

Abstract
Layer-by-layer (LbL) self-assembly is a versatile technique from which multicomponent and stimuli-responsive nanoscale drug carriers can be constructed. Despite the benefits of LbL assembly, the conventional synthetic approach for fabricating LbL nanoparticles requires numerous purification steps that limit scale, yield, efficiency, and potential for clinical translation. In this report, we describe a generalizable method for increasing throughput with LbL assembly by using highly scalable, closed-loop diafiltration to manage intermediate purification steps. This method facilitates highly controlled fabrication of diverse nanoscale LbL formulations smaller than 150 nm composed from solid-polymer, mesoporous silica, and liposomal vesicles. The technique allows for the deposition of a broad range of polyelectrolytes that included native polysaccharides, linear polypeptides, and synthetic polymers. We also explore the cytotoxicity, shelf life and long-term storage of LbL nanoparticles produced using this approach. We find that LbL coated systems can be reliably and rapidly produced: specifically, LbL-modified liposomes could be lyophilized, stored at room temperature, and reconstituted without compromising drug encapsulation or particle stability, thereby facilitating large scale applications. Overall, this report describes an accessible approach that significantly improves the throughput of nanoscale LbL drug-carriers that show low toxicity and are amenable to clinically relevant storage conditions.
Keywords: layer-by-layer nanoparticles, polymer engineering, colloid chemistry, scalable synthesis, biomaterials
Graphical Abstract

The highly scalable preparation of drug-loadable, layer-by-layer nanoparticles is described for a variety of formulations below 150 nm in size. This work demonstrates a method to generate layer-by-layer drug-carriers in a manner that is both facile and much higher throughput than traditional methods. Results also reveal the generated nanoparticles possess clinically relevant shelf lives as well as compatibility with long-term freeze-dried storage.
1. Introduction
Multifunctional nanoscale therapies have the capacity to transform modern healthcare by providing greater control over the spatial and temporal release of drugs.[1] Formulations sized from 15 to 100 nm exhibit unique properties and functions that promote accumulation in target tissues while minimizing nonspecific clearance from the body.[2] These nanotherapies are by their own nature a complex composite of materials that can simultaneously protect, guide, and release biologically active compounds in a desirable manner. Complex materials, in turn, provide significant challenges from a translational point of view – particularly in terms of their eventual scalable fabrication. Effective nanoscale therapies must further exhibit adequate shelf lives under clinically relevant storage conditions in order to facilitate their eventual translation.
LbL assembly is a well-established technique for the solution-phase synthesis of hierarchical and multifunctional nanoscale therapeutics.[3] LbL nanoparticles can possess a range of desirable properties for drug and gene delivery due to the versatility granted by the technique. In its most frequently employed form, LbL assembly is driven by the electrostatic interaction between oppositely charged polyelectrolytes on a charged substrate. Possible substrates include colloids such as gold, polymer, silica, and liposomal nanoparticles, amongst others.[4] Along with the flexibility in the choice of a core substrate, any polyelectrolyte with a multivalent charge can be incorporated into an LbL construct, including native polysaccharides, nucleic acids, linear polypeptides, and synthetic polymers.[4a, 5] The LbL technique has been well characterized and studied by the materials community since the fundamental concept of alternating electrostatic assembly was first described by Iler in 1966.[6] In addition to electrostatics, it is well documented that LbL assembly can be driven by a multitude of intermolecular interactions that include hydrogen bonding, covalent bonding, and biologically specific interactions.[7] More recent research into LbL nanomaterials has yielded important developments for a variety of fields including chemical sensing, catalysis, energy storage, optics, and drug delivery.[4f, 8]
Several advances specifically highlight the promise of LbL nanoparticle systems for drug delivery, including the ability to generate novel hybrid organic-inorganic nanoparticles with tunable cell-particle interactions, and the generation of nanoparticle systems that can release combination drugs and small interfering RNA (siRNA) in a staged and synergistic fashion.[9] Importantly, the thickness of polyelectrolyte multilayers formed by LbL is on the order of nanometers, providing investigators a high degree of control over the final size of an LbL nanoparticle. As generally the polymer film is very thin, the size of an LbL formulation is largely determined by the core substrate, which has been reported to be as small as 10 nm in diameter.[10] Using the LbL platform, researchers have been able to preserve the size of a conventional nanoparticle formulation while providing improved stability and environmentally sensitive functionalities that capitalize on cues from the tumor microenvironment to initiate cellular uptake.[5b, 11] Additional functionalities provided by the multilayer film allows for tuning of drug release and the incorporation of therapeutics into the polymer film.[4f, 9b, 12]
Despite their numerous advantages, both basic research and clinical translation of LbL nanoparticles have been impeded by scalability problems associated with their preparation. Traditional preparation of these materials relies on centrifugal purification to eliminate excess polyelectrolyte, but this approach is both time-intensive and prone to causing irreversible aggregation of LbL particles. Unfortunately, centrifugation becomes much more problematic when transitioning from micro- to nanoscale formulations, due to the need to significantly increase the spin speed and time needed to isolate smaller particles. Work to optimize centrifugal protocols rely on meticulous optimization of polymer molecular weight, charge ratios, centrifugal force, and spin times.[13] Efforts to try and resolve problems with scalability have generated creative techniques including vacuum-filtration, atomization, microfluidic, electrophoresis, and fluidized bed-mediated LbL assembly.[14] While these innovative approaches do offer advances in efficacy and yield during the preparation of colloidal LbL materials, they place limitations on the types and sizes of materials that can be used, reducing LbL versatility. Our group previously described another LbL preparation method, which relies on PRINT® technology. While this approach is robust, it does not allow for the layering of many nanoparticle cores that are of great interest for drug delivery and theranostics, such as quantum dots, liposomes, and responsive self-assembled polymeric colloids.[15]
To address the need for an accessible, robust, and scalable approach for preparing diverse LbL nanoparticles, we incorporated tangential flow filtration (TFF) into our synthetic workflow. TFF works by a process known as diafiltration, where permeable molecules are removed from a solution while passing through a hollow ultrafiltration membrane. Diafiltration for nanoparticle purification has been well characterized for colloidal solutions.[16] When a solution of LbL nanoparticles is pumped through the fiber membrane, the permeable polyelectrolytes rapidly exit through the pores in the membrane and are removed from the system. Meanwhile, the larger nanoparticles are retained and re-circulated through the system until the desired purity is reached. The sample volume can be held constant by the introduction of replacement buffer at the same rate that waste is removed from the system, allowing the purification to be continued for any desired amount of time. Collected waste (permeate) solution can also be recovered and re-concentrated in order to recycle reagents and reduce synthetic costs. Furthermore, these devices are easily scalable to prepare samples ranging anywhere from a single milliliter to more than a liter.
Lohse and colleagues previously described the use of a TFF-based reactor for the synthesis and functionalization of gold nanoparticles, including LbL modification with poly(acrylic acid) and polyallylamine.[17] This early application of TFF towards LbL modification faced issues of efficient purification, requiring washing nanoparticles with upwards of 20 volume equivalents before achieving sufficient purity to perform the next deposition step. Inefficient polyelectrolyte removal also appeared to cause off-target toxicity due to the carryover of free polycations. Here, we employ high surface-area, porous hollow-fiber filtration cassettes that provide improved flux rates that allow TFF purification to occur more rapidly and completely. Indeed, very recently the work by Björnmalm and coworkers demonstrated the effective preparation of LbL particles with this new class of membrane, with more emphasis on the generation of micrometer- and submicrometer-sized particles and capsules.[18] Here, we specifically describe a generalizable method for using TFF to prepare LbL nanoparticles ranging from 40 to 150 nm, which are within the size range important for systemic drug delivery, tumor targeting, and tissue penetration. We document major improvements in efficiency and yield using this method relative to traditional protocols. Furthermore, we demonstrate the compatibility of this method with a broad range of biomedically relevant materials through the fabrication of LbL nanoparticles composed from a variety of substrates and biocompatible polyelectrolytes, and include several novel LbL formulations. To highlight the clinical relevance of these materials, we go on to demonstrate that LbL nanoparticles fabricated using this new technique are biocompatible, and can be stably stored in both refrigerated and lyophilized states.
2. Results and Discussion
2.1. Preparation Of Filter Membranes For LbL Assembly
In LbL assembly, the role of salts is well known to promote the generation of thicker films by shielding the Coulombic repulsion between adsorbed polyelectrolytes.[13, 19] While this phenomenon can provide a measure of control over film thickness, it also promotes particle flocculation by masking the repulsive forces between distinct particles. The inclusion of salt is particularly problematic in LbL formulations where the researcher must balance the electrostatic repulsion between polyelectrolytes that are also subject to intermolecular attractive forces like hydrogen bonding. Consideration for delicate materials aside, the purification of LbL particles requires the eventual removal of salt to halt the gradual re-organization and decomposition of the LbL film.[19c] Normally, salt-removal is accomplished by using pure water during the sequential centrifugal washing steps, where salt-removal actually helps to better stabilize the particles by improving their electrostatic repulsion. Unfortunately, this poses a challenge for efficient purification using filtration, due to the slight negative charge associated with most polymeric filter membranes.[14a, 20] For LbL preparations, the sequential exposure of alternatively charged particle-polymer solutions leads to the build up of an LbL film onto the filter membrane itself, which reduces yields and fouls the membrane.[21]
To overcome this problem, we separate the purification of positively and negatively charged particles to their own filter membranes (Figure 1a). To discourage cationic nanoparticles from adhering to the negative membrane walls, dilute solutions of free polycation (10 mg mL−1) are re-circulated through the filter membrane for 10 minutes. This pre-treatment allows free polycation to bind to anionic sites on the membrane via electrostatic attraction, and reduces the membrane’s capacity for nonspecific adsorption of cationic species during subsequent purification. For the membrane that processes negatively charged particles, no modifications were made as its intrinsic negative charge suitably repels nonspecific adsorption. Separation of charge to specific membranes greatly improved yields with salt-free conditions and allowed us to easily and quickly prepare diverse LbL constructs.
Figure 1.

