

Summary
The surface properties of nanoparticles (NPs) dictate their interaction with the outside world. The use of precisely designed molecular ligands to control NP surface properties provides an important toolkit for modulating their interaction with biological systems, facilitating their use in biomedicine. In this review we will discuss the application of the atom-by-atom control provided by organic synthesis to the generation of engineered nanoparticles, with emphasis on how the functionalization of NPs with these “small” organic molecules (Mw < 1,000) can be used to engineer NPs for a wide range of applications.
Graphical Abstract
Introduction
Fabricating nanoparticles (NPs) with unique biological properties is a challenging but rewarding task [1,2,3]. The combination of multiple NP features such as core size [4,5,6,7], shape [8,9,10], and surface chemistry [11,12] allows the regulation of the biological behavior. The NP surface is the interface with the outside world, and plays a prominent role in the interaction with biomolecules. The relatively large surface area of NPs facilitate the attachment of a wide range of biomacromolecules such as peptides [13,14], proteins [15,16], nucleic acids [17,18], and viruses [19] to dictate NP-protein or NP-cell interactions. Likewise, polymers have been widely employed as NP coverages [20]. The structural complexity and/or potential biodegradability of these macromolecular systems, however, introduce complexity to the interactions between NPs and biomolecules.
The use of non-polymeric “small” organic molecules provides a robust and scalable methodology to tailor the nano-bio interface. The wide variety of moieties available through organic chemistry provides a rich toolkit to provide atom-by-atom control of the NP-biomolecule interactions [21,22,23]. In this review we will present research focusing on controlling the interactions of NPs with proteins and cells by using these “small” molecule (Mw < 1,000) ligands.
Modulating the Interaction between NPs and biological systems
The interaction modes of NPs with proteins and cells can be modified by designing the surface monolayer, concomitantly modulating biological functions.24 This fine-tuned control provides a finely-honed tool for a wide array of biological interactions.
Modulating Enzyme-Nanoparticle Interactions
Engineered interactions between NPs and enzymes provide tools for both enhancing enzyme stability and regulating activity. Decorating NPs with engineered ligands facilitates the ‘dialing in’ of specific modes of interaction including electrostatic, hydrogen bonding, and van der Waals forces [25]. This capability has been demonstrated using anionic gold nanoparticles (AuNPs) and chymotrypsin (ChT), utilizing the positive “patch” around the ChT active site [26,27]. The studies demonstrated that the anionic surface of NPs can selectively recognize this cationic patch, thus inhibiting ChT activity [28,29,30]. Similarly, a variety of amino acid-terminated ligands bearing tunable charge and hydrophobicity (Fig. 1(a)) provided detailed information about NP-ChT interfacial recognition. A thorough examination of the binding constants revealed that AuNPs bearing hydrophobic groups bind more strongly to ChT than AuNPs with hydrophilic groups, indicating the importance of hydrophobic interactions at the interface (Fig. 1(b)) [31]. In addition to hydrophobicity, the chirality of the amino acids is also an important factor for the affinity of NPs and proteins [32]. These results demonstrate that very specific chemical features can be employed to modulate protein recognition. Furthermore, these studies also evidenced that unlike small molecule enzyme regulators, the binding affinity of NPs to enzymes is not the only factor that modifies the catalytic activity. The work from Rotello et al. showed that the reduction of enzymatic activity mediated by NPs was also observed when NPs were functionalized with hydrophilic ligands because of the denaturation of ChT caused by these functional groups (Fig. 1(c) and (d)). Likewise, Hamad-Schifferli et al. reported that irreversible denaturation of Glucose Oxidase (GOx) caused by the interaction with NPs had a negative effect on its enzymatic activity [33]. Recently, Das et al. reported that NPs can also improve enzymatic activity [34]. Their investigation on the activity of mitochondrial membrane Cytochrome c (Cyt c) bound with cationic NPs demonstrated the enhanced peroxidase activity by increasing hydrophobicity of NPs. Structural reorganization caused by hydrophobic NPs exposed the heme group of the enzyme, facilitating substrate access to the catalytic site.
Figure 1.

