtually all amide protons within 1 min of incubation (Fig. S3 and not shown), whereas amino acids 100-120 are extremely rigid, with barely detectable exchange after 130 min of incubation (Fig. S4 and not shown). Importantly, this stabilizing effect was not observed when we analyzed the LI-AA mutant of FLASH 138N (amino acids 118-119) that is unable to interact with Lsm11 (not shown). Altogether, these data support the notion that the region of FLASH, previously identified as the Lsm11-binding site (amino acids 100-135), folds into a stable secondary structure and that this structure is further rigidified in the presence of Lsm11.
The effect of Lsm11 on the C-terminal SANT/Myb-like domain
When monitoring deuteria uptake by MiniFLASH in the presence of Lsm11, we noticed an unexpected change within the α-helical SANT/Myb-like domain (amino acids 139-190). This domain derives from the extreme C-terminus of full length FLASH (amino acids 1931-1982) and in MiniFLASH is directly appended to the N-terminal region of FLASH (amino acids 1-138) as a result of alternative splicing [24]. In contrast to the increased protection of the N-terminal FLASH that was observed in the presence of Lsm11, the C-terminal SANT/Myb-like domain of MiniFLASH in the bound state exchanged hydrogens for deuteria faster (Fig. S3).
The destabilizing effect of Lsm11 on the C-terminal domain of MiniFLASH could be indirect and reflect a conformational change in the SANT/Myb-like domain resulting from the interaction of Lsm11 with its binding site in the N-terminal region (amino acids 100-136). Alternatively, Lsm11 in addition to interacting with the N-terminus may independently recognize the SANT/Myb-like domain, directly affecting the stability of its α-helices. To distinguish between these two possibilities, we monitored H/D exchange in FLASH 60C that corresponds to the last 60 residues of FLASH (amino acids 1923-1982) (Fig. 4A). Unlike MiniFLASH, FLASH 60C contains 8 additional amino acids directly preceding the first α-helix of the SANT/Myb-like domain that improved protein expression and purification (Fig. 4A).
Fig. 4. Different effects of Lsm11 and NPAT binding on the SANT/Myb-like domain.
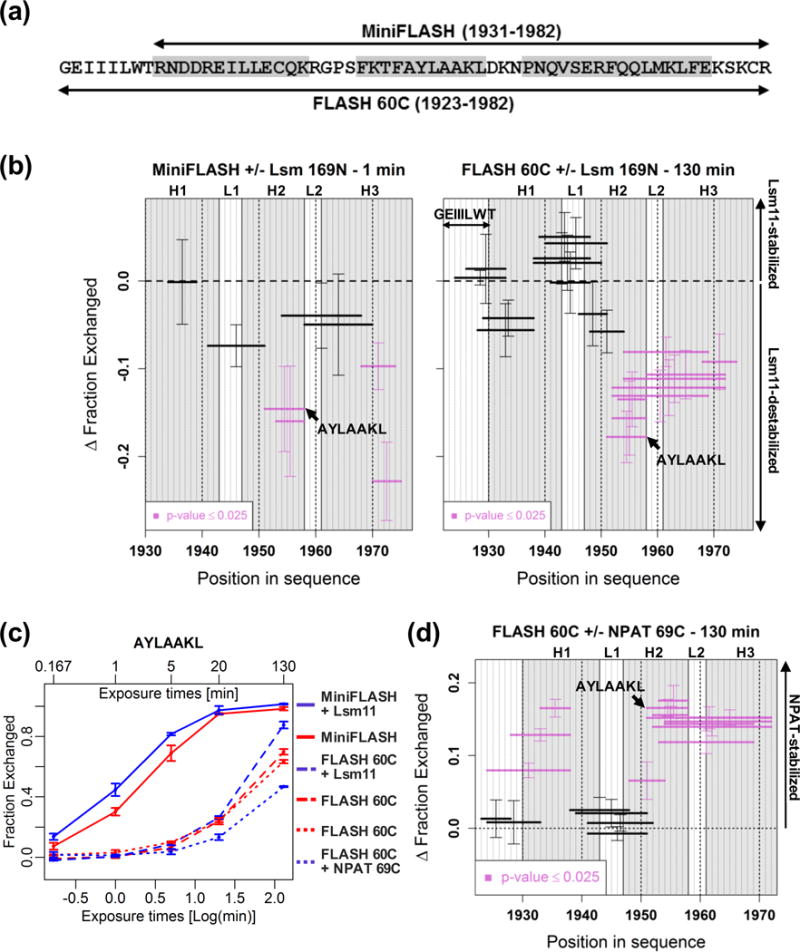
A. Amino acid sequence of the C-terminal region of FLASH in MiniFLASH (top) and FLASH 60C (bottom). Predicted α-helical structures are shown in grey. B. Differential plots of H/D exchange within the C-terminal domain of FLASH in MiniFLASH (left panel) and FLASH 60C (right panel). Δ Fraction Exchanged was calculated as described in Fig. 3B. α-helical structures (H1, H2 and H3) are marked in grey. Loop regions are indicated with L1 and L2. Positive and negative values indicate stabilization and destabilization of the C-terminal FLASH region, respectively. C. Kinetic plots of H/D exchange for the 1952AYLAAKL1958 peptide in MiniFLASH and FLASH 60C, either in the apo-state (red) or in a complex with Lsm 169N or NPAT 69C (blue), as indicated. D. Differential plots of H/D exchange for FLASH 60C bound to equimolar amounts of NPAT 69C after 130 min incubation in D2O buffer.
We first compared H/D exchange in MiniFLASH and FLASH 60C in the absence of Lsm11 and found that the SANT/Myb-like domain in MiniFLASH exchanges amide hydrogens faster than the same domain in FLASH 60C (Fig. S5A). Thus, the SANT/Myb-like domain in MiniFLASH may adopt a less stable conformation, likely due to the presence of the N-terminal FLASH region and/or the lack of the 8-amino acid region directly preceding the first α-helix. As noted above, Lsm11 increases the rate of exchange in the SANT/Myb-like domain in MiniFLASH (Fig. 4B, left panel), indicating that its α-helical structure is further destabilized. Strikingly, the same effect was also observed in the SANT/Myb-like domain of FLASH 60C (Fig. 4B, right panel), which lacks the N-terminal Lsm11-binding site, although it required a much longer incubation time (130 min), consistent with the overall slower H/D exchange in FLASH 60C. Destabilization was observed for peptides in all three α-helices, being most pronounced in helices 2 and 3 (Table S4).