Tangential flow filtration (TFF) facilitates the rapid and controlled fabrication of layer-by-layer nanoparticles. (a) TFF purification schematic depicting continuous diafiltration through a porous membrane. A peristaltic pump drives nanoparticle samples through a circuit containing a filter membrane. Polyelectrolytes, driven by a mild pressure gradient, exit the circuit through the pores in the filter membrane and into a waste reservoir. Removal of solution generates a vacuum within the system, which draws replacement buffer from an attached reservoir to hold sample volume constant. To prevent nonspecific adsorption onto the filter membrane, purification of cationic and anionic nanoparticles was separated to different purification loops, each with its own filtration membrane. The left panel denotes the cationic purification loop, and the right panel denotes the anionic purification loop. (b) Purification of excess poly-L-lysine (PLL) or dextran sulfate (DXS) following LbL deposition can be completed in minutes using TFF. The concentration of PLL extracted using TFF was determined using a BCA assay on samples taken sequentially from the waste stream. DXS concentrations were determined by analyzing permeate samples by gel permeation chromatography. Data were fitted using a one-phase exponential decay model. (c) High yields (68 ± 5% after 10 layers) are reproducibly obtained using TFF-assisted LbL fabrication. Nanoparticle concentration was quantified using a fluorescence plate reader following each purification step during the fabrication of 100 nm, carboxy-modified latex particles coated with five bilayers of PLL and DXS (CML-[PLL/DXS]5). Error bars represent standard deviation of three independent syntheses.
2.2. LbL Nanoparticle Synthesis And Characterization
2.2.1. Purification Kinetics and Product Yields
We assessed purification kinetics using 500 kDa molecular weight cut off (MWCO) membranes and observed the rapid removal of excess 15 kDa poly(L-lysine) (PLL) and 10 kDa dextran sulfate (DXS) from an LbL nanoparticle grown on a fluorescently labeled, 100 nm, carboxy-modified latex (CML) substrate (Figure 1b). During purification, permeate waste was sequentially collected and then analyzed by either the bicinchoninic acid (BCA) assay or by gel permeation chromatography (GPC) to quantify the removal of polyelectrolyte. The results indicated that removal of polyelectrolytes of this size reaches a plateau within four minutes of purification while operating the TFF at a transmembrane pressure between 3–5 PSI. We applied a linear regression analysis to fit the data and calculate the time required to reach a desired purity. The results were similar to the qualitative interpretation of the data, indicating that 95% purity could be expected in 5.1 minutes for PLL and 3.8 minutes for the smaller DXS.
To assess the yield from TFF-assisted LbL synthesis, we repeatedly fabricated ten-layer LbL particles composed of a fluorescently labeled, 100 nm CML core coated with 15 kDa PLL and 10 kDa DXS (100CML-[PLL/DXS]5). After each purification step, a sample was taken to quantify particle recovery. Data from three independent syntheses were combined to determine the typical yield possible using this technique (Figure 1c). The results indicated that a total yield of 68 ± 5% could be expected after ten layer depositions. Linear regression analysis of the data suggested that 96.9 ± 0.4% yield could be expected per layer deposition.
One of the major findings of this work was the rapid timescale of nanoparticle purification using TFF, which reduced purification time to 4–6 minutes per layer. Relative to the 30–60 minutes typically required for a single centrifugal spin during traditional synthesis, TFF-assisted LbL assembly is dramatically more efficient. Analysis of recovered permeates during the purification of either 15 kDa PLL or 10 kDa DXS revealed that free polymers were efficiently removed after washing with 5 volume equivalents of the eluent solution, a significant improvement upon the 20 equivalents required by previous TFF-based approaches for nanoparticles.[17]
Using our diafiltration-based system, rapid purification times are complimented by a reliable, high degree of product yield of 96.9 ± 0.4% per layer, leading to 68 ± 5% yield after ten layer depositions on 100 nm latex cores. This provides 83 ± 2% yield after four layer depositions, which is the number of layers in the formulations we have previously described for applications involving targeted drug delivery to tumors.[9b] These results are a marked improvement over previously published optimized centrifugal protocols that report a 54.1% yield after the deposition of ten layers.[13] Improved yields will facilitate the exploration of LbL drug-carriers that incorporate useful but costly materials, such as siRNA, into the multilayer film. Similarly, recovery of the polyelectrolytes from the permeate solution, as was examined in this study to quantify purification kinetics, could lead to recycling of excess polymer to further reduce costs. Overall, our system allows for faster synthesis of LbL nanoparticles, with a higher yield as compared to previously described methods.
2.2.2. LbL Modification of Solid Carboxy-Modified Latex and Drug-Loadable Nanoparticle Substrates
To confirm that our method retains the desirable versatility of the LbL technique, the hydrodynamic size, uniformity, and surface charge were monitored throughout the preparation of several different LbL formulations. Initial work focused on solid polymer core formulations, such as the 100CML-[PLL/DXS]5 prepared during yield studies (Figure 2a–c). Another latex core formulation was prepared by depositing alternating layers of 15 kDa polyethylenimine (PEI) and unfractionated heparin sulfate (HS) onto a 100 nm CML substrate (100CML-[PEI/HS]3, see Figure S1). Finally, to demonstrate the capacity to prepare smaller sized products, a formulation using 40 nm CML coated with PLL and DXS was prepared, reaching a final hydrodynamic size of 58 ± 3 nm, a low polydispersity index of 0.12 ± 0.01, and a surface charge of −71.6 ± 0.9 mV (40CML-[PLL/DXS]3, Figure S2).
Figure 2.

Diverse layer-by-layer (LbL) nanoparticles are prepared in a controlled manner using the tangential flow filtration (TFF)-assisted method. (a–c) Solid-core LbL particles were prepared by coating 100 nm, carboxy-modified latex particles with five bilayers of poly(L-lysine) and dextran sulfate (100CML-[PLL/DXS]5). (d–f) Liposomal-core LbL particles were prepared by coating negatively charged, doxorubicin-containing liposomes with four bilayers of PLL and DXS followed by a bilayer of PLL and heparin sulfate (Lipo-[PLL/DXS]4-PLL-HS). (a, d) These particles exhibited controlled size increase during layer deposition. (b, e) Nanoparticle uniformity was maintained throughout the layering process as indicated by the low polydispersity index (PDI). (c, f) Complete charge reversal was observed after each layer deposition indicating successful LbL modification. Size and polydispersity data were acquired by dynamic light scattering, and zeta potential data was measured using laser Doppler electrophoresis. Error bars represent standard deviation of three technical replicates. For population-based data see Supplemental Figure S5.
To demonstrate the fabrication of clinically relevant LbL nanoparticles, focus was shifted to the LbL modification of two drug-loadable nanoscale templates, mesoporous silica and liposomal vesicles. We successfully prepared LbL nanoparticles from 30 nm mesoporous silica coated with poly(L-arginine) (PLA) and poly(L-glutamic acid) (PG) (MSN-[PLA/PG]3, Figure S3). The final LbL silica particles possessed a hydrodynamic size of 41 ± 4 nm, a polydispersity index of 0.22 ± 0.02, and a surface charge of −58 ± 2 mV. Subsequent efforts focused on the LbL modification of colloidal liposomal cores, where PLL, DXS, and HS-containing multilayers were formed on the negatively charged surfaces of doxorubicin-loaded liposomes (Lipo-[PLL/DXS]4-PLL/HS, Figure 2d–f). An additional LbL liposome formulation was prepared using PLA and DXS and later used for freeze-dried storage experiments (Lipo-[PLA/DXS]2, Figure S4). Notably, the TFF-assisted method did not need to be altered with any of these formulations, with the exception of changing the MWCO of the filter membrane when working with smaller or more flexible formulations. For particles 100 nm or larger in diameter, 500 kDa MWCO membranes were suitable. For smaller formulations, or liposomal formulations, 100 kDa MWCO membranes were more appropriate. In general, the optimal MWCO was determined by assessing changes in filter flux, where if the MWCO was large enough to permit nanoparticle penetration, then a decay of flux was observed. If flux decay was observed, the MWCO was decreased until stable flux rates were achieved. When changing MWCO of the membrane, the feed flow rate of the peristaltic pump was adjusted to keep the transmembrane pressure between 3–5 PSI and the shear rate below 6,000 s−1.
Hydrodynamic diameter was tracked using dynamic light scattering (DLS) after each purification, and revealed the steady growth from 86 ± 3 nm to a final size of 110 ± 1 nm (Figure 2a) for the case of 100CML-[PLL/DXS]5. The Lipo-[PLL/DXS]4-PLL/HS formulation exhibited thinner film formation, but nonetheless grew steadily from 58 ± 2 nm to 63 ± 2 nm (Figure 2d). The difference between film thicknesses on these two cores was anticipated, given previous experience with these substrates.[9b][22][23] In addition to size measurements, successful layering was confirmed by tracking the complete charge reversal of LbL particles throughout the synthesis. Both 100CML-[PLL/DXS]5 and Lipo-[PLL/DXS]4-PLL/HS alternated between zeta potentials of roughly +60 mV and −60 mV during alternate deposition steps, in accordance to the deposited polyelectrolyte’s charge (Figure 2c,f). Nanoparticle uniformity was well preserved throughout the synthesis, as indicated by the low polydispersity index (PDI) measured by DLS – 0.06 ± 0.01 for 100CML-[PLL/DXS]5 and 0.16 ± 0.02 for Lipo-[PLL/DXS]4-PLL/HS (Figure 2b,c). The other LbL formulations discussed exhibited similar characteristics, and that data can be seen in the online supplemental documentation. Further, population based, documentation on the ten-layer formulation’s size and charge can also be seen in Figure S5.
To further validate the successful LbL assembly of the two principle formulations, cryogenic transmission electron microscopy (Cryo-TEM) was used to evaluate changes in particle morphology after LbL modification (Figure 3). Comparison of the electron micrographs showed the presence of thin films on the LbL nanoparticles that are not visible on the unmodified substrates. These images were further analyzed using ImageJ to quantify size changes from the unmodified and layered formulations. The data reveal statistically significant (P<0.0001) increases in nanoparticle diameter (for 100CML-[PLL/DXS]5) and membrane thickness (for Lipo-[PLL/DXS]4-PLL/HS ) relative to the unmodified substrate (Figure 4a–b). Furthermore, comparison of LbL film thickness calculated from either Cryo-TEM or DLS produced statistically consistent estimates for particles formed on CML (13.3 ± 0.6 nm by Cryo-TEM and 12 ± 1 nm by DLS) and liposomal (2.65 ± 0.09 nm by Cryo-TEM and 2 ± 1 nm by DLS) substrates (Figure 4c).
