

Abstract
Type II non-ribosomal peptide synthetases (NRPS) generate exotic amino acid derivatives that, combined with additional pathways, form many bioactive natural products. One family of type II NRPSs produce pyrrole moieties, which commonly arise from proline oxidation while tethered to a conserved, type II peptidyl carrier protein (PCP), as exemplified by PltL in the biosynthesis of pyoluteorin. We sought to understand the structural role of pyrrole PCPs in substrate and protein interactions through the study of pyrrole analogs tethered to PltL. Solution-phase NMR structural analysis revealed key interactions in residues of helix II and III with a bound pyrrole moiety. Conservation of these residues among PCPs in other pyrrole containing pathways suggests a conserved mechanism for formation, modification, and incorporation of pyrrole moieties. Further NOE analysis provided a unique pyrrole binding motif, offering accurate substrate positioning within the cleft between helices II and III. The overall structure resembles other PCPs but contains a unique conformation for helix III. This provides evidence of sequestration by the PCP of aromatic pyrrole substrates, illustrating the importance of substrate protection and biosynthetic regulation in type II NRPSs.
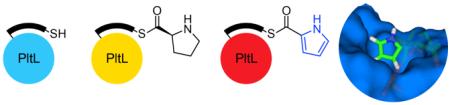
Type II non-ribosomal peptide synthetases (NRPSs) commonly combine with fatty acid and/or polyketide synthases (FAS / PKS) generating structurally diverse secondary metabolites with activities ranging from anti-infective to anti-cancer agents. The products of type II NRPSs are modified amino acids and are frequently aromatic. For example, proline, tyrosine, and salicylic acid are shuttled through type II NRPSs to form pyrrole, hydroxytyrosine, and methoxy-chlorosalicylic acid before incorporation into downstream pathways.1 These moieties are electron-rich rings that can participate in hydrogen bonding and π-stacking interactions that can contribute to the bioactivity of their respective product. The pyrrole containing compounds prodigiosin and chlorizidine A exhibit promising anti-tumor activity,2 for which a prodigiosin derivative recently entered stage II clinical trials (Fig. 1).3 Vancomycin (hydroxytyrosine) and chlorothricin (methoxy-chlorosalicylic acid) are known antibacterial and cholesterol reducing compounds, respectively.4 All type II NRPS components in these compounds are generated while covalently attached to a peptidyl carrier protein (PCP).
Figure 1.
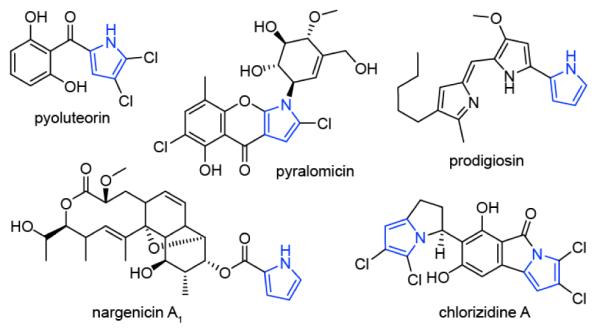
Structures of pyrrole containing natural products.
As shown in Scheme 1, the PCP is responsible for shuttling peptide substrates to partner enzymes for type II NRPS catalyzed modifications. To prime the apo-PCP, a 4′-phosphopantetheine (PPant) arm is first coupled to a conserved serine by a 4′-phosphopantetheinyl transferase (PPTase), generating holo-PCP. In systems that incorporate a pyrrole, an adenlyation domain serves to load proline (Pro) onto the holo-PCP. The resulting prolyl-PCP is then recognized by a flavoprotein dehydrogenase, which catalyzes a four–electron oxidation of Pro to the corresponding pyrrolyl-PCP.5 Further downstream processing can occur while tethered to the PCP, as illustrated by pyrrole halogenation in pyoluteorin biosynthesis (Scheme 1). In this system, the modified pyrrole is then transferred to a three module type I PKS system that facilitates elongation and reduction, followed by cyclization to pyoluteorin.6
Scheme 1.

Biosynthesis of pyoluteorin. Asterisks indicate inactive domain.
Given that PCP attachment is essential for product formation, protein-protein interactions between the PCP and partner enzymes are presumed to be critical, although the structural details of these binding events remain unknown. In pyoluteorin biosynthesis, Pro is loaded onto the PCP PltL by PltF and then oxidized to pyrrole by PltE (Scheme 1). The homologous adenylation and dehydrogenase enzymes in undecylprodigiosin biosynthesis cannot catalyze pyrrole formation with PltL,5 illustrating the importance of PCP identity, substrate demonstration and protein-protein interactions.
