

ABSTRACT
Sirtuins (SIRT) are nicotinamide adenine dinucleotide (NAD+) dependent deacetylases or ADP- ribosyl transferases (ARTs) that deacetylate lysine residues on various proteins regulating a variety of cellular and metabolic processes. These enzymes regulate metabolism, cell survival, differentiation and DNA repair. SIRT proteins play an important role in the survival and drug resistance of cancer cells. The purpose of the present study was to investigate the expression and role of SIRT in chronic lymphocytic leukemia (CLL). We analyzed the expression of SIRT1 and SIRT2 in CLL and normal B cells using the Oncomine database as well as by Western blotting of fresh CLL cells from patients and pro-lymphocytic leukemia (PLL) cell lines, JVM-3 and MEC-2. We showed that both primary CLL cells and JVM-3 and MEC-2 cell lines overexpress high levels of functional SIRT1 and SIRT2. SIRT inhibitors EX-527 and sirtinol impair cell growth, induce ROS production, loss of mitochondrial membrane potential and apoptosis in primary CLL cells and cell lines. Using shRNA knock down of SIRT1 and SIRT2 in JVM-3 and MEC-2 cell lines, we showed that expression of both proteins is crucial for the survival of these cells. Furthermore, studies in nutrient deprived conditions suggest a role of SIRT in metabolism in CLL. These results demonstrate that the inhibition of SIRT1 and SIRT2 activity may be a new therapeutic approach for CLL.
KEYWORDS: Chronic lymphocytic leukemia, pro-lymphocytic leukemia, SIRT1, SIRT2, Sirtuins inhibitor
Introduction
B-cell chronic lymphocytic leukemia (B-CLL) or CLL is the most common form of leukemia in western countries. Currently more than 15,000 newly diagnosed cases/year and ∼4500 deaths/year are estimated. CLL is characterized by abnormal proliferation and accumulation of mature CD5 positive B-lymphocytes in blood, bone marrow, spleen and lymph nodes.1 Many patients with progressive CLL have poor overall outcome and survival is greatly impaired by the presence of 11q deletions and17p deletion/TP53 mutation with complex karyotype.2-5 (HDAC) enzyme activity, including the class III HDACs Sirtuins, has been found to be associated with the development of cancer.6,7
Sirtuins are NAD+ dependent ADP-ribosyl transferases with evolutionary conserved function in cellular metabolism and chromatin regulation.8 Seven sirtuins (SIRT1-SIRT7) have been identified in mammals at distinct subcellular locations and targeting different substrates. SIRT1, 2, 6, and 7 are primarily found in the nucleus, SIRT2 in the cytoplasm and SIRT3, 4, 5 in the mitochondria. Sirtuins are associated with cancer as they deacetylate cancer associated transcription factors, and SIRT1 is overexpressed in acute myeloid leukemia, colon and prostate cancers.9 Several studies reported SIRT2 as a tumor suppressor as it is downregulated in human gliomas.10 SIRT1 and SIRT6 are reported to be significantly increased in CLL.7 Several HDAC inhibitors are currently in clinical trials for the treatment of cancer. Although there are in vitro and in vivo studies using class I and class II HDAC inhibitors (HDACi)11-15 in hematologic malignancies, class III HDACi have not been studied in detail. We hypothesized that sirtuins play an important role in the development and maintenance of CLL and might be a target in CLL. In the present study, we have analyzed the expression of SIRT1 and SIRT2 in fresh CLL cells from patients and in the pro-lymphocytic leukemia (PLL) cell lines JVM-3 and MEC-2, and investigated the effects of sirtuin modulation using pharmacological inhibitors and SIRT1 and SIRT2 shRNA. Our data suggest that both SIRT1 and SIRT2 play an important role in CLL cell proliferation and may be a potential therapeutic target.