To better illustrate time-dependent differences in Lsm11-mediated destabilization of the SANT/Myb-like domain in MiniFLASH and FLASH 60C, we compared incorporation of deuterium by a single peptide from helix 2, AYLAAKL, at five different time points. The accelerated rate of hydrogen/deuterium exchange by the AYLAAKL peptide resulting from binding of Lsm11 to MiniFLASH is readily visible at early time points (1 to 5 min) (Fig. 4C, solid lines, Table S5). In FLASH 60C, the stimulatory effect of Lsm11 on the uptake of deuterium by the AYLAAKL peptide can be readily observed after 130 min of incubation (Fig. 4C, dashed lines, Table S5), with the increase in fraction exchanged reaching nearly 20%, from ~70% for the unbound form to ~90% for the Lsm11-bound form (Fig. 4B, Δ Fraction Exchanged = −0.18). Altogether, our results demonstrate that Lsm11 (amino acids 1-169) partially destabilizes the SANT/Myb-like domain in both MiniFLASH and FLASH 60C, hence suggesting that the N-terminal region of Lsm11 directly interacts with the C-terminal region of FLASH.
H/D exchange in the SANT/Myb-like domain of FLASH bound to NPAT
We previously showed that the SANT/Myb-like domain of FLASH tightly interacts with the last 25 amino acids of NPAT [24]. Here, we tested how this domain in FLASH 60C is affected when it binds the last 69 amino acids of NPAT N-terminally tagged with 6xHis (NPAT 69C). In the unbound FLASH 60C, the fastest deuterium uptake is observed for peptides located within the unstructured GEIIILWT region located in front of the first α-helix, and peptides in the loop 1 that separates α-helices 1 and 2 (Fig. S5A, right panel). Peptides located entirely or largely within the α-helical regions display significantly slower rate of exchange. Importantly, equimolar amounts of NPAT 69C further stabilized the entire SANT/Myb-like domain of FLASH (Fig. 4D, Table S6), thus resulting in the opposite effect to that exerted by Lsm11 (Fig. 4B, right panel). This difference is best illustrated at 130 min time point for the AYLAAAKL peptide, which in the presence of NPAT 69C incorporates ~20% less deuteria than it does in its absence (Fig. 4C, compare blue and red dotted lines, Table S5). The same peptide in a complex with Lsm11 exchanges hydrogens ~20% more efficiently than it does in the apo-state (Fig. 4C, blue and red dashed lines). The stabilizing effect of NPAT 69C on FLASH 60C is even more pronounced when the two proteins are analyzed in a molar ratio of 5:1 (Fig. S5B). For peptides derived largely from the α-helical structures, this effect is most evident after 130 min of incubation, when on average 30% reduction in the level of exchange in the bound versus unbound state is observed (Fig. S5B). Much weaker stabilization during the entire time course of incubation occurs in the region overlapping with the first loop, as exemplified by the QKRGPSFKTF peptide (Fig. S5B), suggesting that the dynamics of this loop is not affected.
We conclude that NPAT stabilizes the α-helical structure of the SANT/Myb-like domain in FLASH and that this sharply contrasts with the effect exerted by Lsm11, which causes partial relaxation of the SANT/Myb-like domain.
Lsm11 directly interacts in vitro with the C-terminal domain of FLASH and competes for the same binding site with NPAT
An unexpected possibility suggested by our H/D exchange results is that Lsm11, in addition to having a primary binding site in the N-terminus of FLASH, may also independently interact with the SANT/Myb-like domain located at the distant C-terminus.
To test whether Lsm11 interacts with the C-terminal SANT/Myb-like domain of FLASH we used GST-pull down assay. In parallel, we tested the C-terminal SANT/Myb-like domain of YARP, which shares with the C-terminal domain of FLASH strong sequence similarity (Fig. 5A) and the ability to bind the C-terminal region of NPAT [24]. 35S-labeled N-terminal region of Lsm11 (Lsm 169N, amino acids 1-169) was synthesized in rabbit reticulocyte lysate and incubated with FLASH 60C fused N-terminally to GST, the same protein that was used in the H/D exchange experiments. In addition to GST-tagged FLASH 60C, we also used a longer C-terminal fragment of FLASH (FLASH 103C, amino acids 1880-1982), the corresponding C-terminal fragment of YARP (YARP 97C), MiniFLASH and FLASH 138N, all N-terminally fused to a GST tag. To control for the specificity of binding, the same GST-tagged proteins were in parallel incubated with 35S-labeled NPAT 131C containing the last 131 amino acids of NPAT [24]. 35S-labeled Lsm 169N or NPAT 131C bound to individual GST-tagged proteins were separated by gel electrophoresis and detected by autoradiography. In agreement with our previous study [24], MiniFLASH and FLASH 138N, both containing the N-terminal region, tightly interacted with Lsm11 (Fig. 5B, top, lanes 5 and 6) and FLASH 138N did not interact with NPAT 131C (Fig. 5B, bottom, lane 5). As expected, FLASH 60C, FLASH 103C and MiniFLASH (all three containing the C-terminal domain) bound NPAT 131C (Fig. 5B, bottom, lanes 3–4 and 6, respectively). Importantly, FLASH 60C and FLASH 103C in spite of lacking the N-terminal region of FLASH also bound Lsm 169N (Fig. 5B, top, lanes 3–4). Thus, both the H/D exchange and GST-pull down experiments support the conclusion that the C-terminal SANT/Myb-like domain of FLASH makes an independent contact with the N-terminal region of Lsm11. In contrast, YARP 97C containing the highly homologous and structurally related C-terminal domain of YARP failed to interact with Lsm 169N, although it efficiently interacted with NPAT 131C (Fig. 5B, lane 2, compare top and bottom panels). This result indicates that the interaction of the FLASH SANT/Myb-like domain with Lsm11 is highly specific.