Substrate sequestration in carrier proteins has been recognized as an important phenomenon in type II fatty acid and polyketide synthases.7 Type II carrier proteins, as opposed to type I, are standalone enzymes that must recognize up to five partner proteins in a particular order. When not interacting with a partner, the carrier protein has been shown to protect the extending substrate from reactive compounds in the complex cytosol. Here the substrate localizes in a hydrophobic cleft between helix II and III of the acyl carrier protein (ACP). NMR structural studies of acylated E. coli fatty acid ACP reveals sequestration of chain lengths greater than C4,7d-e which shields the growing metabolite from nucleophiles other than the cognate partner protein.8 Similar interactions are also seen in type II PKS, where helix II and III of actinorhodin ACP were shown to interact with cyclic and linear intermediate analogs.7f-g While PCPs have been shown to interact with partner enzymes at the helix II/III interface,9 no studies have demonstrated PCP substrate sequestration. The overall helical structure of PCPs is similar in helix II and III, however amino acid distributions are notably diverse, creating significant electrostatic differences.10 These surfaces may be key to protection of various intermediates to discriminate for reactivity with proper partner enzymes. Structural studies of carrier proteins can provide insight into the regulation of substrate modification. Currently, there are no structures of a peptidyl-bound PCP, and there are only two structures of standalone type II PCPs, BlmI and A3404, both of which functions are unknown.10,11
Here we show that pyrrole PCPs actively sequester their pyrrole cargo. Bioinformatic analysis of PltL and other pyrrole PCPs revealed a secluded PCP subfamily with several conserved residues. Using chemoenzymatic techniques developed in our laboratory,12 we attached PPant-Pro and PPant-pyrrole mimetics to PltL to study interactions between the PCP and substrate. NMR analysis revealed direct interactions of the pyrrole with residues in helix II and III not observed in other PCPs.13 Through NOE techniques, the pyrrole acyl substrate was localized in a 3D solution NMR structure of PltL. The results shed light on how type II NRPS PCPs regulate peptidyl substrates and for the first time reveal direct interactions between the substrate and PCP.
Due to the unique nature of pyrrole biosynthesis, we first hypothesized that pyrrole PCPs could encompass a subfamily of PCPs. Sequence homology, generated with MUSCLE,14 was used to compare PCPs that tether pyrrole, Pro, and other amino acids as final products (Fig. S1). The comparison highlights conserved residues and polarity in pyrrole PCPs, particularly at the helix II N-terminus and in helix III. A phylogenetic tree generated with ClustalW15 further illustrates the seclusion of pyrrole PCPs (Fig. S2). By comparison, PCPs that load Pro as a final product are dispersed amongst PCPs that load other amino acids, emphasizing the unique conservations in pyrrole PCPs. Some of the conserved residues play an important part in substrate stabilization (as shown below) and possibly protein-protein recognition.
To analyze the interaction between PltL and peptidyl substrates, NMR experiments were considered. However, the natural thioester between the pantetheine and substrate is susceptible to hydrolysis in aqueous solution, and this instability is further aggravated under conditions for NMR studies.7a,16 Therefore, we prepared pantetheine mimetics with Pro and pyrrole for attachment onto PltL employing an amide linkage in lieu of the thioester. The prolyl-N-pantetheine analog was synthesized by coupling N-Boc-L-Pro to protected pantetheinamine under EDC coupling conditions. Global deprotection with strong acid (20% aq. TFA) yielded the L-prolyl-N-pantetheine analog (SI A.1). The pyrrolyl-N-pantetheine analog was prepared by coupling pyrrole-2-carboxylic acid to protected pantetheinamine under similar conditions. The p-methoxyphenylacetal protecting group was removed under mild acidic conditions (80% aq. AcOH), yielding the pyrrolyl-N-pantetheine analog (SI A.2).