Results
SIRT1 mRNA is upregulated in CLL
We analyzed the differential expression of SIRT1 and SIRT2 mRNA between leukemic cells and normal PBMC by data-mining of the Oncomine microarray gene expression datasets. Oncomine is a bioinformatics initiative that collects, standardizes, analyzes, and delivers cancer transcriptome data to the biomedical research community.16
We found that SIRT1 expression was significantly upregulated in CLL (n = 448) compared with normal PBMC (n = 74) using the Haferlach leukemia data set17 (Fig. 1A). Furthermore SIRT1 was upregulated in other leukemias including acute myeloid leukemia (n = 542), B-cell acute lymphoblastic leukemia (n = 147), B-cell childhood acute lymphoblastic leukemia (n = 359), myelodysplastic syndrome (n = 206) and pre-B acute lymphoblastic leukemia (n = 70) (Fig. 1A). The expression of SIRT1 was found to be lower in T-cell acute lymphoblastic leukemia (n = 174) and chronic myelogenous leukemia (n = 76) (Fig. 1A & B). By contrast, SIRT2 expression remains unchanged in most leukemias, including CLL (Fig. 1C & D).
Figure 1.
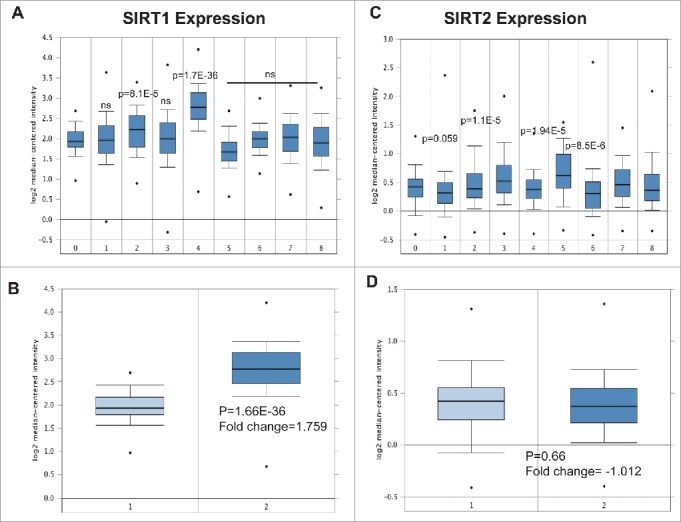
Expression of SIRT1 and SIRT2 mRNA in leukemia revealed by data mining of the Oncomine gene expression database. (A) SIRT1 expression in leukemia cells compared with normal PBMC using the Haferlach leukemia dataset from the Oncomine database (https://www.oncomine.org/resource/login.html). 0) Normal PBMC (n = 74). 1) Acute Myeloid Leukemia (n = 542), 2) B-Cell Acute Lymphoblastic Leukemia (n = 147), 3) B-Cell Childhood Acute Lymphoblastic Leukemia (n = 359). 4) Chronic Lymphocytic Leukemia (n = 448). 5) Chronic Myelogenous Leukemia (n = 76) 6) Myelodysplastic Syndrome (n = 206). 7) Pro-B Acute Lymphoblastic Leukemia (n = 70). 8) T-Cell Acute Lymphoblastic Leukemia (n = 174). (B) Differential expression of SIRT1 in normal PBMC and CLL cells obtained from the Haferlach data set. 1) Peripheral Blood Mononuclear Cell (n = 74) 2) Chronic Lymphocytic Leukemia (n = 448). (C) SIRT2 expression in leukemia cells comparing with normal PBMC using the Haferlach leukemia dataset. (D) Differential expression of SIRT2 in normal PBMC and CLL cells obtained from the Haferlach data set. 1) Peripheral Blood Mononuclear Cell (n = 74) 2) Chronic Lymphocytic Leukemia (n = 448). The error bars represent SDs of mean, and statistically significant differences (by Student t test) between normal PBMC and leukemia are indicated by p-value.