Fig. 5. Binding of Lsm11 and NPAT to the C-terminal region of FLASH is mutually-exclusive.

A. BLASTP-generated amino acid sequence alignment of the C-terminal regions of human FLASH and YARP. Non-identical amino acids that are unlikely to affect the overall structure of each protein are indicated with the plus sign. α-helices are underlined. B. GST-tagged proteins indicated at the top of each lane were incubated with 35S-labeled Lsm11 169N (top panels) or NPAT 131C (bottom panels) and their binding analyzed by the GST pull down assay. Proteins were collected on glutathione beads, separated on a SDS/polyacrylamide gel and detected by either staining with Coomassie blue (GST proteins, bottom panels) or by autoradiography (35S-labeled proteins, top panels). C. Binding of GST-tagged YARP 97C and FLASH 60C, either alone (lanes 2 and 4) or pre-bound to NPAT 69C (lanes 3 and 5), to 35S-labeled Lsm11 169N.
We carried out GST-pull down assays to test whether the FLASH SANT/Myb-like domain can simultaneously interact with NPAT and Lsm11 or whether these interactions are mutually exclusive, with NPAT and Lsm11 competing for a shared binding site at the C-terminus of FLASH. 35S-labeled Lsm 169N was assayed for the interaction with the GST-tagged FLASH 60C in the unbound form or pre-bound to recombinant 6xHis-tagged NPAT 69C. As a specificity control, we used YARP 97C, which binds NPAT 69C but not Lsm 169N (Fig. 5C, lanes 2 and 3). In the absence of NPAT, FLASH 60C and Lsm 169N efficiently interacted with each other, with as much as 20% of the 35S-labeled input protein being immobilized on glutathione beads (Fig. 5C, lane 4). However, when FLASH 60C was pre-bound to NPAT 69C, it lost its ability to interact with Lsm 169N (Fig. 5C, lane 5), indicating that NPAT and Lsm11 compete for the same or overlapping binding site in the SANT/Myb-like domain of FLASH.
Mapping the region in Lsm11 that interacts with the C-terminal FLASH domain
In vitro GST pull down assays [29] and subsequent in vivo studies [30] previously demonstrated that Lsm11 interacts with the N-terminal FLASH through amino acids 20-65 (numbering in human Lsm11, Fig. 6A), with the FD dipeptide (amino acids 31-32, Fig. 6A, underlined) being essential for the interaction. Deleting the first 65 amino acids of Lsm11 [29] or introducing an FD-AA mutation severely affected the interaction of Lsm11 with FLASH 138N (Fig. 6B, bottom, lane 2), while having no detectable effect on the interaction with FLASH 60C (Fig. 6B, bottom, lane 3 and not shown). Thus, the FLASH SANT/Myb-like domain must interact with a region of Lsm11 between 66 and 169. Deleting the last 18 and 27 amino acids from Lsm 169N (Lsm 151N and Lsm 142N, respectively) did not affect the interaction with FLASH 60C (Fig. 6C, middle and bottom panels). However, removing the last 64 amino acids (Lsm 105N) was sufficient to reduce the interaction with the C-terminus of FLASH to the background level observed for YARP 97C (Fig. 6D, lanes 3 and 4, respectively). As expected, Lsm 105N retained the full ability to interact with FLASH 138N (Fig. 6D, lane 2). This mapping analysis indicates that the SANT/Myb-like domain of FLASH interacts in vitro with a region in Lsm11 located between amino acids 106-142.
Fig. 6. Mapping the region in Lsm11 that interacts with the C-terminal FLASH domain.

A. Amino acid sequence of the N-terminal region of human Lsm11 (1–169). Black arrows indicate end points of various deletion mutants used in the pull down experiments. The FD dipeptide critical for the interaction of Lsm11 with the N-terminal FLASH and the highly stable GDGAAGAGRRG peptide (see Fig. 7) are underlined. B. Binding of GST-tagged FLASH 138N or FLASH 60C (lanes 2 and 3, respectively) to 35S-labeled Lsm11 169N or its FD-AA mutant. C. Binding of GST-tagged FLASH 138N or FLASH 60C (lanes 2 and 3, respectively) to 35S-labeled Lsm11 169N (top panels) or its deletion mutants: 151N (middle panels) or 142N (bottom panels). D. Binding of GST-tagged FLASH 138N, FLASH 60C or YARP 97C to 35S-labeled Lsm11 105N.
H/D exchange in Lsm11 bound to FLASH
We used H/D exchange to monitor structural dynamics in Lsm11 before and after binding FLASH. We tested two FLASH variants, FLASH 138N and MiniFLASH that share the same N-terminal region, with only MiniFLASH containing the second potential Lsm11-binding site.
Multiple peptides were identified for the 65-amino acid N-terminal region of Lsm11 that includes FLASH-binding site, with the sequence coverage for this region reaching 100%. The N-terminal 25 amino acids, not involved in FLASH binding [29], exchanged all amide hydrogens within 1 min of incubation and the presence of either FLASH 138N (Fig. 7A) or MiniFLASH (Fig. S6A) did not detectably affect H/D exchange (Fig. 7B and S6B, Table S7). The adjacent 40-amino acid FLASH-binding site (amino acids ~25–65) displayed slower rate of deuterium incorporation, with peptides containing the essential FD motif being most resistant to the exchange (Fig. 7A and C, Table S7). This analysis indicates that the site in Lsm11 interacting with the N-terminal FLASH contains a relatively dynamic structure that is only partially rigidified in the complex.
Fig. 7. H/D exchange in Lsm11 bound to FLASH.