These Pro and pyrrole probes were then analyzed for loading onto PltL via a one pot chemo-enzymatic strategy as previously described.12 Posttranslational modification was verified by urea PAGE and LCMS (Figures S3, S4). To locate which PltL residues are perturbed by addition of the Pro and pyrrole moieties, 1H-15N HSQC experiments were conducted on 15N-holo-PltL, 15N-prolyl-PltL, and 15N-pyrrolyl-PltL, and chemical shift perturbations (CSPs) were calculated (Fig. 2). Analysis revealed several key residues that were altered with the attachment of the Pro and pyrrole moieties (Fig. 2). Residues in helix II and proximal to Ser42 (covalently attached to PPant) showed significant movement, specifically Asn41, Ile45, Lys47, and Val50 (Fig 2a-2c). However the largest movements were seen in helix III, including residues Ile65, Thr66, and Phe70 (Fig. 2b-2c). The sidechains of Asn41, Lys47, and Val50 do not protrude towards the helix II/III cleft and may indicate general movement of helix II, although other residues, as mentioned below, have direct contact with the substrates. The CSP results of PltL coincide with other carrier protein-substrate interactions, where the majority of the interactions occur at helix II and III. Overall, the perturbations were larger for pyrrolyl-PltL compared to prolyl-PltL. This may indicate further sequestration or stabilization of the more hydrophobic pyrrole moiety.
Figure 2.

a, HSQC overlay of 15N-holo-PltL, 15N-prolyl-N-PltL, 15N-pyrrolyl-N-PltL. b, Highlights of 15N-HSQC data, illustrating perturbations of residues I65, T66, and F70. c, CSP plots of 15N-prolyl-PltL and 15N-pyrrolyl-PltL relative to 15N-holo-PltL.
In conjunction with the PltL perturbations, movement was also observed in protons of the pyrrole (Fig. 3). Due to the unique chemical shifts of pyrrole protons, a 1H-NMR experiment could be used to observe the protons in PltL-bound pyrrole. Three isolated peaks of the pyrrole shift significantly upfield when pyrrole-N-pantetheine is tethered to PltL, compared to the probe alone, or in solution but unattached to PltL (Fig. 3). The upfield shift indicates that the pyrrole is located in a more electron rich environment, in this case, the hydrophobic cleft between helix II and III. PltL was also shown to interact with the solvatochromatic compound 4-DMN, which correlates with the pyrrole proton upfield shifts (Fig. S15).17 The NMR and fluorescent data suggests that the pyrrole is solvent protected. A 1H-1H NOESY experiment was then performed on pyrrolyl-PltL to observe any specific proton interactions between the pyrrole and PltL residues. The pyrrole proton NOESY strips contained several peaks corresponding to Leu35 Hδ*, Ile45 Hδ1*, Ile65 Hγ2*/δ1*, and Thr66 Hα, all of which are located in the cleft of helix II and III (Fig. S16). Correspondingly, the pyrrole proton shifts can also be visualized within NOE strips of hydrophobic residues of the PltL cleft (Fig. S17).
Figure 3.

Pyrrole NMR shift analyses. a, 1H-NMR experiment of pyrrolyl-N-pantetheine probe isolated (red), with apo-PltL (blue), and covalently attached to PltL (green). The enlarged spectra reveal perturbations of pyrrole protons, suggesting pyrrole-PltL interactions. b, Structure of pyrrolyl-N-pantetheine probe.
To further visualize the interactions between PltL and peptidyl substrates, the solution NMR structures of both holo-PltL (PDB ID: 2N5H) and pyrrolyl-PltL (PDB ID: 2N5I) were determined with Cyana 3.97 (Fig. 4a-b, S18).18 The overall structure of PltL consists of four bundled helices as seen with all carrier protein structures (Fig. S19).9,10,11 PltL has both positive and negative electrostatic surfaces similar to other PCPs (Fig. S20), but different from FAS and PKS ACPs. which have majority negative surfaces.10 Unlike the multiple states observed for the type I excised PCP TycC3,19 a single state was observed for both PltL structures similar to that seen in other PCPs.10,13,20 Further similarities with PCPs include a linker region and helical turn between helix I and II and a Pro situated on the N-terminus of helix III.10,13 However, PltL contains Gly67 that interrupts the center of helix III, consequently positioning neighboring residues closer to helix II (Fig. 4c). For example, Ile65 and Thr66 are only in position to interact with peptidyl substrates due to the helical interruption. Several other pyrrole carrier proteins contain Gly and/or Pro in the middle of helix III, likely interrupting these helices (Fig. S1), while other PCPs do not contain a helix interrupter in helix III (Fig. S19). Interruption of helix III may be important for substrate and partner protein recognition in some of the pyrrole carrier proteins.
Figure 4.