(a) Structures of amino acid functionalized AuNPs. (b) Correlation between Gibbs free energy changes and hydrophobicity index of amino acid side chains. (c) Normalized activity of ChT (3.2 μM) with nanoparticles (0.8 μM) bearing various amino acid side chains. (d) Correlation between the denaturation rate constants (k) of ChT and the hydrophobicity index of amino acid side chains in nanoparticles. Reprinted with permission from [31].
In stark contrast to their ability to denature protein, functionalized NPs have been used as synthetic chaperones that assist in the refolding of denatured enzymatic proteins. For instance, fully recovery of enzymatic activity of thermally denatured cationic proteins such as ChT and papain has been observed using malonic acid-functionalized anionic AuNPs followed by disruption of the protein-NP complex with aqueous sodium chloride [35].
Enzymatic activity can also be modified by using the NPs functionalized with specific small molecule enzyme inhibitors or activators. Supuran and co-workers coated AuNPs with an inhibitor of carbonic anhydrase (CA), and the NP displayed higher selectivity toward tumor associated isoform IX over the cytosolic isozymes I and IIIX [36]. Likewise, CA showed increased enzymatic activity when mixed with NPs bearing a CA activator [37]. These studies show functionalization on NP surface can modulate the activity of small molecule enzyme regulators, providing a new platform for the enzyme regulation.
Modulation of Protein-Protein Interactions
Protein-protein interactions (PPI) play a central role in numerous cellular processes including apoptosis and angiogenesis, thus targeting the interface between proteins provides a potential way for treating diseases [38,39]. However, due to the large contact area between proteins in these complexes [40,41], effective inhibition of PPIs is quite challenging. Knapp and Rotello reported that charged AuNPs can inhibit the recognition of Cyt c with cytochrome c peroxidase (CPP) (Fig. 2) [42]. Since this PPI is defined by salt-bridges between the cationic surface of Cyt c and the anionic surface of CPP, they designed AuNPs with cationic, neutral, or anionic AuNPs, and examined charge dependent inhibition of PPI. As a result, effective disruption of the Cyt c-CPP complex was observed only when charged NPs were used, while neutral NPs had no effect. Notably, because of the multivalency of NPs, a single NP could interact with more than one protein, implying that NPs could be effective modulators that can disrupt multiple PPIs.
Figure 2.
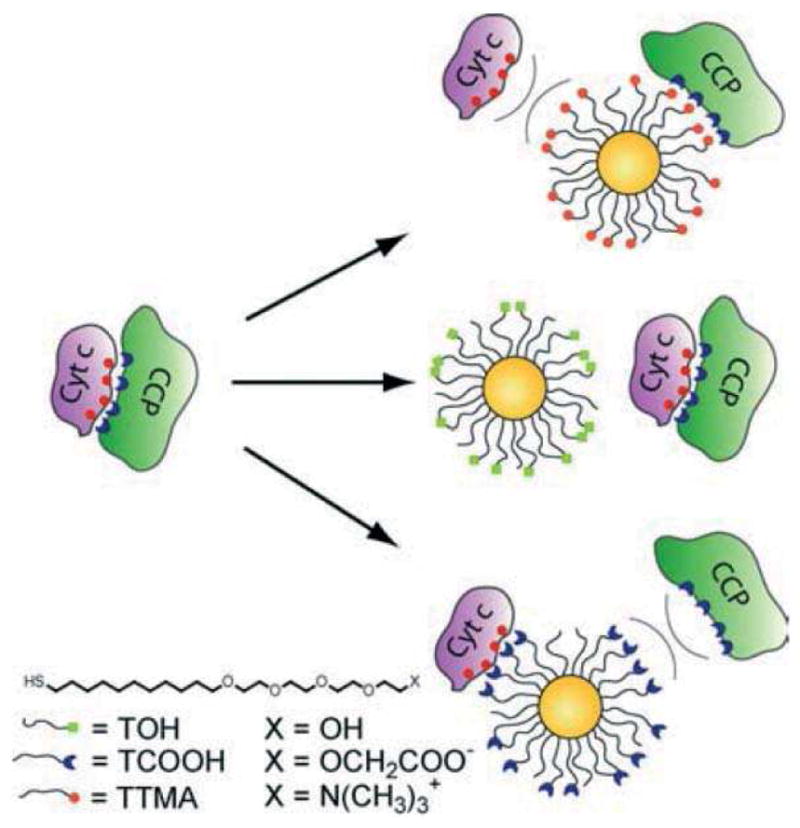
Schematic illustration of the interaction between Cyt c, CCP, and AuNPs with different surface charge. Reprinted with permission from [42].