Figure 3.

Cryogenic TEM images of core particles before and after layering confirm the presence of a thin film. Cryogenic TEM of (a) uncoated, carboxy-modified latex particles; (b) purified, LbL-coated carboxy-modified latex (CML-[PLL/DXS]5); (c) uncoated, doxorubicin-loaded liposome; (d) purified, LbL-coated, doxorubicin-loaded liposome (Lipo-[PLL/DXS]4-PLL-HS).
Figure 4.

The quantification of Cryo-TEM images confirm statistically significant increases in size consistent with LbL modification. (a) Comparison of the diameters of bare, carboxy-modified latex (CML) particles and LbL-modified CMLs indicate a statistically significant (P<0.0001) shift in the size distribution. Dashed line denotes the mean size. N = 103 for bare and 153 for LbL CML. (b) Similarly, comparison of the liposome membrane thickness reveals statistically significant (P<0.0001) increase in thickness after LbL modification. Dashed lines denote the mean thickness. N = 113 for bare and 298 for LbL liposomes. (c) Calculation of LbL film thickness from either Cryo-TEM data or dynamic light scattering data give statistically consistent results for both CML (13.3 ± 0.6 nm by Cryo-TEM and 12 ± 1 nm by DLS) and liposomal (2.65 ± 0.09 nm by Cryo-TEM and 2 ± 1 nm by DLS) substrates. Error bars represent SEM. These data highlight important changes in LbL film thickness due to substrate selection. Individual particle measurements were plotted as a histogram and the kernel density function was estimated using R Statistical Software. All statistical tests were performed using one-way ANOVA (alpha = 0.01), with the Bonferroni post-test, on PRISM graphing software.
These results indicate the highly efficient and scalable fabrication of LbL nanoparticles smaller than 150 nm using TFF. Another major finding of this study was that the TFF-assisted method could be generalized to a diverse number of nanoscale formulations including solid latex, mesoporous silica, and liposomal cores. The breadth of polyelectrolytes used to construct LbL nanoparticles shows that TFF-assisted LbL assembly is compatible with many different material systems. Careful tracking of the nanoparticle size, surface charge, and uniformity during synthesis indicated that this method provided a great deal of control over these important nanomaterial characteristics. The results from Cryo-TEM validated the presence of intact and uniform films on LbL-modified latex nanoparticles and liposomes. Furthermore, the agreement between film thicknesses calculated from Cryo-TEM or DLS measurements further support the presence of a thin, but measurable, film on the nanoparticle surface. These small changes in size are representative of the highly controllable growth of the polyelectrolyte multilayer afforded by the LbL technique. The generalizable nature and excellent quality control of this method has important implications for the future of basic research into LbL nanomedicines, as TFF is a highly accessible technology that could be employed by most laboratories.
2.3. Generation of an LbL Small Library
The generation of a small library of LbL nanoparticles demonstrated the potential of TFF-assisted LbL assembly to increase experimental throughput, allowing researchers to better evaluate the contribution of different variables in their nanoparticle formulations. Since one of the greatest strengths of the LbL platform is its ability to generate diverse material systems, there is a large parameter space to be explored. It has been well documented that nanoparticle surface chemistry plays an important role in particle-cell interactions.[24] The work by Murphy and colleagues has highlighted several biological effects of LbL nanoparticles that depend on surface chemistry, demonstrating the importance of broadly studying different terminal coatings.[9a, 25] To this end, ten unique LbL nanoparticle formulations were generated from 100 nm CML cores. Each particle was coated with PLA and then terminated with one of ten polyanions described in Table 1. These terminal coats include a variety of polysaccharides, linear polypeptides, and synthetic or modified polymers. In particular, the hyaluronic acid and heparin-folate conjugate coated formulations possess known targeting properties.[26] Of note, the inclusion of hyaluronic acid into LbL particles can be challenging due to the polymer’s propensity to form secondary and tertiary structures that can drive aggregation.[27] However, hyaluronic acid is a highly desirable material that benefits biomedical applications with its unique biological behaviors such as mucoadhesion, receptor targeting, and antifouling.[4a, 11, 28] Use of the TFF-assisted method greatly increased the throughput of nanoparticles functionalized with hyaluronic acid (ca. 10-fold per particle). Each particle prepared for the library exhibited excellent size, uniformity, and surface charge, consistent with the descriptions of the more highly layered formulations.
Table 1.
Ten formulations of LbL nanoparticles with unique terminal polymer layers were fabricated using the TFF method. Each particle is a bilayer formed on carboxy-modified polystyrene substrates (CML-[poly(L-arginine)/polymer X], where polymer X is one of the terminal layers described below). The size, uniformity and zeta potential of each nanoparticle formulation was determined by dynamic light scattering and laser Doppler electrophoresis. Error represents the standard deviation of three technical replicates.
| Terminal Layer | Average Hydrodynamic Size [nm] | Polydispersity Index | Zeta Potential [mV] |
|---|---|---|---|
| Carboxy-modified latex | 95 ± 5 | 0.10 ± 0.03 | −63 ± 1 |
| Poly(L-arginine) | 105 ± 2 | 0.03 ± 0.01 | 66 ± 1 |
| Poly(L-aspartic acid) | 101 ± 9 | 0.08 ± 0.02 | −63 ± 1 |
| Poly(L-glutamic acid) | 106 ± 3 | 0.04 ± 0.04 | −61 ± 1 |
| Poly(L-glutamic acid)-b-polyethylene glycol | 105 ± 9 | 0.08 ± 0.01 | −51 ± 3 |
| Poly(acrylic acid) | 99 ± 7 | 0.05 ± 0.03 | −68 ± 2 |
| Hyaluronic acid | 153 ± 8 | 0.05 ± 0.02 | −56 ± 2 |
| Dextran sulfate | 109 ± 9 | 0.13 ± 0.04 | −75 ± 1 |
| Heparin-folate conjugatea) | 120 ± 9 | 0.07 ± 0.03 | −61 ± 2 |
| Sulfated beta cyclodextrin polymer | 110 ± 8 | 0.05 ± 0.06 | −50 ± 2 |
| Heparin sulfate | 107 ± 4 | 0.11 ± 0.04 | −73 ± 2 |
| Fucoidan | 126 ± 4 | 0.12 ± 0.05 | −61 ± 1 |
Heparin sulfate was conjugated to folic acid using NHS addition chemistry. See supplemental information for more detail.
Importantly, this work describes the manufacture of several novel LbL formulations that incorporated sulfated beta-cyclodextrin polymer, the algae-derived polysaccharide fucoidan, and a heparin sulfate-folate conjugate. The fabrication of stable LbL particles composed of a cyclodextrin polymer opens the door to generating LbL films with drug-loaded polyelectrolytes for the inclusion of hydrophobic small molecules. LbL nanoparticles coated with fucoidan present opportunities to investigate emergent reports of this molecule’s involvement in cellular differentiation and immune modulation.[29] The inclusion of a heparin-folate conjugate also raises opportunities to leverage both molecules’ biological interactions towards an effective targeted therapy. Furthermore, given the success of this technique with a variety of material systems, it is likely that this approach could be adapted to prepare LbL formulations that utilize alternative attractive forces such as hydrogen bonding and hydrophobic interactions.
2.4. Evaluation of LbL Nanoparticle Toxicity
To address the concern that TFF-made LbL nanoparticles may have unintended cytotoxic effects due to the carryover of excess polycation, a viability assay was performed on SKOV3 cells with three LbL formulations. Poly(L-aspartic acid), hyaluronic acid, and heparin sulfate terminated nanoparticles were prepared by either the TFF-method or the conventional centrifugal method and incubated with SKOV3 cells over the course of 72 hours. Cell viability was measured at 24, 48, and 72 hours and compared to the viability of untreated cells and cells exposed to the bare latex core (Figure 5 and Figure S6). None of the nanoparticle formulations exhibited statistically significant changes in viability compared to untreated controls, indicating that TFF-made LbL nanoparticles show low toxicity in vitro.
Figure 5.
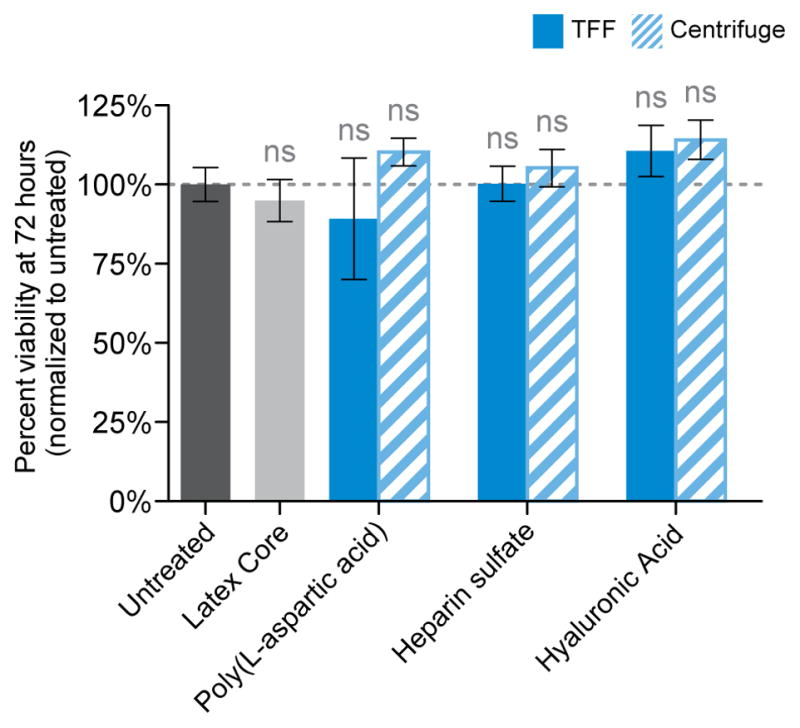
LbL nanoparticles prepared using the TFF-assisted method do not exhibit nonspecific cytotoxic effects in vitro. SKOV3 cells were incubated with poly(L-aspartic acid), herparin sulfate, and hyaluronic acid terminated LbL nanoparticles that were prepared by either the TFF method (solid, blue bars) or the conventional centrifugal method (striped bars). After 72 hours, the cellular viability was determined using the Cell-Titer Glo luminescence assay. The results were normalized relative to untreated controls, and subsequent analysis by one-way ANOVA failed to find any statistically significant changes in cellular viability for any formulation, regardless of preparation method.