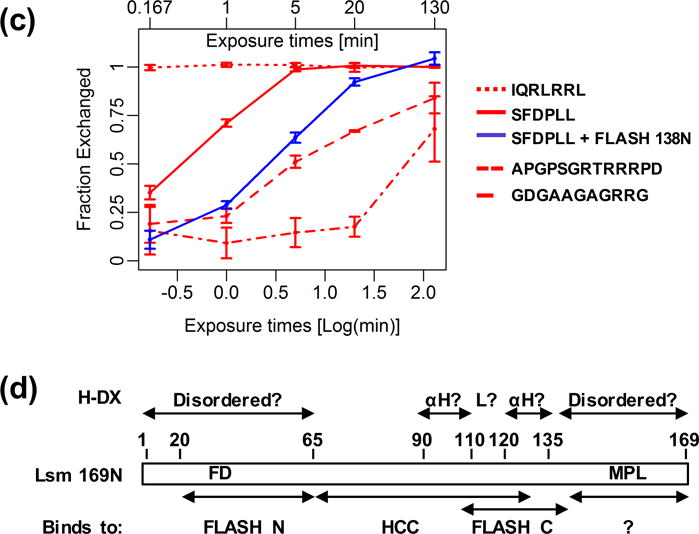
A. H/D exchange for Lsm11 (amino acids 1-169) in the apo-state (red) and bound to an equimolar amount of FLASH 138N (blue) after 1 min incubation in D2O buffer. Peptides analyzed in panel C are indicated with the arrows. B. Differential plot of H/D exchange presented in panel A. C. Time-dependent H/D exchange for indicated Lsm11 peptides. The 30SFDPLL35 peptide is shown in both the apo-state (red) and in a complex with FLASH 138N (blue). D. Localization of potential functional and structural elements within the N-terminal region of Lsm11 (amino acids 1-169), as predicted based on the H/D exchange and GST-pull down assays.
The central region of Lsm11 located between amino acids 65 and 140 yielded only few peptides and the reason for this limited sequence coverage is not clear. The entire region contains low complexity sequences and a high concentration of arginines and may be poorly digested with proteases. amino acids 80-140, with the exception of a short segment located between amino acids 110-120, exchange hydrogens for deuteria relatively slowly and they may fold into two stable α-helices that are separated by a less protected loop region. This region of Lsm11 is virtually unaffected when tested in the complex with FLASH 138N (Fig. 7B, Table S7), consistent with the N-terminal FLASH binding more upstream residues (amino acids 25-65). Interestingly, amino acids 80-140 overlap with a region that is essential for the interaction of Lsm11/FLASH complex with polyadenylation factors of the HCC [10] and, as indicated by our current study, also contains an element that interacts with the SANT/Myb-like domain of FLASH in vitro (Fig. 6).
To better illustrate differences in structural dynamics between different regions of Lsm11, we directly compared H/D exchange for 4 selected peptides in a time frame spanning 130 min (Fig. 7C). The SFDPLL peptide (amino acids 30-35) that is essential for binding the N-terminal FLASH, requires 5 min to exchange nearly all hydrogens with deuteria, whereas the IQRLRRL peptide (amino acids 110-116) achieves the same level of exchange after ~10 sec (Fig. 7C). The two remaining and most stable peptides that flank the IQRLRRL peptide, APGPSGRTRRRPD (amino acids 89-101) and GDGAAGAGRRG (amino acids 123-133), undergo only ~50 and 80% exchange, respectively, following 130 min of incubation in deuterated buffer. Notably, this exchange is slower than that observed for the SFDPLL peptide in a complex with the N-terminal FLASH (Fig. 7C), further arguing that Lsm11 amino acids 80-140 fold into a stable structure (Fig. 7D).
We did not observe any stabilizing effect of MiniFLASH on amino acids 65-142 of Lsm11 (Fig. S6) that in vitro mediates the interaction with the SANT/Myb-like domain of FLASH. However, a complete analysis of this region was hampered by a very few peptides recovered from the complex and the overall poor sequence coverage obtained in few independent experiments.
Discussion
Biosynthesis of replication-dependent histone mRNAs is strictly regulated during cell cycle and includes two major steps: transcription of histone genes and 3′ end processing of the resultant nascent histone pre-mRNAs. These steps are tightly coupled in vivo and simultaneously activated at the G1/S-phase transition. Transcription of all five classes of histone genes is controlled by NPAT, a transcriptional co-activator [2,4], whose function is likely mediated by number of histone subtype-specific transcription factors [31], including HiNF-P for histone H4 genes [5] and Oct-1 for histone H2B genes [32]. The activity of NPAT is under control of cyclin E/Cdk2, which also controls other aspects of cell metabolism during S-phase, including generation of the DNA replication machinery [33]. Hyper-phosphorylation of NPAT at multiple sites within the C-terminal half by this kinase complex at the G1/S-phase transition results in an increase in transcription of histone genes, although the details of this regulatory mechanism are largely unknown [2,4].
The co-transcriptional 3′ end processing of histone pre-mRNAs into mature mRNAs is mediated by a dedicated multi-subunit complex [6,8]. The holo-U7 snRNP is a key component in this complex and functions as a site-specific endonuclease that cleaves histone pre-mRNA 4–5 nucleotides downstream from a conserved stem-loop structure. The N-terminal region of Lsm11, the largest protein of the U7-specifc Sm ring, interacts with the N-terminal region of FLASH, and the two proteins recruit the HCC composed of a number of polyadenylation factors to the U7 snRNP, including the catalytic component CPSF73 [10,11].
We recently showed that the last 25 amino acids of NPAT interact with the SANT/Myb-like domain located at the C-terminal region of FLASH and that this interaction is essential for formation of Histone Locus Bodies (HLBs) [24]. The same region of NPAT also interacts with a homologous SANT/Myb-like domain located near the C-terminus of YARP, an HLB resident, which likely functions as a repressor of histone gene transcription [25].