Minimizing Nonspecific NP-Protein Interactions
Minimizing interactions between nanoparticles and proteins is also an important challenge for a diversity of biological applications. For example, when NPs are intended for biomedical imaging or as therapeutic agents, the adsorption and denaturation of proteins over the NP surface induces a rapid elimination of the from the body [43]. This phenomenon critically reduces the efficacy of the engineered delivery vehicles and targeted NPs [44]. The classical approach to provide “noninteracting” capabilities to the NPs is the use of oligo(ethylene glycol) (OEG) functionalities, which reduces protein denaturation over the NP surface [45]. This is translated into longer blood circulation times comparative to other systems, achieving higher tumor accumulation rates [46]. However, OEG functionalized NPs undergo a process known as accelerated blood clearance (ABC), a phenomenon that increases the elimination of the NPs after the second dose due to the formation of antibodies against the probe [47].
An alternative to OEG functionalization is the use of zwitterionic molecules based principally on two chemical structures, namely betaines and amino acids [48]. The zwitterionic nature of those structures confers strong non-fouling properties to NPs. As an example, it has been reported that small AuNPs (~5 nm, high area/volume ratio) coated with a mixture of citrate and cysteine do not absorb proteins even at physiological serum concentrations (Fig. 3) [49]. In addition, functionalized quantum dots (3.4 nm) with cysteine and tumor targeting groups have exhibited significantly lower renal clearance rates as well as the high tumor targeting efficiency (Fig. 4) [50]. The derivatization of ligands using amino acids, however, is limited by the fact that neutral charge is desirable for the stealth capabilities, and the addition of more amino acids may modify this property. Betaines, on the other hand, come in a myriad of structures (carboxybetaines, phosphobetaines, sulfobetaines) and they have been shown to enhance the stealth properties of NPs such as reduced protein adsorption, longer blood circulation time and low clearance [51,52]. In addition, betaines are easily modified by changing the substituents over the ammonium group without affecting the overall neutral charge [53]. A final method to obtain zwitterionic properties at the NP surface is the use of a mixed monolayer NPs containing both cationic and anionic ligands. By adjusting the ratio of those ligands during the place exchange reaction, the zero net charge can be achieved. Although this technique has also rendered NPs with strong non-fouling properties [54], reproducibility is still a challenge.
Figure 3.

(a) Chemical structure of citrate and lysine coated AuNP. (b) Chemical structure of citrate and cysteine coated AuNP. (c) DLS distributions of citrate and lysine coated AuNP (coverage; lysine/citrate = 1.4) in water (green curve) and FBS (red curve), and citrate AuNP in FBS (black curve). (d) DLS distributions of citrate and cysteine coated AuNP (cysteine/citrate = 1.6) in water (green curve) and FBS (red curve). Reprinted with permission from [49].
Figure 4.

(a) Chemical structure of zwitterionic quantum dots bearing targeting groups. (b) Fluorescent images of prostate-specific membrane antigen (PSMA)-positive LNCaP and PSMA-negative prostate cancer cells after incubation with functionalized zwitterionic quantum dots. Reprinted with permission from [50].
Effect of the Properties of NPs on their Interaction with Cells
The cellular adsorption and internalization of NPs are predominantly determined by their surface charge [55]. Multiple reports demonstrate a significant difference on the cellular uptake rate between positively charged NPs and other charged NPs (negative, neutral, and zwitterionic) (Fig. 5) [56,57,58,59]. The strong electrostatic interaction between the negatively charged cell membranes and cationic NPs is the underlying factor that increases the amount of NPs adsorbed onto the cell surface, thus enhancing cellular uptake [60]. However, in some case, the strong cellular uptake was observed with negatively charged nanoparticles. The studies demonstrated that iron oxide (Fe2O3) nanoparticles functionalized with carboxylate group can be efficiently taken up by cancer cell lines [61] presumably because of the direct diffusion through membrane or the pinocytosis of anionic nanoparticles.
Figure 5.

Schematic illustration of the interaction between cell membrane and AuNPs with different surface charge.