SIRT1 protein is upregulated in CLL primary cells and cell lines
To address the change in protein expression, we quantified SIRT1 and SIRT2 protein expression in fresh CLL samples from 9 patients and 2 cell lines derived from B-prolymphocytic leukemia (JVM-3 and MEC-2) using western blotting. As shown in Fig. 2A, both SIRT1 and SIRT2 proteins were overexpressed in all the CLL samples examined, however, we observed that SIRT2 levels were lower than SIRT1 in most samples (Fig. 2B). By contrast, there was little difference in the protein levels of SIRT1 and SIRT2 in the 2 PLL cell lines JVM-3 and MEC-2 as measured by Western blotting (Fig. 2C) followed by quantitation of relative protein expression (Fig. 2D). Interestingly SIRT1 expression was higher in primary cells than in the cell lines, while SIRT2 expression was lower in primary cells compared with cell lines (Fig. 2 E). These results demonstrate that both SIRT1 and SIRT 2 are overexpressed in CLL patients and PLL cell lines.
Figure 2.
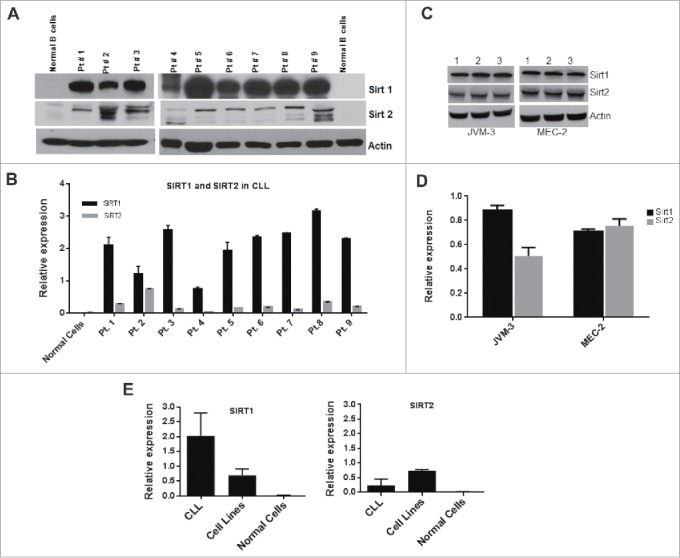
SIRT1 and SIRT2 is overexpressed in CLL primary cells and cell lines. (A) Expression level of SIRT1 and SIRT2 proteins was determined by Western blotting of PBMC obtained from CLL patients and normal B cells. (B) Quantitative estimation of SIRT1 and SIRT2 obtained by measuring band density of Western blots in Fig. 1A using Image-J software. Relative expression denotes values of SIRT1 or SIRT2 in relation to Actin. (C) Expression level of SIRT1 and SIRT2 protein in JVM-3 and MEC-2 cell lines. Western blot of cell lysates obtained from JVM-3 and MEC-2 cell line. Samples were loaded in triplicate for statistical analysis. (D) Quantitative estimation of SIRT1 and SIRT2 protein in JVM-3 and MEC-2 cell lines obtained from Fig. 2C. (E) Graphic presentation of SIRT1 and SIRT2 in grouped samples of CLL and cell lines. Actin is used as an internal control on all Western blots. The error bars represent standard deviation (SD) of mean.
Pharmacological inhibition of sirtuins has anti-proliferative effects on CLL cells and cell lines
To investigate the possible role of sirtuins on CLL cell growth, we tested the effect of SIRT inhibitors, sirtinol and EX-527 in PBMC obtained from 9 CLL patients (data from 3 representative samples are presented) by analyzing cell viability by WST1. EX-527 and sirtinol treatment inhibited cell growth in all the samples tested with an IC50 ranging from 50–100 µM for Ex-527 and 10–20 µM for sirtinol. Cell viability was significantly inhibited in the presence of both inhibitors in a dose dependent manner at 24–72 hr. (Fig. 3A). Similar effects of EX-527 and sirtinol were observed on JVM-3 and MEC-2 cell lines (Fig. 3B). To investigate whether apoptosis was triggered following EX-527 or sirtinol treatment, we incubated cells with the inhibitors for 48 hr. followed by annexin V/PI staining and analyzed by flow cytometry. Both drugs induced significant apoptosis at all the concentrations tested in most of the samples (Fig. 3C) as well as in the cell lines (Fig. 3D). Taken together, these data suggest that inhibition of SIRT1 results in cellular apoptosis in CLL patient samples and relevant cell lines.