These results provide evidence that LbL nanoparticles prepared using the TFF method are cell-compatible by performing a head-to-head comparison of toxicities between several formulations. The results further prove that TFF purification provides highly pure LbL nanoparticles free of cytotoxic contaminants such as free polyelectrolytes.
2.5. LbL Nanoparticle Refrigerated Shelf-Life
The capability to increase the scale, yield and throughput of LbL preparations is an important step towards realizing clinical translation. Another important factor for translation, which to our knowledge has not been addressed previously for LbL nanoparticles, is their shelf life. We evaluated whether appreciable destabilization would occur with nanoparticles from our small library during a three-month storage period in pure water at 4°C. We found that LbL nanoparticles broadly retained their number-average and z-average sizes, as well as their low PDI and high zeta potentials (Figure 6). Relative to the freshly prepared nanoparticles, nine out of ten formulations exhibited statistically consistent hydrodynamic sizes. Using the more aggregate-sensitive size determinant, z-average size, seven out of ten formulations retain their original size. Interestingly, the PDI of all formulations remained statistically consistent during storage, which may indicate that size changes are due more to swelling of the film than to aggregation events. Furthermore, the zeta potential of the LbL nanoparticles remained very high during storage, although some statistically significant changes were noted. Two formulations (HA and DXS-terminated) exhibited minor but significant (P<0.05) loss of charge. On the other hand, sulfated beta-cyclodextrin-terminated particles exhibited a modest (14.9 mV) and significant (P<0.001) increase in magnitude of surface charge during storage. Overall, these data suggest that LbL formulations are stable on the order of at least 3 months when stored under refrigerated, aqueous conditions. Stability is important because it indicates that these formulations could be kept at-the-ready in clinical settings, and furthermore, could facilitate use in clinical settings by minimizing the need for constant reconstitution prior to administration.
Figure 6.

LbL nanoparticles have long refrigerated shelf lives as indicated by the preservation of their size, uniformity and charge characteristics. Ten different LbL particles consisting of a carboxy-modified latex core and a bilayer of poly(L-arginine) and a unique polyanion were stored for three months at 4 degrees Celsius. (a) We compared the hydrodynamic number average size for the particles after storage and compared them to the particle’s original size. With the exception of the Fucoidan-coated particle, all others maintained statistically consistent sizes during storage. (b) These particles likewise exhibited statistically consistent polydispersity index, suggesting that particle uniformity is maintained during storage. (c) The z-average size measurement, which is more sensitive to aggregates, demonstrated that 7 out 10 formulations exhibited statistically consistent sizes during storage. (d) Generally, the zeta potential of the nanoparticles was conserved during storage with a few exceptions. Sulfated beta cyclodextrin-coated particles exhibited a significant charge increase during storage. On the other hand, hyaluronic acid and dextran sulfate-coated particles exhibited a small, but statistically significant decrease in zeta potential. Overall these data indicate that LbL particles would be amenable to months-long refrigerated storage without concern of loss of colloidal stability. One-way ANOVA with the Bonferroni post-test (alpha = 0.01) was used to determine statistical significance between the indicated samples.
2.6. Freeze-dried, Room Temperature Storage of Drug-loaded LbL Liposomes
Refrigerated storage can pose its own problems for clinical translation in terms of transportation and accessibility. Moreover, storing LbL nanoparticles in an aqueous state can allow for the slow release of encapsulated small molecule drugs. With this in mind, lyophilization and room temperature storage of freeze-dried powders is ultimately a far more convenient and manageable option for clinical and industrial scale operations.[30] To determine if LbL nanoparticles can survive such storage conditions, the doxorubicin-loaded Lipo-[PLA/DXS]2 formulation was used as a model system for cryogenic storage. LbL liposomes were combined with several different cryoprotectants (5% glucose, 10% sucrose, 11% trehalose), and with or without a poly(vinyl alcohol) preservative (PVP). LbL-modified and bare liposomes were flash-frozen in liquid nitrogen and lyophilized to produce a fluffy, cotton-like powder (Figure 7a). The powder was kept at room temperature for a week prior to being reconstituted in pure water.[31] The reconstituted particles were dialyzed overnight and then analyzed to determine if particle integrity or drug encapsulation had been compromised.
Figure 7.

Liposomal-core layer-by-layer nanoparticles can be lyophilized and reconstituted for long-term storage. Doxorubicin-loaded liposomes were coated with two bilayers of poly(L-arginine) and dextran sulfate (Lipo-[PLA/DXS]2), using the TFF-method. (a) Lipo-[PLA/DXS]2 particles were lyophilized with different cryopreservatives. (b) TEM of particles before lyophilization (left panel) and after being reconstituted from freeze-dried powder (right panel). (c) LbL modified liposomes retained more drug than bare liposomes regardless of choice of cryoprotectant, though several protectants facilitate nearly 100% drug retention during storage and reconstitution. (d) The presence of cryoprotectants prevents aggregation upon LbL liposome reconstitution. (e) Reconstituted LbL liposomes exhibit lower polydispersity index than reconstituted bare liposomes, indicating improved uniformity. (f) Cryopreservation with 10% sucrose or 11% trehalose prevented decreases in the zeta potential of LbL liposomes. Size and polydispersity data were acquired by dynamic light scattering, and zeta potential data was measured using laser Doppler electrophoresis. Error bars represent standard deviation of three technical replicates.
The results indicated that LbL liposomes protected their drug payload better than bare liposomes under these storage conditions (Figure 7c). In five out of seven conditions, including the cryopreservative-free control, statistically significant (P<0.05 or P<0.01) improvements in retention relative to bare liposomes were observed. Notably, preservation by 5% glucose or 11% trehalose led to virtually no drug loss in LbL liposomes (97 ± 3% and 97 ± 2% encapsulation, respectively). The rest of the formulations retained at least 89% of the drug payload, whereas the liposomes only retained 79–89% of their encapsulated drug after reconstitution.
Generally, we observed that inclusion of PVP was detrimental to overall stability of these nanoparticles. These results are consistent with previous work, which demonstrated that poly(vinyl acid)-based preservatives fail to adequately stabilize liposomes unless modified.[32] In contrast, it has been seen that PVP is sufficient for protection of polymeric nanoparticles, such as those composed of poly(dl-lactide-co-glycolide), poly (D,L-lactic acid), and poly(lactic acid-co-ethylene oxide).[33] Therefore, the presence of a polymeric film on the LbL-coated liposomes may help to explain their better stability relative to bare liposomes when lyophilized in the presence of PVP.
Cryoprotectants were found to be essential for LbL nanoparticle survival under long-term storage conditions. Without cryoprotectants, LbL particles undergo irreversible aggregation into micron-sized particles. With protectants, there is no statistically significant impact on LbL nanoparticle size after freeze-dried storage (Figure 7d, Figure S7). On the other hand, the use of cryoprotectants led to statistically significant (P<0.001) increases in the z-average diameter of bare liposomes (Figure S7). Inclusion of PVP partially rescued liposomes protected with either glucose or sucrose, but the resulting z-average size was still significantly larger than the original formulation. Bare liposomes also undergo statistically significant (P=0.001) increases in PDI after reconstitution, in contrast to LbL liposomes, which retain low PDIs that are statistically consistent with their original PDI (Figure 7e).
Zeta potential measurements of the reconstituted particles indicated that the choice of cryoprotectant was important in preserving the original surface charge on LbL nanoparticles (Figure 7f). Either 10% sucrose or 11% trehalose managed to prevent any decrease in zeta potential for LbL particles. These results are consistent with prior work, which demonstrated that disaccharides are more suitable cryoprotectants for lipid-based nanoparticles than monosaccharides.[34] Overall this choice of cryoprotectant is an important characteristic to optimize, as loss of surface charge may compromise the reconstituted particle’s shelf life or further in vivo stability.
Overall, these results strongly indicate that LbL liposomes can be stably stored under freeze-dried, room-temperature conditions. This finding has major implications for the clinical and industrial translatability of this technology, as it would facilitate the stockpiling, shipping and distribution of future nanomedicines. Our work compared the efficacy of several cryoprotectants head-to-head, and revealed that 11% trehalose was the most consistent protectant and maintained the size, uniformity, and charge of our LbL liposomes. Furthermore, preservation by trehalose facilitated high drug payload retention, with a 97 ± 2% encapsulation of doxorubicin. Notably, LbL liposomes exhibited superior drug cargo retention compared to bare doxorubicin-containing liposomes, and could represent a safe and straightforward means towards the improvement of already clinically approved nanotherapies that may suffer from suboptimal drug-retention under storage.
3. Conclusion
LbL nanoparticles represent a desirable class of materials that are modular, hierarchical, and multifunctional. These characteristics are critical for the advancement of potent next-generation nanomedicines. Until now, these materials were difficult to prepare, despite their straightforward assembly behavior. So far, intermediate purification steps between layer depositions presented the greatest barrier for both lab scale research and clinical translation. Here we described a solution to this bottleneck by using tangential flow filtration to handle the cumbersome and time-consuming washing steps. This approach did not restrict the hallmark versatility of LbL assembly, allowing us to prepare nanoparticles ranging from 40 to 150 nm from solid polymer, mesoporous silica, and liposomal substrates. Furthermore, this technique was compatible with several distinct LbL formulations composed of over a dozen biologically relevant polyelectrolytes. This versatility was accompanied by rapid purification/processing times, excellent product yields, and control over important nanoparticle characteristics (e.g., size, uniformity, and surface charge). This approach has the potential to become fully automated, which would benefit the colloidal LbL community in a similar way that robotic dipping machines have enhanced research into the LbL modification of macroscopic substrates. In particular, future work that focuses on incorporating programmable, robotic operation of TFF devices would provide hands-off synthesis of LbL nanomaterials.