In addition to the surface charge, the hydrophobicity of NPs can also be employed to modify the cellular uptake of these materials. A study using cationic NPs demonstrated that an increase of the AuNP surface hydrophobicity enhances cellular uptake in the absence of serum, while it decreases uptake in the presence of serum [62]. The binding of serum proteins to AuNPs is influenced by the surface charge and hydrophobicity, compromising the NP uptake. There are some efforts to enhance the cellular uptake of zwitterionic AuNPs by using hydrophobicity, demonstrating slightly higher cellular uptake of hydrophobic AuNPs compared with hydrophilic counterparts, either in the presence or absence of serum proteins [43].
The binding mode between ligand and nanoparticle surface can affect the cellular interaction. Cell experiments using cationic quantum dots bearing monothiol or dithiol coordination groups revealed a significant difference on the cellular uptake of nanoparticles (Fig. 6) [63]. These results indicate the importance of molecular design that considers not only effects from the headgroups but also effects from the overall ligand design.
Figure 6.

(a) Chemical structure of mono- and dithiol capped CdSe/ZnS quantum dots. (b) Uptake of quantum dots after the incubation with HeLa cells. Reprinted with permission from [63].
Engineering Responsive Biological Interactions
In addition to “permanent” physicochemical properties, a chemical designing approach can also construct “responsive” surfaces that can alter the physicochemical features of the NPs by endogenous stimuli. Such chemical surfaces generate unique interaction modes with biological systems.
pH-responsive Aggregate Formation
Enhancing cellular uptake with stimuli responsive NP surfaces was initially reported by the Kim group. Negatively charged citraconic amide functionalized “smart” AuNPs form aggregates upon exposure to weakly acidic environments (pH = 5.5) (Fig. 7(a) and (b)) [64]. Acid induced partial cleavages of citraconic amide group and subsequent exposure of cationic primary amine [65, 66, 67] neutralize the surface charge of NPs, inducing aggregation. AuNPs are exposed to acidic endosomal environments [68] during cell-internalization process. When an aggregate is formed inside an endosome, the exocytosis of AuNPs is blocked [69], resulting in a larger amount of AuNPs accumulated in B16F10 cells compared to negatively charged AuNPs used as controls (Fig. 7(c)).
Figure 7.

(a) Chemical structure of the AuNPs capable of aggregating in acidic pH. (b) TEM images of AuNPs in pH 5.5 acetate buffer with different incubation times of (i) 0, (ii) 10, (iii) 30, (iv) 60, (v) 90 min. (c) Dark field microscope images of B16 F10 cells incubated with “smart” AuNPs (first column), citrate AuNPs (second column), and MUA (11-mercaptoundecanoic acid)-capped AuNPs (last column). Reprinted with permission from [64]. Figure 3a is based on information obtained in the literature.
AuNPs bearing a mixed monolayer of quaternary ammonium and carboxylic acid ligands can provide high tunable pH-induced aggregation systems [70]. Partial protonation of carboxylate functionality at acidic pH can induce the aggregate formation by the neutralization of surface charge. The pH value at which NPs form aggregates can be tuned by changing the surface ligand coverage ratio and core size. Ji et al. applied this system for tumor targeting [54], utilizing the pH difference between tumor tissue (pH 6.0–6.6) and normal tissue (pH 7.4) as a tumor environment signal [71,72]. Mixing the same mole ratio of cationic and anionic ligands with 16 nm AuNPs, they constructed AuNPs that form aggregates at pH 6.5 (Fig. 8(a) and (b)). In vitro cellular uptake studies demonstrated that a pH decrease enhanced NP internalization (Fig. 8(c)) due to the accelerated sedimentation of AuNPs [73,74]. Subsequent in vivo studies indicated the enhancement of tumor accumulation and retentions of these AuNPs (Fig. 8(d) and (e)).
Figure 8.