Figure 3.

Pharmacological inhibition of Sirtuins effects cell viability and induces apoptosis (A) PBMC purified from CLL patients were treated with indicated concentrations of EX-527 or sirtinol for up to 72 hr. Cell viability was measured by WST-1 assay following 24, 48 and 72 hr. incubation. Representative 3 patients are shown. (B) JVM3 and MEC-2 cell lines were treated with indicated concentrations of EX-527 and Sirtinol for up to 72 hr. Cell viability was measured by MTT assay following 24, 48 and 72 hr. (C) PBMC purified from CLL patients and (D) JVM-3 and MEC-2 cell lines were treated with indicated concentrations of EX-527 or sirtinol for 48 hr. Apoptosis was measured by annexinV/PI staining followed by flow cytometry and analyzed by FCS express software. The error bars represent SDs of mean and statistically significant differences (by Student t test) in between control and treatment are indicated by an asterisk (*, P< 0.01, **, P < 0.001; ***, P < 0.0001).
SIRT inhibitors increase tubulin acetylation
Since we have shown that SIRT inhibitors induce apoptosis and inhibit cell proliferation, we then examined the effect of SIRT inhibitors on SIRT1 and SIRT2 protein expression and acetylation of α-tubulin. As shown in Figs. 4A and B, treatment of cells with EX-527 or sirtinol slightly reduces the expression of SIRT1 protein in CLL primary cells and more so in the representative cell lines. Expression of SIRT2 protein remains unaffected by treatment of cells with any of these inhibitors. To investigate the functional consequences of SIRT inhibition, we investigated the effect of EX-527 and Sirtinol on deacetylating activity of SIRT1 in CLL cells and in cell lines by measuring α-tubulin acetylation by Western blot using acetyl α-tubulin antibody. While EX-527 induced only a 1.5-fold increase in α-tubulin acetylation, a 4–5-fold increase was observed in CLL cells following treatment with sirtinol (a representative patient is shown in Fig. 4C). Fold change in tubulin acetylation over time was determined by quantitation of band densities in Western blots using the Image-J program and represented by histogram. Interestingly, the effect of these inhibitors was more robust in the cell lines. As shown in Fig. 4D, tubulin acetylation was as high as 6-fold in JVM-3 cells and more than 20-fold in MEC-2 cells following treatment with EX-527. By contrast, sirtinol showed a 2-fold and 4-fold increase in α-tubulin acetylation in JVM-3 and MEC-2 cell lines respectively (Fig. 4E). Taken together these data suggest that both EX-527 and sirtinol inhibit deacetylase activity in CLL cells and cell lines albeit with varying intensity resulting in an increase in acetylation.
Figure 4.

Sirt inhibitors impair SIRT1 protein expression and de-acetylation activity. (A) Western blots showing expression of SIRT1 and SIRT2 proteins in CLL cells following exposure to indicated concentrations of EX-527 and Sirtinol for 24 hr. (B) JVM3 and MEC-2 cell lines were treated with EX-527 or sirtinol followed by Western blotting using specific antibodies for SIRT1 and SIRT2. (C) CLL cells were treated with indicated concentrations of EX-527 or sirtinol for 6 hr. followed by cell lysis and Western blotting using specific antibodies for acetyl-α tubulin and total tubulin. Bar graph shows the fold change in tubulin acetylation as measured by quantitation of band densities from the Western blot in Fig. 3C using image-J. (D) JVM-3 and MEC-2 cell lines were treated with 50 µM EX-527 or 20 µM sirtinol (E) and samples were collected at the indicated time points. Western blot was performed using specific antibodies for acetyl-α tubulin and total tubulin. Bar graph shows the fold change in tubulin acetylation. Total tubulin is used for normalization. The error bars represent SDs of mean and statistically significant differences (by Student t test) in between control and treatment over time are indicated by an asterisk (*, P< 0.01, **, P < 0.001; ***, P < 0.0001).