This report also highlights that LbL materials can be stored in aqueous, refrigerated conditions on the order of months. Additionally, we demonstrate that LbL liposomes loaded with small molecule drugs, namely doxorubicin, can be stored under freeze-dried, room temperature conditions without compromising drug encapsulation or nanoparticle characteristics. Overall, this work provides a new path towards ramping up basic research into this important class of drug-delivery vehicles, as well as a means to scale-up existing and future technologies to the clinic.
4. Experimental Section
Colloidal Substrate And Polyelectrolyte Preparation
The polyelectrolytes poly(L-lysine) HBr (PLL, 4–15 kDa; Sigma Aldrich), dextran sulfate sodium salt (DXS, 6.5–10 kDa; Sigma Aldrich), poly(acrylic acid) (PAA, 15 kDa; Sigma Aldrich), poly(L-arginine) (PLA, 9.6 kDa; Alamanda Polymers), poly(L-glutamic acid) (PG, 15 kDa; Alamanda Polymers), poly(L-aspartic acid) (PD, 14 kDa; Alamanda Polymers), poly(L-glutamic acid)-b-polyethylene glycol (PG-b-PEG, 30 kDa PG, 5 kDa PEG; Alamanda Polymers), linear polyethylenimine (PEI, 25 kDa, Polysciences), hyaluronic acid (HA, 40 kDa; LifeCore Biomedical), sulfated beta-cyclodextrin polymer (18kDa; Cyclodextrin Technologies), and fucoidan (57 kDa; Santa Cruz Biotechnology) were used without modifications. Heparin sodium salt (HS, unfractionated; Celsus Labs) was used both unmodified and as a conjugate with folic acid (Sigma Aldrich) as described previously.[35] For additional detail on the synthesis see supplemental section online.
The fluorescent, carboxylate-modified latex nanoparticles (100 nm, blue fluorescent (350/440) and 40 nm yellow-green fluorescent (505/515); Life Technologies) were used without modification.
Mesoporous silica nanoparticles were prepared according to the protocol for 40 nm particles described by Zhang and coworkers.[36] The procedure was as follows: (i) 1.52 g of hexadecyltrimethylammonium bromide (CTAB, Sigma Aldrich), 0.35 g triethanolamine (0.311 mL, Sigma Aldrich), and 100 mL Millipore water were mixed and heated at 80ºC for 1 hour, until dissolved. (ii) 14.6 g of tetraethyl orthosilicate (Sigma Aldrich) was added quickly to the solution, which was left stirring for another 2 hours. (iii) Particles were isolated by high-speed centrifugation (30,000 × g for 1 hour) and washed once with water and then once more with methanol. Particles were resuspended in methanol for CTAB extraction (iv) Template removal was accomplished by first adding 1 mL of 12 M HCl followed by refluxing the solution at 80 ºC for 14 hours. (v) The silica particles were recovered by centrifugation and washed twice with methanol prior to being dried under vacuum. For LbL preparations, dried silica was reconstituted in Millipore water. The size of these particles was measured to be 30 ± 10 nm by dynamic light scattering. For the porosity of these nanoparticles, the reader is referred to the original paper describing this protocol.[36]
Doxorubicin-loaded liposomes were prepared as follows: (i) 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC; Avanti Polar Lipids), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) sodium salt (POPG; Avanti Polar Lipids), and cholesterol (Sigma Aldrich) prepared at a 56:5:39 weight ratio were dissolved in a cosolvent of cholesterol and methanol (2:1 volume ratio). (ii) The solution was evaporated using a rotary evaporator at 40°C at 150 mbar to make a thin lipid film. (iii) The film was hydrated in 300 mM citric acid buffer (pH 4) under sonication at 65°C until uniform liposomes are generated. (iv) 300 mM sodium carbonate buffer was added into the liposome solution to adjust the pH of the solution to 6.5, which generated a pH gradient between the buffer solution outside liposomes and the internal environment of liposomes. (v) Doxorubicin HCl salt (Dox; LC Laboratories) was dissolved at 3 mg mL−1 in sodium chloride solution (154 mM, 0.9 wt/vol %), and the Dox solution was added into the liposome solution under sonication at 65°C for 5 min. Dox-loaded liposomes were purified using the TFF system (500 kDa MWCO filter module; Spectrum Labs) as described below.
Tangential Flow Filtration Assisted LbL Fabrication
Colloidal substrates (CS) and polyelectrolytes (PE) were dissolved in equal volumes of purified water at a 1:5 mass ratio of CS to PE. Prior to the first layer deposition, the CS solution was sonicated for 5 minutes and the PE solution for 15 minutes to disrupt any pre-existing aggregates. The CS solution was added to the PE solution under sonication to generate the PE film. Once mixed, the CS-PE mixture was sonicated for 5 more seconds and incubated on an orbital shaker for 15 minutes. The CS-PE mixture was then transferred to a 50 mL conical flask (Corning) and connected to a tangential flow filtration (TFF) device (KrosFlo Research IIi; Spectrum Labs). All connections were made using MasterFlex size 14 Pharma-Pure tubing, with the exception of the tubing that runs through the peristaltic pump, which is size 16. The TFF was connected to a suitable filtration membrane composed of modified polyethersulfone, either a 100 kDa MWCO or a 500 kDa MWCO membrane (Spectrum Labs, MidiKros Class) depending on the size of the CS being used. Refer to the supplemental information for a video showing the experimental set up.
The filter membranes were used exclusively with one charge type to prevent product loss and membrane fouling. The membrane used to process positively charged nanoparticles is pre-processed by recirculating salt-free, cationic PE solution for 10 minutes to mask anionic sites. The negative-particle membrane is used without further modification.
The CS-PS mixture was pumped with a constant feed rate (190 mL min−1 for 500 kDa membrane and 170 mL min−1 for 100 kDa membranes) through the appropriate TFF membrane using a peristaltic pump. Feed rate was chosen to maintain a transmembrane pressure between 3–5 PSI and a shear rate below 6,000 s−1. These values were calculated using the provided software and sensors within the TFF device.
To maintain a constant sample volume during purification, a 2 L buffer reservoir filled with Millipore water was connected to the sample container. The CS-PE mixture was washed until 5 complete volumes were eluted (for a 40 mL sample that would be 200 mL of permeate) when using a 500 kDa membrane. With the 100 kDa membrane, 7 complete volumes were eluted. Permeate solution was collected in sequential 20 mL aliquots to determine purification kinetics as described below.
After adequate purification, the nanoparticle sample was disconnected from the buffer reservoir in order to concentrate the pure product down to 20 mL in anticipation for the next layer deposition. At this stage, samples were taken for characterization of the nanoparticles as described below.
After removing the pure CS sample from the TFF device, another layer was deposited as before, with an oppositely charged PE solution. During this incubation, filter membranes were briefly rinsed with water and switched to the appropriate charge-specific membrane for the next purification. This process was repeated until reaching the desired number of layers.
Note that the mass ratio of 1:5 for substrate to polyelectrolyte generally yielded stable layer depositions, but for certain polyelectrolytes the ratio was shifted until a zeta potential measurement with a magnitude of at least 40 mV was observed in the unpurified mixture. For example, our CML cores were used at a 1:5 mass ratio with PLL, whereas we used a 1:3 mass ratio with DXS, which equates to 10 mg of PLL and 6 mg of DXS being used per layer deposition during the modification of 2 mg of CML substrates.
Characterizing Purification Kinetics and Yield
Permeate samples were taken from purifications involving PLL and DXS. To quantify the amount of PLL extracted using the TFF, we directly analyzed the PLL permeate samples as well as PLL standards using the bicinchoninic assay (BCA; Pierce). Absorbance measurements were taken with a TECAN Infinite M200 Pro plate reader and used to calculate the mass of PLL removed from the system.
The concentration of DXS in the permeate was determined by gel permeation chromatography (GPC). DXS permeate samples and standards were lyophilized and redissolved in the appropriate GPC eluent. The measurements of DXS was performed on an Viscotek GPCmax system (Malvern) equipped with Agilent PL aquagel-OH columns (PL aquagel-OH Guard 8 μm (50 mm × 7.5 mm), and PL aquagel-OH MIXED-M 8 μm (300 mm × 7.5 mm, exclusion limit > 6×105 g mol−1)) thermostated to 35°C and a refractive index detector maintained at 35°C. 100 mM NaNO3 and 10 mM NaH2PO4 in aqueous solution at pH 7.4 containing 4:1 (v/v) MeOH was used as mobile phase with a flow rate of 1.0 mL min−1. The refractive index area (mVmL) of each sample (injection volume constant at 50 μL) was measured and calibrated against a DXS standard curve.
To determine nanoparticle yields, purified product was weighed in tared conical flasks to determine recovered sample volume. Then, 10 μL samples of the product were taken and diluted to 100 μL in pure water and analyzed using the Infinite M200 plate reader. Sample fluorescence measurements (350 nm excitation, 440 nm emission) were compared to standards of known CML concentration in order to calculate the product yield.
For LbL nanoparticles prepared by centrifugation, instead of processing with the TFF the nanoparticle solution was centrifuged at 15,000 rcf for 15 minutes. Supernatant was removed and spun again until no nanoparticle pellet was formed, about 6 spins total per layer. Recovered pellets were gently resuspended in pure water, to avoid dislodging any insoluble aggregates from the tube wall. HA terminated nanoparticles was spun at 10,000 rcf for 5 minutes to minimize product loss.