(a) Chemical structure of mixed monolayer AuNPs capable of aggregating in acidic conditions. (b) TEM images of AuNPs at pH 7.4 and 6.5 [scale bar 50 nm (upper image), 20 nm (lower image)]. (c) Effect of pH on the cellular uptake of AuNPs by HepG2 cells. (d) Comparison of tumor accumulation in KB tumor in BALB/c nude mice between pH-responsive AuNP and PEG-functionalized AuNP. (e) Comparison of tumor tissue retention pattern between pH-responsive AuNP and PEG-functionalized AuNP. Reprinted with permission from [54]. Figure 3a is based on information obtained in the literature.
pH-responsive Charge Conversion
Rotello and coworkers recently developed a new pH responsive surface which can reversibly switch from zwitterionic into cationic. The chemical moiety at the surface features alkoxyphenyl acylsulfonamide groups that is protonated under weakly acidic conditions (Fig. 9(a)) [75]. AuNPs functionalized with this pH-responsive zwitterionic group exhibited neutral zeta-potential at pH 7.4 and showed a sharp response to pH decrease, switching into cationic at pH 6.6 and 6.0 without forming aggregates (Fig. 9(b)). As discussed previously, AuNPs with cationic ligands are prone to be adsorbed by the negatively charged cell membranes, resulting in higher cellular uptake than that of zwitterionic AuNPs. Cell studies indicated that the pH-responsive zwitterionic surface does induce an enhancement in cellular uptake of AuNPs by cancerous cells at tumor pH (6.6–6.0), achieving a four-fold increase at pH 6.0 compared to the cellular uptake at pH 7.4. On the other hand, a non-responsive alkyl analogue did not show differences over the pH range studied (Fig. 9(c)). This result indicates that subtle structural differences of zwitterionic moieties could generate a targeting function for selective accumulation of AuNPs in tumor tissues.
Figure 9.

(a) Chemical structures of the acylsulfonamide-functionalized pH-responsive and non-responsive zwitterionic AuNPs, and the mechanism of acidic pH enhanced cellular uptake. (b) pH dependence of zeta-potential of acylsulfonamide functionalized AuNP (c) Effects of pH difference on cellular uptake of AuNPs. Reprinted with permission from [75].
Esterase-induced Cellular Uptake of Amphiphilic AuNP
pH is not the only stimulus to enhance the interaction between NPs and cells. Niikura and coworkers demonstrated that an esterase-catalyzed hydrolysis of ester moieties can be used as to enhance the cellular uptake of amphiphilic AuNPs [76]. 10 nm AuNPs were functionalized with ester or ether-headed amphiphilic PEG ligands which enable NPs to dissolve both of polar and nonpolar solvents through systematic conformational changes (Fig. 10(a) and (b)) [77]. Investigation of the cellular internalization of those AuNPs revealed that AuNPs with ester functionalities were taken up by cells in a greater extent than the corresponding ether-AuNPs (Fig. 10(c)). The authors assumed that even though both ester and ether-headed AuNPs have a high affinity to hydrophobic cell membranes, the exposure of alkyl moieties during the interaction with cells generated an esterase-mediated hydrolysis that enabled longer retention of AuNPs at the cell membrane, accelerating endocytosis (Fig. 10(d)).
Figure 10.

(a) Conformational change of the amphiphilic PEG ligands on AuNPs in polar and nonpolar solvents. (b) Chemical structures of the ester and ether-headed amphiphilic PEG ligands. (c) Difference of the number of AuNPs taken up by HeLa cells (1.0 × 105). (d) Esterase-induced cell internalization of AuNPs. Reprinted with permission from [76,77]. Figure 10d is based on information obtained in the report.
Conclusions and prospective
As shown above, surface chemistry defines the interactions of NPs with biological systems. Building the surface with proper chemical structures has the potential to overcome many challenges currently present in nanomedicine. The examples highlighted in this review clearly show the power of chemical approaches for tailoring the physicochemical properties of NPs, providing fruitful biological features.
Organic chemistry can be utilized as a “palette” containing an unlimited range of functional groups for the modulation of the NP functions. Desired features and functions can be constructed along with the synthesis of functional groups, providing an opportunity of the atom by atom level regulation. This approach results in the information of structure-function relationships, providing a deeper understanding of the molecular recognition between NPs and biological systems as well as bringing along clues on how to control desired effects at specific places. Such methods will help to maximize the potential of nanomaterials for the creation of novel therapeutics, diagnostics and imaging agents.
Highlights.
Surface properties are a key determinant of the biological properties of nanomaterials
Organic synthesis provides atom-by-atom control over nanomaterial surfaces
The control of nanoparticle surfaces is an important tool for the creation of diagnostics and therapeutics
Acknowledgments
This work was supported by NSF (CHE- 1307021) and the NIH (#GM077173, EB014277). T. M. is grateful to the Japan Society for the Promotion of Sciences (JSPS, Postdoctoral Fellowship for Research Abroad and Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation).