Structural “landscape” of the N-terminal FLASH and Lsm11 predicted by H/D exchange coupled to MS
We used mass spectrometry to monitor exchange of hydrogens for deuteria in FLASH and Lsm11 individually or in their complex. Peptides that display slow exchange typically coincide with regions of dense hydrogen bonding that is indicative of local secondary structures, although occasionally they may derive from buried parts of the protein where amide protons are less accessible to solvent or from rigidified exposed loops. As such, H/D exchange is a method of choice for mapping regions of restricted dynamics in studied proteins that will need to be further characterized by more direct methods of structural analysis, including NMR or X-ray crystallography. Note that for technical reasons, we studied H/D exchange in fragments of FLASH and Lsm11 and as a result we were unable to detect long distance interactions that may occur in full length proteins and their potential effects on the interaction between FLASH and Lsm11.
Using H/D exchange combined with mass spectrometry we demonstrate that the FLASH region required to bind Lsm11 (amino acids 100-136) exchanges amide protons with deuteria at the lowest rate, indicating that it folds into a stable structure. A relatively slow exchange also occurs within the preceding 25 amino acids, suggesting that amino acids 75-136 form a continuous and well defined structural unit. The entire region located between amino acids 90-136 becomes further stabilized in the presence of Lsm11, with the strongest reduction in H/D exchange being observed for amino acids 100-120, which are likely at the core of the Lsm11-binding site. We previously showed that an alanine substitution of the LI dipeptide located in this region abolished the interaction between human FLASH and Lsm11 in vitro [29]. Moreover, a point mutation of the corresponding dipeptide in Drosophila FLASH had the same effect on binding Drosophila Lsm11 in vitro and abolished processing in vivo [30]. Based on H/D exchange, the Lsm11-binding site acquires the most stable conformation in FLASH 159N, i.e. when it is followed by ~25 residues of the natural FLASH sequence (amino acids 137-159). This downstream region shows rapid H/D exchange, suggesting lack of well-defined structure. Nevertheless, it may provide an optimal sequence context for the most efficient folding of the Lsm11-binding site.
GST-pull downs assays and mutagenesis studies mapped the FLASH-binding site in Lsm11 to amino acids 20-65 [29]. This Lsm11 region in the unbound form rapidly exchanges hydrogens, suggesting that it lacks a stable secondary structure but gains significant protection in the presence of FLASH (Fig. 7A and B, Table S7). A lower rate of hydrogen/deuterium exchange was observed for amino acids 80-138 of Lsm11, with the exception of a short segment located near amino acid 110 that rapidly incorporates deuteria. The stable regions may correspond to α-helical structures separated by a dynamic loop region (Fig. 7D). The region of Lsm11 located between amino acids 105-130 was previously shown to be indispensable for the interaction with HCC [10]. In contrast to this structured domain, the majority of the N-terminal region of FLASH exchanges all amide hydrogens within 1 min and is likely either disordered or folds into a highly dynamic structure. Interestingly, a noticeable although relatively minor reduction in deuterium uptake within this region is observed for the YEEIL peptide (amino acids 58-62) that directly precedes the LDLY motif required for the interaction of FLASH with the HCC. This slower exchange may indicate that the YEEIL peptide alone or in conjunction with the immediately upstream sequence folds into a highly dynamic secondary structure.
The C-terminal region of FLASH interacts with both NPAT and Lsm11
In MiniFLASH, a natural splice variant of FLASH, the N-terminal region of 138 amino acids that interacts with Lsm11 and the HCC is directly juxtaposed to the last 52 amino acids that fold into a SANT/Myb-like domain and interact with NPAT [24]. The SANT/Myb-like domain of FLASH consists of three α-helices and our H/D exchange studies indicate that binding of NPAT to this domain stabilizes all three α-helices but has no major effect on the loop 1 located between helices 1 and 2. Our H/D exchange studies also raise an intriguing possibility, independently supported by GST pull-down assays that the SANT/Myb-like domain of FLASH, in addition to interacting with NPAT, also interacts with the first 169 amino acids of Lsm11. However, in contrast to NPAT, Lsm11 upon interacting with the SANT/Myb-like domain of FLASH results in a significant increase in H/D exchange by this domain, likely reflecting partial destabilization of its α-helical structure. This suggests that formation of a complex between Lsm11 and the C-terminal FLASH primarily involves side chain interactions. Importantly, Lsm11 does not bind in vitro to SANT/Myb-like domain of YARP. Thus, the interaction of the SANT/Myb-like domain of FLASH with Lsm11 depends on residues and/or biochemical properties that are not conserved in the YARP domain.
Our mapping experiments indicate that the interaction of Lsm11 with the C-terminal region of FLASH requires a region in Lsm11 located between amino acids 105-142. Thus, this additional FLASH-binding site in Lsm11 may be directly adjacent to or overlapping with the site that interacts with the HCC (Fig. 7D).
Possible role of Lsm11 binding to the C-terminus of FLASH
We previously showed that the interaction between Lsm11 and the N-terminal region of FLASH is sufficient to recruit the HCC to the U7 snRNP in vitro [10]. In the linear sequence of full length FLASH, the two Lsm11-binding sites are located at the opposite ends, nearly 1,800 amino acids away, but in the FLASH/Lsm11 complex they may form a continuous platform for binding Lsm11 (Fig. 8). In MiniFLASH, these two regions are physically linked due to a splicing event that results in skipping of a large internal part of the protein.
Fig. 8. A possible set of events that occur at the G1/S phase transition, resulting in activation of histone gene expression.

FLASH and NPAT interact through their C-terminal regions (indicated by the double-headed arrow), forming a repressive complex that prevents each protein from performing its function in histone gene expression. Phosphorylation of NPAT by Cyclin E/CDK2 at the end of the G1 phase disrupts this complex and activates both transcription of histone genes by liberating NPAT and 3′ end processing of histone transcripts by promoting FLASH-dependent assembly of the active U7 snRNP. The Lsm11-binding sites in the N- and C-terminal region of FLASH are indicated with a long and short vertical arrow, respectively.
Binding of Lsm11 and NPAT to the SANT/Myb-like domain of FLASH are mutually exclusive, suggesting that they compete for the same or overlapping binding sites at the C-terminus of FLASH. Alternatively, binding of one protein to its target site may result in a conformational shift of a separate site in the SANT/Myb-like domain, rendering it incompetent for the interaction with the other protein. This latter allosteric mechanism is consistent with our H/D exchange results, which revealed that binding of Lsm11 and NPAT to the SANT/Myb-like domain of FLASH results in opposite effects on its α-helical structure, with NPAT making all three α-helices more compact and Lsm11-causing their partial relaxation.