Lbl Liposome Lyophilization And Storage
Samples were prepared with cryoprotectant (5% glucose, 10% sucrose or 11% trehalose; Sigma Aldrich), cryoprotectant plus poly(vinyl alcohol) preservative (PVP, Mowiol 4–88; Sigma Aldrich), or without any stabilizer. Samples were flash frozen in liquid nitrogen for 15 minutes and lyophilized for 48 hours. The freeze-dried powders were backfilled with N2 and stored at room temperature on the bench top for a week prior to reconstitution in pure water (reverse osmosis, MilliQ) followed by an overnight equilibrium dialysis (8 kDa MWCO, 4C).
Nanoparticle Characterization
Hydrodynamic size and polydispersity index were measured using dynamic light scattering (Malvern ZS90 particle analyzer, λ = 633 nm, material/dispersant RI 1.590/1.330). Zeta potential measurements were made using laser Doppler electrophoresis with the Malvern ZS90 as well. All measurements were conducted in water (reverse osmosis, MilliQ). Cryo-TEM was performed with a JEOL 2100 FEG instrument, and quantitative image analysis was performed using ImageJ software. For Cryo-TEM, 3 uL of the sample solution was dropped onto a lacey copper grid coated with a continuous carbon film. The sample was blotted to remove excess liquid by Gatan Cryo Plunge III. The grid was mounted on a Gatan 626 cryo-holder, and then the specimen and holder tip were cooled using liquid nitrogen. Imaging was performed using the minimum dose method to prevent damage to the sample by the electron beam. The microscope was operated at 200 kV with a magnification setting of 10,000–30,000 for assessing particle size and distribution. All images were then recorded on a Gatan 2kx2k UltraScan CCD camera. Conventional TEM was conducted using either a FEI Tecnai Multipurpose TEM (120 kV) or JEOL 2011 High Contrast Digital TEM (120 kV). Specimens for conventional TEM were prepared by drop-casting nanoparticle solutions onto mesh copper grid coated with a continuous carbon film.
Drug encapsulation was determined for Dox-loaded nanoparticles by measuring Dox fluorescence (500 nm excitation, 600 nm emission) from particle and diafiltrate solutions (50% DMSO) following an overnight equilibrium dialysis (8 kDa MWCO, 4C) against lyophilization solutions. Fluorescence measurements were made using the Infinite M200 plate reader.
Cytotoxicity Characterization
SKOV3 (ATTC) ovarian cancer cells were maintained in regular Dulbecco’s modified Eagle medium (DMEM, Corning) supplemented with 10% fetal bovine serum (Seradigm) and 1% penicillin-streptomycin (Corning). Cells were seeded in tissue-culture grade 96-well plates (Greiner) at a density of 5,000 cells per well the night before treatment with LbL nanoparticles. The next day, the cells were incubated in media containing a 20 pM concentration of LbL nanoparticles. At the reported time points, the cellular viability was determined using the Cell-Titer Glo luminescence assay (Promega).
Data Processing And Statistical Analysis
Purification kinetics data were fit to a one-phase exponential decay model with PRISM software. The model constrained y0 (set to 0), plateau (set to known amount introduced into the system), and K (must be greater than 0). PRISM was also used to fit a basic linear model to yield data.
One-way ANOVA with the Bonferroni post-test was used to determine statistically significant differences in most cases (results from this test are annotated in gray on all plots). To assess statistically significant changes between original nanoparticles and the various cryogenic storage conditions, one-way ANOVA with the Dunnet post-test was performed. Determinations from this particular test are denoted in blue (for LbL particles) and red (for bare liposomes) in Figure 7. Unless noted otherwise, alpha was set to 0.01 (99% confidence). All tests were conducted in PRISM software.
Size measurements taken from ImageJ were imported and analyzed in R studio. The data were visualized as probability density histograms, and overlaid with their kernel density distribution estimate using the ggplot2 package. Size and charge distribution data exported from the Malvern Zetasizer software were processed in Microsoft excel to convert the raw data into a frequency table for import into R studio. The distributions were then plotted using the violinplot functionality of the ggplot2 package to produce the figures seen in the supplemental documentation.
Supplementary Material
Supplemental Figure S1. Solid-core LbL particles were prepared by coating 100 nm, carboxy-modified latex particles with three bilayers of polyethylenimine and heparin sulfate (100CML-[PEI/HS]3). This formulation was prepared using the TFF-assisted protocol. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Gray denotes latex core, orange denotes PEI layers, and dark blue denotes HS layers.
Supplemental Figure S2. Solid-core LbL particles were prepared by coating 40 nm, carboxy-modified latex particles with three bilayers of poly(L-lysine) and dextran sulfate (40CML-[PLL/DXS]3). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with smaller LbL substrates. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Gray denotes latex core, orange denotes PLL layers, and light blue denotes DXS layers.
Supplemental Figure S3. Mesoporous-core LbL particles were prepared by coating 50 nm, mesoporous silica nanoparticles with three bilayers of poly(L-arginine) and poly(L-glutamic acid) (50MSN-[PLL/DXS]3). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with a solid, drug-loadable substrate. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. We observed improvement in the size distribution during LbL modification which maybe due to the introduction of stronger electrostatic stabilization thanks to the polymer coating. The initial increase in size at the first layer is thought to be the generation of a relatively thick “fuzzy” polymer surface, which is then compacted by deposition of the next layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. The weak charge on the initial MSN substrate led to relatively broader and less uniform charge distributions for the earlier layers, which eventually gives way to more uniform, tight charge distribution by layer 4. (c) The polydispersity index was measured by dynamic light scattering, and decreased throughout LbL assembly indicating improving uniformity. This is consistent with the observations made in the shape of the size distribution. Error represents SD of three technical replicates. Green denotes mesoporous silica core, orange denotes PLA layers, and purple denotes PG layers.
Supplemental Figure S4. Hollow-core LbL particles were prepared by coating 70 nm, doxorubicin-loaded liposomes with two bilayers of poly(L-arginine) and dextran sulfate (Lipo-[PLA/DXS]2). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with liposomal substrates, which are important for drug delivery. This formulation was also used to assess long-term storage options for LbL liposomes (see Main Figure 7). (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Red denotes liposomal core, orange denotes PLA layers, and light blue denotes DXS layers.
Supplemental Figure S5. This figure displays the size and charge distribution data for the two formulations featured in the main document; 100CML-[PLL/DXS]5 and Lipo-[PLL/DXS]4-PLL-HS. (a, c) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b, d) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. For polydispersity index data see the main document. Gray denotes latex core, red denotes liposomal core, orange denotes PLL layers, light blue denotes DXS layers, and dark blue denotes HS layers.
Supplemental Figure S6. Cellular viability of SKOV3 cells for 24, 48, and 72 hours following treatment by LbL nanoparticles indicate that these materials are not nonspecifically toxic. No statistically significant differences in viability were detected for any formulation, at any of the tested incubation times. Significance was evaluated using one-way ANOVA with the Dunnet post-test, comparing each formulation to the untreated control group for each incubation time. Error represents SD of three biological replicates.
Supplemental Figure S7. The z-average size measurement, which is more sensitive to aggregation, further indicates that LbL liposomes can be lyophilized and stored at room temperature without loss of colloidal stability. This requires the use of a cryoprotectant, otherwise large-scale aggregation occurs as indicated above. Notably, bare liposomal formulations stored with cryoprotectants exhibit statistically significant z-average size increases that were not observed by the number average measurements, indicating the presence of an aggregating population. This finding supports the observation from the main document that polydispersity index increases for bare liposomes during long-term storage with cryoprotectants. Error represents SD of three technical replicates. Red denotes bare liposomes and blue represents LbL liposomes, Lipo-[PLL/DXS]2.
Supplemental Figure S8. Further detail of the surface of LbL nanoparticles is provided by the magnification of the Cryogenic TEM images of LbL nanoparticles. The region indicated by the dashed red line is magnified 200% in the right panel. (a) LbL-coated carboxy-modified latex (CML-[PLL/DXS]5); (b) LbL-coated, doxorubicin-loaded liposome (Lipo-[PLL/DXS]4-PLL-HS).
Supplemental Figure S9. Convention TEM images of the LbL nanoparticle library, taken after three months of storage at 4 degrees Celsius in pure water, provide evidence that the morphology of these nanoparticles are preserved. Specimens were drop-casted from a 10 μg/mL solution onto a mesh copper grid coated with a continuous carbon film and then imaged on a JEOL 2011 High Contrast Digital TEM (120 kV). The red scale bar denotes 500 nm in each image. The inset shows a high-magnification view of a single nanoparticle, and the black scale bar from panel (a) represents 100 nm for all insets.