Biographies

Tsukasa Mizuhara received his Ph.D. in Pharmaceutical Science from Kyoto University where he worked in the field of organic chemistry and medicinal chemistry. Currently, he is working as a postdoctoral researcher in the Department of Chemistry at the University of Massachusetts Amherst under the guidance of Professor Vincent M. Rotello. His research interests focus on the fabrication of well-designed chemical surfaces and its application for nanomedicine.

Daniel F. Moyano received his B.Sc. (honors) in Chemistry at the National University of Colombia, Bogotá, Colombia in 2009. He is currently a Ph.D. candidate in the Chemistry Department at the University of Massachusetts Amherst, under the supervision of Prof. Vincent M. Rotello. His current research focuses on the chemical design of nanoparticle surfaces for modulation of immune responses.

Vincent Rotello is Goessmann Professor of Chemistry and Distinguished University Professor at the University of Massachusetts-Amherst. He joined the University of Massachusetts in 1993, receiving the NSF CAREER, Cottrell Scholar and Camille Dreyfus Teacher-Scholar awards, the Sloan Fellowship, and the Langmuir Lectureship, and is a Fellow of the AAAS and the Royal Society of Chemistry (U.K.). He is the Editor-in-Chief of Bioconjugate Chemistry, and on the Editorial Board of 12 other journals. His research program focuses on engineering the interface between hard and soft materials for applications in nanotechnology and nanomedicine, with > 450 papers published to date.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, Medintz IL. Chem Rev. 2013;113:1904–2074. doi: 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]
- 2.Mout R, Rotello VM. Isr J Chem. 2013;53:521–529. [Google Scholar]
- 3.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Chem Rev. 2011;111:5610–5637. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 4.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. J Am Chem Soc. 2004;126:6520–6521. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 5.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Bu L, Xie J, Chen K, Cheng Z, Li X, Chen X. ACS Nano. 2010;4:7151–7160. doi: 10.1021/nn101643u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun S, Zhou C, Chen S, Liu J, Yu J, Chilek J, Zhao L, Yu M, Vinluan R, Huang B, Zheng J. Bioconjugate Chem. 2014;25:453–459. doi: 10.1021/bc500008a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 9.Arnida, Malugina A, Ghandeharia H. J Appl Toxicol. 2010;30:212–217. doi: 10.1002/jat.1486. [DOI] [PubMed] [Google Scholar]
- 10.Huang X, Teng X, Chena D, Tanga F, He J. Biomaterials. 2010;31:438–448. doi: 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 11.Albanese A, Tang PS, Chan WCW. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 12.Saha K, Bajaj A, Duncan B, Rotello VM. Small. 2011;7:1903–1918. doi: 10.1002/smll.201100478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao M, Kircher MF, Josephson L, Weissleder R. Bioconjugate Chem. 2002;13:840–844. doi: 10.1021/bc0255236. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang PH, Sun X, Chiu JF, Sun H, He QY. Bioconjugate Chem. 2005;16:494–496. doi: 10.1021/bc049775d. [DOI] [PubMed] [Google Scholar]
- 16.Kocbek P, Obermajer N, Cegnar M, Kos J, Krist J. J Control Release. 2007;120:18–26. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 17.Cutler JI, Auyeung E, Mirkin CA. J Am Chem Soc. 2012;134:1376–1391. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 18.Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA. Nano Lett. 2007;7:3818–3821. doi: 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huh YM, Lee ES, Lee JH, Jun YW, Kim PH, Yun CO, Kim CJH, Suh JS, Cheon J. Adv Mater. 2007;19:3109–3112. [Google Scholar]