Solution NMR structures of PltL species. a, holo-PltL. b, pyrrolyl-PltL. c, Expanded view of pyrrolyl-PltL with colors corresponding to CSP values from Fig. 2. Key residues are labelled. CSPs not determined are show in grey. d, Expanded view of surface-filled pyrrolyl-PltL structure illustrating pyrrole sequestration between the helix II/III cleft.
Spectroscopic observation of the PPant arm of holo-PltL was facilitated by 15N and 13C isotope labeling of the tethered molecule (Fig. S8, S10). This was achieved by co-transformation of PltL and Sfp constructs for in vivo production of holo-PltL (SI B.3). NOEs from the PPant to the protein were observed on both ends, but only intra-NOEs were observed in the middle of the PPant arm. These NOEs provided constraints between the thiol end and residues in both helix II and III, yielding a loop conformation with the thiol positioned between helix II and III.
The solution NMR structure of pyrrolyl-PltL was determined containing the solvent protected pyrrole localized between helix II and III (Fig. 4d, S21). The structure of pyrrolyl-PltL differs slightly with the holo-PltL, where the hydrophobic cleft between helix II and III expands to accommodate the pyrrole. The PPant in holo-PltL has more constraints than in pyrrolyl-PltL, possibly due to the pyrrole displacing the PPant further into solution. The position of the pyrrole protects the reactive 4 and 5 positions of the aromatic ring, which are eventually chlorinated (Scheme 1).21 In this position, the pyrrole is inaccessible to the halogenase PltA and, therefore, the interaction between PltL and PltA must induce a conformational change in PltL to display the pyrrole to PltA. This structural data highlights substrate sequestration by PltL in the biosynthesis of pyrrole and points to a similar role for other PCPs in type II NRPS pathways.
In this report, the structure of the first pyrrole carrier protein was determined, and the protein’s interaction with substrate mimetics is revealed. This work illustrates the features of PCPs that are responsible for binding and protection of substrate intermediates and suggests the existence of similar PCP-dependent regulation in other type II NRPS biosynthetic pathways. For example, the methoxy-chlorosalicylic acid in chlorothricin is generated while attached to the type II PCP ChlB2,1a which has high sequence identity to PltL (36%) and therefore may have similar sequestration events. These studies also provide important information toward understanding the protein interfaces that facilitate specificity within pyrrole biogenesis. Future work with type II NRPS enzymes will provide a more complete understanding of protein-protein recognition and offer the prospect of enhanced bioactive compounds via synthetic biology.
Supplementary Material
ACKNOWLEDGMENT
Funding was provided from NIH GM095970, and NIH/NCI T32 CA009523. We thank J. Beld for the solvachromatic probe, C. T. Walsh for the plasmid constructs, Drs. X. Huang and A. Mrse for NMR assistance, and Dr. Y. Su for MS services.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04525.
REFERENCES
- (1) (a).Jia X-Y, Tian Z-H, Shao L, Qu X-D, Zhao Q-F, Tang J, Tang G-L, Liu W. Chem. Biol. 2006;13:575. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]; (b) Thomas MG, Burkart MD, Walsh CT. Chem. Biol. 2002;9:171. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]; (c) Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13843. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hubbard BK, Walsh CT. Angew. Chem. Int. Ed. 2003;42:730. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- (2) (a).Ho T-F, Ma C-J, Lu C-H, Tsai Y-T, Wei Y-H, Chang J-S, Lai J-K, Cheuh P-J, Yeh C-T, Tang P-C, Tsai Chang J, Ko J-L, Liu F-S, Yen HE, Chang C-C. Toxicol. Appl. Pharm. 2007;225:318. doi: 10.1016/j.taap.2007.08.007. [DOI] [PubMed] [Google Scholar]; (b) Alvarez-Mico X, Jensen PR, Fenical W, Hughes CC. Org. Lett. 2013;15:988. doi: 10.1021/ol303374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Parikh SA, Kantarjian H, Schimmer A, Walsh W, Asatiani E, El-Shami K, Winton E, Verstovsek S. Clin. Lymphoma Myeloma Leuk. 2010;10:285. doi: 10.3816/CLML.2010.n.059. [DOI] [PubMed] [Google Scholar]