Determining whether the interaction between Lsm11 and the C-terminal region of FLASH is biologically significant will require detailed in vivo studies. We propose that in early G1, de novo synthesized FLASH and NPAT form a tight complex that is localized to the nucleus, where it primarily functions to initiate the assembly of Histone Locus Bodies (Fig. 8). In support of this possibility, removing the extreme C-terminal region in FLASH or NPAT prevents formation of HLBs [24,34]. In addition, RNAi-mediated downregulation of one protein results in co-depletion of the binding partner [34,35], indicating that each protein is unstable outside the heterodimer. In early G1, the U7 snRNP may enter the HLB as a separate entity, consisting solely of the U7 snRNA and the Sm ring (core U7 snRNP), as suggested by our studies in Drosophila [30].
FLASH and NPAT bound together may be functionally inert in transcription and 3′ end processing, respectively, actively preventing synthesis of histone mRNAs outside S-phase. Phosphorylation of NPAT by cyclin E/Cdk2 at the end of the G1 phase may result in disrupting this repressive complex, triggering simultaneous activation of histone gene transcription by NPAT and 3′ end processing of histone pre-mRNAs through promoting the assembly of the functional U7 snRNP containing FLASH and the CPSF73 endonuclease (Fig. 8). This hypothetical regulatory mechanism is reminiscent of the Cyclin E/Cdk2-mediated liberation of the E2F transcriptional factor from its repressive complex with pRB and transcriptional activation of genes encoding components of the replication machinery at the onset of the S phase [36]. Interaction of Lsm11 with both the N-terminal and C-terminal FLASH may facilitate appropriate folding of FLASH and prevent re-association of FLASH with NPAT via their C-terminal regions, as envisioned in Fig. 8. Alternatively, unlike the in vitro situation, both terminal domains of FLASH must be bound to Lsm11 to promote the assembly of the complete U7 snRNP particle that contains the CPSF73 endonuclease and is active in 3′ end processing of histone pre-mRNAs in vivo.
Materials and Methods
Protein cloning and expression
Recombinant proteins used in this study were expressed in bacteria, depending on the construct at 18°C, 25°C or 37°C, with 0.5 mM IPTG for 5 to 16 hours and purified on nickel beads (Qiagen) or Glutathione Sepharose (GE Healthcare Life Sciences), as described by the manufacturers. The following proteins were cloned in the pET-42a vector and expressed as fusions with an N-terminal 6xHis tag followed by glutathione S-transferase (GST): FLASH 138N (amino acids 29-138, with C83 changed to alanine to prevent disulfide bond-mediated protein aggregation), FLASH 159N (amino acids 1-159, also containing the C83A mutation), MiniFLASH (amino acids 1-138 fused to amino acids 1931-1982), FLASH 60C (amino acids 1923-1982), FLASH 103C (amino acids 1879-1982), YARP 97C (amino acids 2149-2246), NPAT 69C (amino acids 1359-1427) and NPAT 131C (amino acids 1296-1427). YARP 61C (amino acids 2149-2209) and NPAT 69C were cloned in pET-28a and expressed as fusions with an N-terminal 6xHis tag. The following proteins were cloned in the pDEST566 vector and expressed, as previously described [10], as fusions with an N-terminal 6xHis tag followed by maltose binding protein (MBP): Lsm 169N (Lsm11 amino acids 1-169), Lsm 151N (amino acids 1-151), Lsm 142N (amino acids 1-142) and Lsm 105N (amino acids 1-105). For 35S-labeling, proteins were cloned into a vector containing a promoter for SP6 RNA polymerase [10] and synthesized in the presence of [35S] methionine using rabbit reticulocyte or wheat germ lysate (Promega), as recommended by the manufacturer.
GST Pull down assay
Appropriate pairs of GST-tagged and 35S-labeled proteins were briefly incubated on ice in a binding buffer (20 mM Hepes pH 7.9, 100 mM KCl, 10% glycerol, 0.01% NP-40) and rotated with glutathione beads for 1 hour. Immobilized proteins were extensively washed with the same buffer, separated on SDS-polyacrylamide gels, and detected by Coomassie staining (recombinant proteins) and autoradiography (35S-labeled proteins). In the competition experiment, FLASH 60C was pre-bound to NPAT 69C for 30 min and subsequently incubated with 35S-labeled Lsm 169N.
Hydrogen Deuterium (H/D) exchange coupled to mass spectrometry (MS)
The method was performed as described previously [37], with minor modifications. Initially, the list of peptides was established by diluting each analyzed protein to 5–10 μM in a non-deuterated buffer (20 mM Hepes pH 6, 100mM KCl). Each sample (50 μL) was acidified by adding formic acid to final 1%, combined with 10 μL of protease from Aspergillus saitoi (Sigma-Aldrich) in 1% formic acid and offline digested in a thermos shaker (Ditabis) for 2 minutes at 2 °C followed by online digestion in an immobilized pepsin resin column (Poroszyme, ABI). Peptides were passed directly to a trapping column (Acquity BEH C18, Waters) and eluted onto a reversed phase column (Acquity UPLC BEH C18, Waters) using an 8–40 % gradient of acetonitrile in 0.1 % formic acid with a total running time of 13.5 minutes. The C18 column outlet was directly coupled to the ion source of SYNAPT G2 HDMS mass spectrometer (Waters) working in Ion Mobility mode. Mass spectra were acquired in MSE mode over the m/z range of 50–2000 and peptides were identified using ProteinLynx Global Server software (Waters) and analyzed using the DynamX 2.0 program (Waters). For H/D exchange experiments, proteins samples were diluted in a buffer containing 99.8% D2O (Cambridge Isotope Laboratories, Inc.) and incubated at room temperature for 10 sec, 1 min, 5 min, 20 min and 130 min before reducing pH to 2.5. Two control experiments were carried out to account for in- and out-exchange artifacts. In-time point controls were performed by adding a protein to D2O buffer acidified to 1% formic acid with 10 μL of protease from Aspergillus saitoi to obtain minimum exchange for each peptide. Out-time point controls were performed by incubation of the protein in D2O buffer for 20 hours to obtain maximum exchange for each peptide. Each experiment was repeated at least three times and the results represent the mean of all replicates.