- 20.Voigt J, Christensen J, Shastri VP. Proc Natl Acad Sci USA. 2014;111:2942–2947. doi: 10.1073/pnas.1322356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 22.Mout R, Moyano DF, Rana S, Rotello VM. Chem Soc Rev. 2012;41:2539–2544. doi: 10.1039/c2cs15294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lévy R. Chem Bio Chem. 2006;7:1141–1145. [Google Scholar]
- 24.Lynch I, Dawson KA. Nano Today. 2008;3:40–47. [Google Scholar]
- 25.Moyano DF, Rotello VM. Langmuir. 2011;27:10376–10385. doi: 10.1021/la2004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blow DM. Acc Chem Res. 1976;9:145–152. [Google Scholar]
- 27.Cassidy CS, Lin J, Frey PA. Biochemistry. 1997;36:4576–4584. doi: 10.1021/bi962013o. [DOI] [PubMed] [Google Scholar]
- 28.Fischer NO, McIntosh CM, Simard JM, Rotello VM. Proc Natl Acad Sci USA. 2002;99:5018–5023. doi: 10.1073/pnas.082644099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blow DM. Acc Chem Res. 1976;9:145–152. [Google Scholar]
- 30.Tsukada H, Blow DM. J Mol Biol. 1985;184:703–711. doi: 10.1016/0022-2836(85)90314-6. [DOI] [PubMed] [Google Scholar]
- 31.You CC, De M, Han G, Rotello VM. J Am Chem Soc. 2005;127:3822–3823. doi: 10.1021/ja0512881. [DOI] [PubMed] [Google Scholar]
- 32.You CC, Agasti SS, Rotello VM. Chem Eur J. 2008;14:143–150. doi: 10.1002/chem.200701234. [DOI] [PubMed] [Google Scholar]
- 33.Tellechea E, Wilson KJ, Bravo E, Hamad-Schifferli K. Langmuir. 2012;28:5190–5200. doi: 10.1021/la2050866. [DOI] [PubMed] [Google Scholar]
- 34.Maiti S, Das K, Dutta S, Das PK. Chem Eur J. 2012;18:15021–15030. doi: 10.1002/chem.201202398. [DOI] [PubMed] [Google Scholar]
- 35.De M, Rotello VM. Chem Commun. 2008;30:3504–3506. doi: 10.1039/b805242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stiti M, Cecchi A, Rami M, Abdaoui M, Barragan-Montero V, Scozzafava A, Guari Y, Winum JY, Supuran CT. J Am Chem Soc. 2008;130:16130–16131. doi: 10.1021/ja805558k. [DOI] [PubMed] [Google Scholar]
- 37.Saada MC, Montero JL, Vullo D, Scozzafava A, Winum JY, Supuran CT. J Med Chem. 2011;54:1170–1177. doi: 10.1021/jm101284a. [DOI] [PubMed] [Google Scholar]
- 38.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 39.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 40.Jones S, Thornton JM. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conte LL, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 42.Bayraktar H, Ghosh PS, Rotello VM, Knapp MJ. Chem Commun. 2006:1390–1392. doi: 10.1039/b516096k. [DOI] [PubMed] [Google Scholar]
- 43.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Adv Drug Deliv Rev. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Aberg C, Mahon E, Dawson KA. Nat Nanotechnol. 2013;8:137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 45.You CC, De M, Rotello VM. Org Lett. 2005;7:5685–5658. doi: 10.1021/ol052367k. [DOI] [PubMed] [Google Scholar]
- 46.Hoffman ASJ. J Control Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Dams ETM, Laverman P, Oyen WJG, Storm G, Scherphof GL, Van der Meer JWM, Corstens FHM, Boerman OC. J Pharmacol Exp Ther. 2000;292:1071–1079. [PubMed] [Google Scholar]
- 48.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murthy AK, Stover RJ, Hardin WG, Schramm R, Nie GD, Gourisankar S, Truskett TM, Sokolov KV, Johnston KP. J Am Chem Soc. 2013;135:7799–7802. doi: 10.1021/ja400701c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, Robertson JD, Rotello VM, Reid JM, Mukherjee P. Plos One. 2011;6:e24374. doi: 10.1371/journal.pone.0024374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhan N, Palui G, Safi M, Ji X, Mattoussi H. J Am Chem Soc. 2013;135:13786–13795. doi: 10.1021/ja405010v. [DOI] [PubMed] [Google Scholar]