Supplemental Figure S10: UV-Visible spectrum of folic acid decorated heparin (after dialysis) indicating the immobilization of folic acid as evident by the characteristic shoulder of folate at 363 nm.[1]
Acknowledgments
The authors thank the MIT Koch Institute Swanson Biotechnology Center, which is supported by the Koch Institute Core Grant P30-CA14051 from the NCI, for the use of facilities, and specifically the Peterson (1957) Nanotechnology and Biopolymer Core Facilities. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374 for S.C. Additionally, this work was supported by the National Institutes of Health (1F32EB017614–02; E.C.D.) and the Swiss National Science Foundation Postdoctoral Fellowship for K. R. The authors wish to express their appreciation to Shawn Musgrave, Frances Liu, Griffin Clausen, Kelly Moynihan, Ryan Truby, Brittany Goods, and Marianna Sofman for helpful discussions.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Santiago Correa, Institute for Integrative Cancer Research Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Dr. Ki Young Choi, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Dr. Erik C. Dreaden, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Kasper Renggli, Koch Institute for Integrative Cancer Research Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Aria Shi, Koch Institute for Integrative Cancer Research Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Dr. Li Gu, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Dr. Kevin E. Shopsowitz, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Dr. Mohiuddin A. Quadir, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Elana Ben-Akiva, Koch Institute for Integrative Cancer Research Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Dr. Paula T. Hammond, Email: hammond@mit.edu, Koch Institute for Integrative Cancer Research Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
References
- 1.a) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; b) Torchilin VP. Adv Drug Deliv Rev. 2012;64:302–315. doi: 10.1016/j.addr.2012.08.003. [DOI] [PubMed] [Google Scholar]; c) Bao G, Mitragotri S, Tong S. Annu Rev Biomed Eng. 2013;15:253–282. doi: 10.1146/annurev-bioeng-071812-152409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG. Nat Rev Cancer. 2012;12:39–50. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- 3.a) Caruso F. Adv Mater. 2001:13. [Google Scholar]; b) Hammond PT. Adv Mater. 2004;16:1271–1293. [Google Scholar]; c) Hammond PT. Nanomedicine. 2012;7:619–622. doi: 10.2217/nnm.12.47. [DOI] [PubMed] [Google Scholar]
- 4.a) Poon Z, Lee JB, Morton SW, Hammond PT. Nano Lett. 2011;11:2096–2103. doi: 10.1021/nl200636r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morton SW, Shah NJ, Quadir MA, Deng ZJ, Poon Z, Hammond PT. Adv Healthcare Mater. 2014;3:867–875. doi: 10.1002/adhm.201300465. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang Y, Yan Y, Cui J, Hosta-Rigau L, Heath JK, Nice EC, Caruso F. Adv Mater. 2010;22:4293–4297. doi: 10.1002/adma.201001497. [DOI] [PubMed] [Google Scholar]; d) Fujimoto K, Toyoda T, Fukui Y. Macromolecules. 2007;40:5122–5128. [Google Scholar]; e) Gittins DI, Caruso F. J Phys Chem B. 2001;105:6846–6852. [Google Scholar]; f) Morton SW, Poon Z, Hammond PT. Biomaterials. 2013;34:5328–5335. doi: 10.1016/j.biomaterials.2013.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Elbakry A, Zaky A, Liebl R, Rachel R, Goepferich A, Breunig M. Nano Lett. 2009;9:2059–2064. doi: 10.1021/nl9003865. [DOI] [PubMed] [Google Scholar]; b) Poon Z, Chang D, Zhao X, Hammond PT. ACS Nano. 2011;5:4284–4292. doi: 10.1021/nn200876f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shutava TG, Balkundi SS, Vangala P, Steffan JJ, Bigelow RL, Cardelli JA, O'Neal DP, Lvov YM. ACS Nano. 2009;3:1877–1885. doi: 10.1021/nn900451a. [DOI] [PubMed] [Google Scholar]
- 6.a) Iler RK. J Colloid Interface Sci. 1966;21:569–594. [Google Scholar]; b) Decher G, Hong JD. Makromol Chem, Macromol Symp. 1991;46:321–327. [Google Scholar]; c) Decher G, Hong JD, Schmitt J. Thin Solid Films. 1992;210:831–835. [Google Scholar]; d) Keller SW, Johnson SA, Brigham ES, Yonemoto EH, Mallouk TE. J Am Chem Soc. 1995;117:12879–12880. [Google Scholar]; e) Donath E, Sukhorukov GB, Caruso F, Davis SA, Möhwald H. Angew Chem Int Ed. 1998;37:2201–2205. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2201::AID-ANIE2201>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]; Angw Chem. 1998;110:2323–2327. [Google Scholar]; f) Sukhorukov GB, Donath E, Davis S, Lichtenfeld H, Caruso F, Popov VI, Möhwald H. Polym Adv Technol. 1998;9:759–767. [Google Scholar]; g) Caruso F, Caruso RA, Mohwald H. Science. 1998;282:1111–1114. doi: 10.1126/science.282.5391.1111. [DOI] [PubMed] [Google Scholar]
- 7.Borges J, Mano JF. Chem Rev. 2014;114:8883–8942. doi: 10.1021/cr400531v. [DOI] [PubMed] [Google Scholar]
- 8.a) Del Mercato LL, Abbasi AZ, Ochs M, Parak WJ. ACS Nano. 2011;5:9668–9674. doi: 10.1021/nn203344w. [DOI] [PubMed] [Google Scholar]; b) Del Mercato LL, Abbasi AZ, Parak WJ. Small. 2011;7:351–363. doi: 10.1002/smll.201001144. [DOI] [PubMed] [Google Scholar]; c) Kazakova LI, Shabarchina LI, Sukhorukov GB. Phys Chem Chem Phys. 2011;13:11110–11117. doi: 10.1039/c1cp20354a. [DOI] [PubMed] [Google Scholar]; d) Shchukin DG, Sukhorukov GB. Adv Mater. 2004;16:671–682. [Google Scholar]; e) Ariga K, Hill JP, Ji Q. Phys Chem Chem Phys. 2007;9:2319–2340. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]
- 9.a) Alkilany AM, Boulos SP, Lohse SE, Thompson LB, Murphy CJ. Bioconjug Chem. 2014;25:1162–1171. doi: 10.1021/bc500174b. [DOI] [PubMed] [Google Scholar]; b) Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. ACS Nano. 2013;7:9571–9584. doi: 10.1021/nn4047925. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dreaden EC, Kong YW, Morton SW, Correa S, Choi KY, Shopsowitz KE, Renggli K, Drapkin R, Yaffe MB, Hammond PT. Clin Cancer Res. 2015;21:4410–4419. doi: 10.1158/1078-0432.CCR-15-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Chanana M, Gliozzi A, Diaspro A, Chodnevskaja I, Huewel S, Moskalenko V, Ulrichs K, Galla HJ, Krol S. Nano Lett. 2005;5:2605–2612. doi: 10.1021/nl0521219. [DOI] [PubMed] [Google Scholar]; b) Schneider G, Decher G. Nano Lett. 2004;4:1833–1839. [Google Scholar]; c) Schneider G, Decher G, Nerambourg N, Praho R, Werts MH, Blanchard-Desce M. Nano Lett. 2006;6:530–536. doi: 10.1021/nl052441s. [DOI] [PubMed] [Google Scholar]; d) Hong X, Li J, Wang M, Xu J, Guo W, Li J, Bai Y, Li T. Chem Mater. 2004;16:4022–4027. [Google Scholar]
- 11.Dreaden EC, Morton SW, Shopsowitz KE, Choi JH, Deng ZJ, Cho NJ, Hammond PT. ACS Nano. 2014;8:8374–8382. doi: 10.1021/nn502861t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider GF, Subr V, Ulbrich K, Decher G. Nano Lett. 2009;9:636–642. doi: 10.1021/nl802990w. [DOI] [PubMed] [Google Scholar]
- 13.Schneider G, Decher G. Langmuir. 2008;24:1778–1789. doi: 10.1021/la7021837. [DOI] [PubMed] [Google Scholar]
- 14.a) Voigt A, Lichtenfeld H, Sukhorukov GB, Zastrow H, Donath E, Bäumler H, Möhwald H. Ind Eng Chem Res. 1999;38:4037–4043. [Google Scholar]; b) Qi A, Chan P, Ho J, Rajapaksa A, Friend J, Yeo L. ACS Nano. 2011;5:9583–9591. doi: 10.1021/nn202833n. [DOI] [PubMed] [Google Scholar]; c) Shchukin DG, Kommireddy DS, Zhao Y, Cui T, Sukhorukov GB, Lvov YM. Adv Mater. 2004;16:389–393. [Google Scholar]; d) Kantak C, Beyer S, Yobas L, Bansal T, Trau D. Lab Chip. 2011;11:1030–1035. doi: 10.1039/c0lc00381f. [DOI] [PubMed] [Google Scholar]; e) Priest C, Quinn A, Postma A, Zelikin AN, Ralston J, Caruso F. Lab Chip. 2008;8:2182–2187. doi: 10.1039/b808826h. [DOI] [PubMed] [Google Scholar]; f) Richardson JJ, Ejima H, Lorcher SL, Liang K, Senn P, Cui J, Caruso F. Angew Chem Int Ed. 2013;52:6455–6458. doi: 10.1002/anie.201302092. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:6583–6586. [Google Scholar]; g) Richardson JJ, Teng D, Bjornmalm M, Gunawan ST, Guo J, Cui J, Franks GV, Caruso F. Langmuir. 2014;30:10028–10034. doi: 10.1021/la502176g. [DOI] [PubMed] [Google Scholar]
- 15.Morton SW, Herlihy KP, Shopsowitz KE, Deng ZJ, Chu KS, Bowerman CJ, Desimone JM, Hammond PT. Adv Mater. 2013;25:4707–4713. doi: 10.1002/adma.201302025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Dalwadi G, Benson HA, Chen Y. Pharm Res. 2005;22:2152–2162. doi: 10.1007/s11095-005-7781-z. [DOI] [PubMed] [Google Scholar]; b) Sweeney SF, Woehrle GH, Hutchison JE. J Am Chem Soc. 2006;128:3190–3197. doi: 10.1021/ja0558241. [DOI] [PubMed] [Google Scholar]
- 17.Lohse SE, Eller JR, Sivapalan ST, Plews MR, Murphy CJ. ACS Nano. 2013;7:4135–4150. doi: 10.1021/nn4005022. [DOI] [PubMed] [Google Scholar]
- 18.Bjornmalm M, Roozmand A, Noi KF, Guo J, Cui J, Richardson JJ, Caruso F. Langmuir. 2015;31:9054–9060. doi: 10.1021/acs.langmuir.5b02099. [DOI] [PubMed] [Google Scholar]
- 19.a) Dubas ST, Schlenoff JB. Langmuir. 2001;17:7725–7727. [Google Scholar]; b) Burke SE, Barrett CJ. Langmuir. 2003;19:3297–3303. [Google Scholar]; c) Parveen N, Schonhoff M. Macromolecules. 2013;46:7880–7888. [Google Scholar]
- 20.Shields PA, Farrah SR. Appl Environ Microbiol. 1983;45:526–531. doi: 10.1128/aem.45.2.526-531.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang GJ, Yan HH, Ji SL, Liu ZZ. J Membr Sci. 2007;292:1–8. [Google Scholar]
- 22.We hypothesize that increased film thicknesses observed from CML particle cores relative to that from liposomes arises due to increased quantities of polycation required for both initial and subsequent surface charge reversal steps. Given the headgroup areas of DSPC and POPG in lipid bilayers (66 and 65 Å2, respectively), one could estimate the net anionic parking area for a 56:5 mol DSPC:POPG vesicle as approaching 804 Å2. In contrast, commercial carboxylate modified latex beads exhibit significantly lower parking areas, ranging from 10 to 125 Å2, which necessitate increasingly dense polyelectrolyte layers to achieve charge neutralization and reversal. See reference 23 for further sources.