Data analysis
Mass of each peptide was calculated with the DynamX 2.0 software (Waters), using the list of peptides created in the PLGS program and the following criteria: randomized database and false discovery rate parameter below 4%, minimum intensity threshold of 3000, minimum fragmentation products per amino acids in precursor of 0.3, mass difference between measured and theoretical value for parent ions below 10 ppm. Analyses of the isotopic envelopes were carried out using the following parameters: retention time deviation ± 18 sec, m/z deviation ± 15 ppm, drift time deviation ± 2 time bins. The values reflecting experimental mass of each peptide in all possible states, replicates, time points and charge states were exported from the DynamX 2.0 and further data analysis was carried out using in house scripts written in R language (http://www.R-project.org). Fraction Exchanged (D) was calculated with the following formula:
where (Mex0) and (Mex100) indicate the minimum and maximum exchange, respectively. Error bars for fraction exchanged represent standard deviations calculated from at least three independent experiments. The difference in the Fraction Exchanged (Δ Fraction Exchanged) was calculated by subtracting the values for peptides in the complex from the values for the same peptides in the apo-state, with the error bars being calculated as the square root of the sum of the variances from compared states. Student’s t-test for two independent samples with unequal variances and sample sizes (also known as Welsh t-test) was carried out to evaluate differences between the same peptides in two different states (bound and unbound form) or from two different protein constructs.
Supplementary Material
Highlights.
Proteins involved in processing of histone pre-mRNAs form a complex network of interactions.
Several interactions within the complex were characterized by hydrogen/deuterium exchange.
Our study reveals an unexpected interaction of Lsm11 with the C-terminal region of FLASH.
This interaction may play a role in regulating histone gene expression in vivo.
Acknowledgments
This work was supported by the grants POIG.02.02.00-00-025/09, CEPT and TEAM/2011-7/1 to M.D. and NIH grant GM29832 to W.F.M. and Z.D.
Abbreviations
- FLASH
FLICE-Associated Huge protein
- NPAT
Nuclear Protein, Ataxia-Telangiectasia locus
- YARP
Yin-Yang 1-Associated protein-Related Protein
- H/D exchange
Hydrogen/Deuterium exchange
- MS
Mass Spectrometry
References
- 1.Marzluff WF. Metazoan replication-dependent histone mRNAs: a distinct set of RNA polymerase II transcripts. Curr Opin Cell Biol. 2005;17:274–280. doi: 10.1016/j.ceb.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 2.Ma TL, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW. Cell cycle-regulated phosphorylation of p220NPAT by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes and Development. 2000;14:2298–2313. doi: 10.1101/gad.829500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye X, Wei Y, Nalepa G, Harper JW. The cyclin E/Cdk2 substrate p220(NPAT) is required for S-phase entry, histone gene expression, and Cajal body maintenance in human somatic cells. Mol Cell Biol. 2003;23:8586–8600. doi: 10.1128/MCB.23.23.8586-8600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao JY, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes and Development. 2000;14:2283–2297. [PMC free article] [PubMed] [Google Scholar]
- 5.Miele A, Braastad CD, Holmes WF, Mitra P, Medina R, Xie R, Zaidi SK, Ye X, Wei Y, Harper JW, Van Wijnen AJ, Stein JL, Stein GS. HiNF-P directly links the cyclin E/CDK2/p220NPAT pathway to histone H4 gene regulation at the G1/S phase cell cycle transition. Mol Cell Biol. 2005;25:6140–6153. doi: 10.1128/MCB.25.14.6140-6153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominski Z, Carpousis AJ, Clouet-d’Orval B. Emergence of the beta-CASP ribonucleases: Highly conserved and ubiquitous metallo-enzymes involved in messenger RNA maturation and degradation. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbagrm.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Schumperli D, Pillai RS. The special Sm core structure of the U7 snRNP: far-reaching significance of a small nuclear ribonucleoprotein. Cell Mol Life Sci. 2004;61:2560–2570. doi: 10.1007/s00018-004-4190-0. [DOI] [PubMed] [Google Scholar]
- 8.Dominski Z, Marzluff WF. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene. 2007;396:373–390. doi: 10.1016/j.gene.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang XC, Burch BD, Yan Y, Marzluff WF, Dominski Z. FLASH, a proapoptotic protein involved in activation of caspase-8, is essential for 3′ end processing of histone pre-mRNAs. Mol Cell. 2009;36:267–278. doi: 10.1016/j.molcel.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang XC, Sabath I, Debski J, Kaus-Drobek M, Dadlez M, Marzluff WF, Dominski Z. A Complex Containing the CPSF73 Endonuclease and Other Polyadenylation Factors Associates with U7 snRNP and Is Recruited to Histone Pre-mRNA for 3′-End Processing. Mol Cell Biol. 2013;33:28–37. doi: 10.1128/MCB.00653-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabath I, Skrajna A, Yang XC, Dadlez M, Marzluff WF, Dominski Z. 3′-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. RNA. 2013;19:1726–1744. doi: 10.1261/rna.040360.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominski Z, Yang XC, Marzluff WF. The Polyadenylation Factor CPSF-73 Is Involved in Histone-Pre-mRNA Processing. Cell. 2005;123:37–48. doi: 10.1016/j.cell.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Kolev NG, Steitz JA. Symplekin and multiple other polyadenylation factors participate in 3′-end maturation of histone mRNAs. Genes Dev. 2005;19:2583–2592. doi: 10.1101/gad.1371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner EJ, Burch BD, Godfrey AC, Salzler HR, Duronio RJ, Marzluff WF. A genome-wide RNA interference screen reveals that variant histones are necessary for replication-dependent histone pre-mRNA processing. Mol Cell. 2007;28:692–699. doi: 10.1016/j.molcel.