- 53.Moyano DF, Saha K, Prakash G, Yan B, Kong H, Yazdani M, Rotello VM. ACS Nano. 2014;8:6748–6755. doi: 10.1021/nn5006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Chen Y, Li H, Huang N, Jin Q, Ren K, Ji J. ACS Nano. 2013;7:6244–6253. doi: 10.1021/nn402201w. [DOI] [PubMed] [Google Scholar]
- 55.Cho EC, Xie JW, Wurm PA, Xia YN. Nano Lett. 2009;9:1080–1084. doi: 10.1021/nl803487r. [DOI] [PubMed] [Google Scholar]
- 56.Arvizo RR, Miranda OR, Thompson MA, Pabelick CM, Bhattacharya R, Robertson JD, Rotello VM, Prakash YS, Mukherjee P. Nano Lett. 2010;10:2543–2548. doi: 10.1021/nl101140t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim CK, Ghosh P, Pagliuca C, Zhu ZJ, Menichetti S, Rotello VM. J Am Chem Soc. 2009;131:1360–1361. doi: 10.1021/ja808137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du JZ, Sun TM, Song WJ, Wu J, Wang J. Angew Chem Int Ed. 2010;49:3621–3626. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 59.Baier G, Costa C, Zeller A, Baumann D, Sayer C, Araujo PHH, Mailänder V, Musyanovych A, Landfester K. Macromol Biosci. 2011;11:628–638. doi: 10.1002/mabi.201000395. [DOI] [PubMed] [Google Scholar]
- 60.Wilhelm C, Gazeau F, Roger J, Pons JN, Bacri JC. Langmuir. 2002;18:8148–8155. [Google Scholar]
- 61.Shi XY, Thomas TP, Myc LA, Kotlyar A, Baker JR. Phys Chem Chem Phys. 2007;9:5712–5720. doi: 10.1039/b709147h. [DOI] [PubMed] [Google Scholar]
- 62.Zhu ZJ, Posati T, Moyano DF, Tang R, Yan B, Vachet RW, Rotello VM. Small. 2012;8:2659–2663. doi: 10.1002/smll.201200794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeh Y-C, Saha K, Yan B, Miranda OR, Yu X, Rotello VM. Nanoscale. 2013;5:12140–12143. doi: 10.1039/c3nr04037b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nam J, Won N, Jin H, Chung H, Kim S. J Am Chem Soc. 2009;131:13639–13645. doi: 10.1021/ja902062j. [DOI] [PubMed] [Google Scholar]
- 65.Brinegar AC, Kinsella JE. J Agric Food Chem. 1980;28:818–824. doi: 10.1021/jf60230a016. [DOI] [PubMed] [Google Scholar]
- 66.Shetty JK, Kinsella JE. Biochem J. 1980;191:269–272. doi: 10.1042/bj1910269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee Y, Fukushima S, Bae Y, Hiki S, Ishii T, Kataoka K. J Am Chem Soc. 2007;129:5362–5363. doi: 10.1021/ja071090b. [DOI] [PubMed] [Google Scholar]
- 68.Bae Y, Fukushima S, Harada A, Kataoka K. Angew Chem Int Ed. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- 69.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 70.Pillai PP, Huda S, Kowalczyk B, Grzybowski BA. J Am Chem Soc. 2013;135:6392–6395. doi: 10.1021/ja4001272. [DOI] [PubMed] [Google Scholar]
- 71.Randolph LM, Chien MP, Gianneschi NC. Chem Sci. 2012;3:1363–1380. doi: 10.1039/C2SC00857B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mura S, Nicolas J, Couvreur P. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 73.Limbach LK, Li Y, Robert N, Brunner TJ, Hintermann MA, Muller M, Gunther D, Stark XJ. Environ Sci Technol. 2005;39:9370–9376. doi: 10.1021/es051043o. [DOI] [PubMed] [Google Scholar]
- 74.Cho EC, Zhang Q, Xia Y. Nat Nanotechnol. 2011;6:385–391. doi: 10.1038/nnano.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizuhara T, Saha K, Moyano DF, Kim CS, Yan B, Rotello VM. Angew Chem Int Ed. 2015;54:6567–6570. doi: 10.1002/anie.201411615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kobayashi K, Niikura K, Takeuchi C, Sekiguchi S, Ninomiya T, Hagiwara K, Mitomo H, Ito Y, Osada Y, Ijiro K. Chem Commun. 2014:1265–1267. doi: 10.1039/c3cc48532c. [DOI] [PubMed] [Google Scholar]
- 77.Sekiguchi S, Niikura K, Matsuo Y, Ijiro K. Langmuir. 2012;28:5503–5507. doi: 10.1021/la300299x. [DOI] [PubMed] [Google Scholar]