- (4) (a).Williams DH, Bardsley B. Angew. Chem. Int. Ed. 1999;38:1172. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (b) Kawashima A, Nakamura Y, Ohta Y, Akama T, Yamagishi M, Hanada K. J Antibiot (Tokyo) 1992;45:207. doi: 10.7164/antibiotics.45.207. [DOI] [PubMed] [Google Scholar]
- (5).Thomas MG, Burkart MD, Walsh CT. Chem. Biol. 2002;9:171. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]
- (6).Gross H, Loper JE. Nat. Prod. Rep. 2009;26:1408. doi: 10.1039/b817075b. [DOI] [PubMed] [Google Scholar]
- (7) (a).Zornetzer GA, Fox BG, Markley JL. Biochemistry. 2006;45:5217. doi: 10.1021/bi052062d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roujeinikova A, Baldock C, Simon WJ, Gilroy J, Baker PJ, Stuitje AR, Rice DW, Slabas AR, Rafferty JB. Structure. 2002;10:825. doi: 10.1016/s0969-2126(02)00775-x. [DOI] [PubMed] [Google Scholar]; (c) Mercer AC, Burkart MD. Nat. Prod. Rep. 2007;24:750. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]; (d) Kosa NM, Haushalter RW, Smith AR, Burkart MD. Nat. Methods. 2012;9:981. doi: 10.1038/nmeth.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Roujeinikova A, Simon WJ, Gilroy J, Rice DW, Rafferty JB, Slabas AR. J. Mol. Biol. 2007;365:135. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]; (f) Haushalter RW, Filipp FV, Ko KS, Yu R, Opella SJ, Burkart MD. ACS Chem. Biol. 2011;6:413. doi: 10.1021/cb200004k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shakya G, Rivera H, Lee DJ, Jaremko MJ, La Clair JJ, Fox DT, Haushalter RW, Schaub AJ, Bruegger J, Barajas JF, White AR, Kaur P, Gwozdziowski ER, Wong F, Tsai S-C, Burkart MD. J. Am. Chem. Soc. 2014;136:16792. doi: 10.1021/ja5064857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nguyen C, Haushalter RW, Lee DJ, Markwick PRL, Bruegger J, Caldara-Festin G, Finzel K, Jackson DR, Ishikawa F, O'Dowd B, McCammon JA, Opella SJ, Tsai S-C, Burkart MD. Nature. 2014;505:427. doi: 10.1038/nature12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9) (a).Lai JR, Fischbach MA, Liu DR, Walsh CT. J. Am. Chem. Soc. 2006;128:11002. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]; (b) Lai JR, Fischbach MA, Liu DR, Walsh CT. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5314. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou Z, Lai JR, Walsh Christopher T. Chem. Biol. 2006;13:869. doi: 10.1016/j.chembiol.2006.06.011. [DOI] [PubMed] [Google Scholar]
- (10).Lohman JR, Ma M, Cuff ME, Bigelow L, Bearden J, Babnigg G, Joachimiak A, Phillips GN, Shen B. Proteins: Struct., Funct., Bioinf. 2013;82:1210. doi: 10.1002/prot.24485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Allen CL, Gulick AM. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2014;70:1718. doi: 10.1107/S1399004714008311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Worthington AS, Burkart MD. Org. Biomol. Chem. 2006;4:44. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- (13).Tufar P, Rahighi S, Kraas FI, Kirchner DK, Löhr F, Hen-rich E, Köpke J, Dikic I, Güntert P, Marahiel MA, Dötsch V. Chem. Biol. 2014;21:552. doi: 10.1016/j.chembiol.2014.02.014. [DOI] [PubMed] [Google Scholar]
- (14).Edgar RC. Nucleic Acids Res. 2004;32:1792. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- (16).Goodrich AC, Frueh DP. Biochemistry. 2015;54:1154. doi: 10.1021/bi501433r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Beld J, Cang H, Burkart MD. Angew. Chem. Int. Ed. 2014;53:14456. doi: 10.1002/anie.201408576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Güntert P. Eur Biophys J. 2009;38:129. doi: 10.1007/s00249-008-0367-z. [DOI] [PubMed] [Google Scholar]
- (19) (a).Koglin A, Mofid MR, Löhr F, Schäfer B, Rogov VV, Blum M-M, Mittag T, Marahiel MA, Bernhard F, Dötsch V. Science. 2006;312:273. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]; (b) Koglin A, Lohr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Guntert P, Wagner G, Walsh CT, Marahiel MA, Dotsch V. Nature. 2008;454:907. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Haslinger K, Redfield C, Cryle MJ. Proteins: Struct., Funct., Bioinf. 2015;83:711. doi: 10.1002/prot.24758. [DOI] [PubMed] [Google Scholar]
- (21).Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13843. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.