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Romeo V, Griesbach E, Schumperli D. CstF64: cell cycle regulation and functional role in 3′ end processing of replication-dependent histone mRNAs. Mol Cell Biol. 2014;34:4272–4284. doi: 10.1128/MCB.00791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruepp MD, Schweingruber C, Kleinschmidt N, Schumperli D. Interactions of CstF-64, CstF-77, and symplekin: implications on localisation and function. Mol Biol Cell. 2011;22:91–104. doi: 10.1091/mbc.E10-06-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan D, Marzluff WF, Dominski Z, Tong L. Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3′hExo ternary complex. Science. 2013;339:318–321. doi: 10.1126/science.1228705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dominski Z, Zheng LX, Sanchez R, Marzluff WF. Stem-loop binding protein facilitates 3′-end formation by stabilizing U7 snRNP binding to histone pre-mRNA. Mol Cell Biol. 1999;19:3561–3570. doi: 10.1128/mcb.19.5.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitfield ML, Kaygun H, Erkmann JA, Townley-Tilson WH, Dominski Z, Marzluff WF. SLBP is associated with histone mRNA on polyribosomes as a component of the histone mRNP. Nucleic Acids Res. 2004;32:4833–4842. doi: 10.1093/nar/gkh798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nizami Z, Deryusheva S, Gall JG. The Cajal body and histone locus body. Cold Spring Harb Perspect Biol. 2010;2:a000653. doi: 10.1101/cshperspect.a000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghule PN, Dominski Z, Yang XC, Marzluff WF, Becker KA, Harper JW, Lian JB, Stein JL, Van Wijnen AJ, Stein GS. Staged assembly of histone gene expression machinery at subnuclear foci in the abbreviated cell cycle of human embryonic stem cells. ProcNatlAcadSciUS A. 2008;105:16964–16969. doi: 10.1073/pnas.0809273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajendra TK, Praveen K, Matera AG. Genetic analysis of nuclear bodies: from nondeterministic chaos to deterministic order. Cold Spring Harb Symp Quant Biol. 2010;75:365–374. doi: 10.1101/sqb.2010.75.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White AE, Burch BD, Yang XC, Gasdaska PY, Dominski Z, Marzluff WF, Duronio RJ. Drosophila histone locus bodies form by hierarchical recruitment of components. J Cell Biol. 2011;193:677–694. doi: 10.1083/jcb.201012077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang XC, Sabath I, Kunduru L, Van Wijnen AJ, Marzluff WF, Dominski Z. A conserved interaction that is essential for the biogenesis of histone locus bodies. J Biol Chem. 2014;289:33767–33782. doi: 10.1074/jbc.M114.616466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulchand S, Menon SD, George SE, Chia W. Muscle wasted: a novel component of the Drosophila histone locus body required for muscle integrity. J Cell Sci. 2010;123:2697–2707. doi: 10.1242/jcs.063172. [DOI] [PubMed] [Google Scholar]
- 26.Lu P, Hankel IL, Hostager BS, Swartzendruber JA, Friedman AD, Brenton JL, Rothman PB, Colgan JD. The developmental regulator protein Gon4l associates with protein YY1, co-repressor Sin3a, and histone deacetylase 1 and mediates transcriptional repression. J Biol Chem. 2011;286:18311–18319. doi: 10.1074/jbc.M110.133603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engen JR, Smith DL. Investigating protein structure and dynamics by hydrogen exchange MS. Anal Chem. 2001;73:256A–265A. doi: 10.1021/ac012452f. [DOI] [PubMed] [Google Scholar]
- 29.Yang XC, Xu B, Sabath I, Kunduru L, Burch BD, Marzluff WF, Dominski Z. FLASH is required for the endonucleolytic cleavage of histone pre-mRNAs but is dispensable for the 5′ exonucleolytic degradation of the downstream cleavage product. Mol Cell Biol. 2011;31:1492–1502. doi: 10.1128/MCB.00979-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burch BD, Godfrey AC, Gasdaska PY, Salzler HR, Duronio RJ, Marzluff WF, Dominski Z. Interaction between FLASH and Lsm11 is essential for histone pre-mRNA processing in vivo in Drosophila. RNA. 2011;17:1132–1147. doi: 10.1261/rna.2566811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeRan M, Pulvino M, Greene E, Su C, Zhao J. Transcriptional activation of histone genes requires NPAT-dependent recruitment of TRRAP-Tip60 complex to histone promoters during the G1/S phase transition. Mol Cell Biol. 2008;28:435–447. doi: 10.1128/MCB.00607-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng L, Roeder RG, Luo Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell. 2003;114:255–266. doi: 10.1016/s0092-8674(03)00552-x. [DOI] [PubMed] [Google Scholar]
- 33.Zhao J. Coordination of DNA synthesis and histone gene expression during normal cell cycle progression and after DNA damage. Cell Cycle. 2004;3:695–697. [PubMed] [Google Scholar]
- 34.Kiriyama M, Kobayashi Y, Saito M, Ishikawa F, Yonehara S. Interaction of FLASH with arsenite resistance protein 2 is involved in cell cycle progression at S phase. Mol Cell Biol. 2009;29:4729–4741. doi: 10.1128/MCB.00289-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barcaroli D, Bongiorno-Borbone L, Terrinoni A, Hofmann TG, Rossi M, Knight RA, Matera AG, Melino G, De L V. FLASH is required for histone transcription and S-phase progression. Proc Natl Acad Sci USA. 2006;103:14808–14812. doi: 10.1073/pnas.0604227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes and Development. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 37.Kacprzyk-Stokowiec A, Kulma M, Traczyk G, Kwiatkowska K, Sobota A, Dadlez M. Crucial role of perfringolysin O D1 domain in orchestrating structural transitions leading to membrane-perforating pores: a hydrogen-deuterium exchange study. J Biol Chem. 2014;289:28738–28752. doi: 10.1074/jbc.M114.577981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.