- 23.a) Petrache HI, Dodd SW, Brown MF. Biophys J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Henin J, Shinoda W, Klein ML. J Phys Chem B. 2009;113:6958–6963. doi: 10.1021/jp900645z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hermanson GT. Bioconjugate Techniques. 2. Academic Press; Amsterdam: 2008. [Google Scholar]
- 24.a) Grabinski C, Schaeublin N, Wijaya A, D'Couto H, Baxamusa SH, Hamad-Schifferli K, Hussain SM. ACS Nano. 2011;5:2870–2879. doi: 10.1021/nn103476x. [DOI] [PubMed] [Google Scholar]; b) Hauck TS, Ghazani AA, Chan WC. Small. 2008;4:153–159. doi: 10.1002/smll.200700217. [DOI] [PubMed] [Google Scholar]
- 25.Alkilany AM, Lohse SE, Murphy CJ. Acc Chem Res. 2013;46:650–661. doi: 10.1021/ar300015b. [DOI] [PubMed] [Google Scholar]
- 26.a) Choi KY, Yoon HY, Kim JH, Bae SM, Park RW, Kang YM, Kim IS, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. ACS Nano. 2011;5:8591–8599. doi: 10.1021/nn202070n. [DOI] [PubMed] [Google Scholar]; b) Salazar MD, Ratnam M. Cancer Metastasis Rev. 2007;26:141–152. doi: 10.1007/s10555-007-9048-0. [DOI] [PubMed] [Google Scholar]
- 27.a) Scott JE, Cummings C, Brass A, Chen Y. Biochem J. 1991;274:699–705. doi: 10.1042/bj2740699. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Scott JE, Heatley F. Proc Natl Acad Sci USA. 1999;96:4850–4855. doi: 10.1073/pnas.96.9.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scott JE, Heatley F. Biomacromolecules. 2002;3:547–553. doi: 10.1021/bm010170j. [DOI] [PubMed] [Google Scholar]
- 28.Lim ST, Martin GP, Berry DJ, Brown MB. J Control Release. 2000;66:281–292. doi: 10.1016/s0168-3659(99)00285-0. [DOI] [PubMed] [Google Scholar]
- 29.a) Kim BS, Kang HJ, Park JY, Lee J. Exp Mol Med. 2015;47:e128. doi: 10.1038/emm.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sharma G, Kar S, Basu Ball W, Ghosh K, Das PK. Cell Mol Immunol. 2014;11:263–274. doi: 10.1038/cmi.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Adv Drug Delivery Rev. 2006;58:1688–1713. doi: 10.1016/j.addr.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 31.Because the lyophilization and reconstitution stages of the freeze-dried storage process are most likely to destabilize the nanoparticles, we expect that these particles could be stored for longer periods than demonstrated here. Nonetheless, a full week was chosen so as to demonstrate compatibility with timeframes common to shipping needs.
- 32.Takeuchi H, Yamamoto H, Toyoda T, Toyobuku H, Hino T, Kawashima Y. Int J Pharm. 1998;164:103–111. [Google Scholar]
- 33.a) Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Int J Pharm. 1999;187:143–152. doi: 10.1016/s0378-5173(99)00187-8. [DOI] [PubMed] [Google Scholar]; b) Berton M, Allémann E, Stein CA, Gurny R. Eur J Pharm Sci. 1999;9:163–170. doi: 10.1016/s0928-0987(99)00049-4. [DOI] [PubMed] [Google Scholar]; c) De Jaeghere F, Allemann E, Leroux JC, Stevels W, Feijen J, Doelker E, Gurny R. Pharm Res. 1999;16:859–866. doi: 10.1023/a:1018826103261. [DOI] [PubMed] [Google Scholar]
- 34.Kamiya S, Nozawa Y, Miyagishima A, Kurita T, Sadzuka Y, Sonobe T. Chem Pharm Bull. 2006;54:181–184. doi: 10.1248/cpb.54.181. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Bae BC, Tran TH, Yoon KH, Na K, Huh KM. Carbohydr Polym. 2011;86:708–715. [Google Scholar]
- 36.Zhang K, Xu LL, Jiang JG, Calin N, Lam KF, Zhang SJ, Wu HH, Wu GD, Albela B, Bonneviot L, Wu P. J Am Chem Soc. 2013;135:2427–2430. doi: 10.1021/ja3116873. [DOI] [PubMed] [Google Scholar]
- 37.Hirsjarvi S, Qiao Y, Royere A, Bibette J, Benoit JP. Eur J Pharm Biopharm. 2010;76:200–207. doi: 10.1016/j.ejpb.2010.07.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Solid-core LbL particles were prepared by coating 100 nm, carboxy-modified latex particles with three bilayers of polyethylenimine and heparin sulfate (100CML-[PEI/HS]3). This formulation was prepared using the TFF-assisted protocol. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Gray denotes latex core, orange denotes PEI layers, and dark blue denotes HS layers.
Supplemental Figure S2. Solid-core LbL particles were prepared by coating 40 nm, carboxy-modified latex particles with three bilayers of poly(L-lysine) and dextran sulfate (40CML-[PLL/DXS]3). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with smaller LbL substrates. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Gray denotes latex core, orange denotes PLL layers, and light blue denotes DXS layers.
Supplemental Figure S3. Mesoporous-core LbL particles were prepared by coating 50 nm, mesoporous silica nanoparticles with three bilayers of poly(L-arginine) and poly(L-glutamic acid) (50MSN-[PLL/DXS]3). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with a solid, drug-loadable substrate. (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. We observed improvement in the size distribution during LbL modification which maybe due to the introduction of stronger electrostatic stabilization thanks to the polymer coating. The initial increase in size at the first layer is thought to be the generation of a relatively thick “fuzzy” polymer surface, which is then compacted by deposition of the next layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. The weak charge on the initial MSN substrate led to relatively broader and less uniform charge distributions for the earlier layers, which eventually gives way to more uniform, tight charge distribution by layer 4. (c) The polydispersity index was measured by dynamic light scattering, and decreased throughout LbL assembly indicating improving uniformity. This is consistent with the observations made in the shape of the size distribution. Error represents SD of three technical replicates. Green denotes mesoporous silica core, orange denotes PLA layers, and purple denotes PG layers.
Supplemental Figure S4. Hollow-core LbL particles were prepared by coating 70 nm, doxorubicin-loaded liposomes with two bilayers of poly(L-arginine) and dextran sulfate (Lipo-[PLA/DXS]2). This formulation was prepared using the TFF-assisted protocol to demonstrate compatibility with liposomal substrates, which are important for drug delivery. This formulation was also used to assess long-term storage options for LbL liposomes (see Main Figure 7). (a) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. (c) The polydispersity index was measured by dynamic light scattering, and remained small throughout LbL assembly indicating good uniformity. Error represents SD of three technical replicates. Red denotes liposomal core, orange denotes PLA layers, and light blue denotes DXS layers.
Supplemental Figure S5. This figure displays the size and charge distribution data for the two formulations featured in the main document; 100CML-[PLL/DXS]5 and Lipo-[PLL/DXS]4-PLL-HS. (a, c) We tracked the hydrodynamic size using dynamic light scattering and plotted the particle size distribution on a violin plot to demonstrate controlled growth. Violin plots display the estimated kernel density of the size or charge distribution, and allow the visualization of population-wide evolution of LbL nanoparticle characteristics during synthesis. The black squares denote the mean hydrodynamic size for each layer. (b, d) The zeta potential reversal at each stage of the layering process confirms successful LbL assembly. This was tracked with laser Doppler electrophoresis, and the charge distributions were plotted as a violin plot. The blacks squares denote the mean zeta potential for each layer. For polydispersity index data see the main document. Gray denotes latex core, red denotes liposomal core, orange denotes PLL layers, light blue denotes DXS layers, and dark blue denotes HS layers.
Supplemental Figure S6. Cellular viability of SKOV3 cells for 24, 48, and 72 hours following treatment by LbL nanoparticles indicate that these materials are not nonspecifically toxic. No statistically significant differences in viability were detected for any formulation, at any of the tested incubation times. Significance was evaluated using one-way ANOVA with the Dunnet post-test, comparing each formulation to the untreated control group for each incubation time. Error represents SD of three biological replicates.
Supplemental Figure S7. The z-average size measurement, which is more sensitive to aggregation, further indicates that LbL liposomes can be lyophilized and stored at room temperature without loss of colloidal stability. This requires the use of a cryoprotectant, otherwise large-scale aggregation occurs as indicated above. Notably, bare liposomal formulations stored with cryoprotectants exhibit statistically significant z-average size increases that were not observed by the number average measurements, indicating the presence of an aggregating population. This finding supports the observation from the main document that polydispersity index increases for bare liposomes during long-term storage with cryoprotectants. Error represents SD of three technical replicates. Red denotes bare liposomes and blue represents LbL liposomes, Lipo-[PLL/DXS]2.
Supplemental Figure S8. Further detail of the surface of LbL nanoparticles is provided by the magnification of the Cryogenic TEM images of LbL nanoparticles. The region indicated by the dashed red line is magnified 200% in the right panel. (a) LbL-coated carboxy-modified latex (CML-[PLL/DXS]5); (b) LbL-coated, doxorubicin-loaded liposome (Lipo-[PLL/DXS]4-PLL-HS).
Supplemental Figure S9. Convention TEM images of the LbL nanoparticle library, taken after three months of storage at 4 degrees Celsius in pure water, provide evidence that the morphology of these nanoparticles are preserved. Specimens were drop-casted from a 10 μg/mL solution onto a mesh copper grid coated with a continuous carbon film and then imaged on a JEOL 2011 High Contrast Digital TEM (120 kV). The red scale bar denotes 500 nm in each image. The inset shows a high-magnification view of a single nanoparticle, and the black scale bar from panel (a) represents 100 nm for all insets.
Supplemental Figure S10: UV-Visible spectrum of folic acid decorated heparin (after dialysis) indicating the immobilization of folic acid as evident by the characteristic shoulder of folate at 363 nm.[1]