Superoxide production, dissipation of Mitochondrial transmembrane potential (ΔΨm) and caspase activation
Since generation of ROS has been shown to accelerate cell death, we examined the effects of SIRT inhibitors on intracellular ROS formation. Mito-SOX Red, which produces fluorescence when it is oxidized by mitochondrial superoxide, was used to detect ROS production in mitochondria. A significant increase in mitochondrial superoxide formation was observed in both JVM-3 and MEC-2 cell lines following incubation with EX-527 and sirtinol (Fig. 5)
Figure 5.

Generation of intracellular ROS. JVM3 and MEC-2 cells were treated with EX-527 for 16 hr. and sirtinol for 30 minutes followed by 30 min incubation with 2.5 µM MitoSOX red and flow cytometry. Histograms next to the graph overlays show mean fluorescence intensity (MFI). The error bars represent SDs of mean and statistically significant differences (by Student t test) in between control and treatment are indicated by an asterisk (*, P < 0.01, **, P < 0.001; ***, P < 0.0001).
Next, we examined the effect of SIRT inhibitors on mitochondrial transmembrane potential following staining with JC-1, a cationic dye that exhibits a potentially dependent accumulation in mitochondria. Mitochondrial depolarization was indicated by a decrease in the red to green fluorescence intensity ratio. The increase in the green JC-1 monomeric form is indicative of collapse of transmembrane potential and was quantitatively determined using flow cytometry. As can be seen in Fig. 6A the treated CLL cells predominantly showed a shift in the mean fluorescence intensity of green JC-1 monomers compared with controls. Quantitation of FACS data indicated up to ∼90% increase in the mean fluorescence intensity of drug-treated JC-1-stained cells compared with controls. EX-527 and sirtinol treatment caused a statistically significant increase in the percentage of cells with green fluorescence, indicating collapse of the mitochondrial membrane potential, as shown by decrease in red/green fluorescence intensity ratio (Fig. 6B).
Figure 6.

Sirtuin inhibitors induce mitochondrial depolarization and caspase activation. (A) CLL cells were treated with indicated concentrations of EX-527 and sirtinol for 16 hr. followed by JC-1 staining and flow cytometry to measure MMP as described in materials and methods. Figures are representatives of 3 independent experiments. Values in the left quadrant represent cells with intact MMP. CCCP is used as positive control. (B) The bar graph is obtained from Fig. 6A, and represents the mean value of 3 replicates ± SD of A. * indicates significant difference from the corresponding control. The error bars represent SDs of mean and statistically significant differences (by Student t test) in between control and treatment is indicated by an asterisk (***, P < 0.0001). (C) CLL cells and (D) JVM-3 and MEC-2 cells were treated with indicated concentrations of EX-527 and sirtinol for 6 hr. followed by cell lysis and protein gel blotting using specific antibodies for caspase 3 and PARP. Actin is used as an internal control.
Mitochondrial depolarization was accompanied by caspase 3 and PARP cleavage. In CLL cells, EX-527 at 100 µM at 6 hr. caused a significant increase in cleaved caspase 3 and PARP while the effect of sirtinol was minimal (Fig. 6C). Unlike in CLL cells, EX-527 and sirtinol both induced significant caspase 3 and PARP activation in JVM-3 and MEC-2 cell lines (Fig. 6D). Together these results suggest that although both EX-527 and sirtinol induce apoptosis in CLL cells and cell lines, their mode of action might be different in cell lines and primary CLL cells due to higher expression of SIRT1 in CLL cells.
Nutrient deprivation enhances the anti-proliferative effect of SIRT inhibitors
Sirtuins, and SIRT1 in particular have been implicated in the regulation of glucose and lipid metabolism during cellular stress induced by caloric restriction.18 We therefore investigated the activity of SIRT inhibitors in nutrient-deprived medium. JVM-3 and MEC-2 cell lines were cultured in either normal or nutrient deprived (glucose/essential amino acid deficient) medium containing 25 µM of EX-527 or 10 µM sirtinol. Apoptosis was measured after 24 hr. Fig. 7A and B show that the apoptotic effect of EX-527 and sirtinol was enhanced in starving nutrient deprived conditions in both JVM-3 and MEC-2 cell lines.
Figure 7.
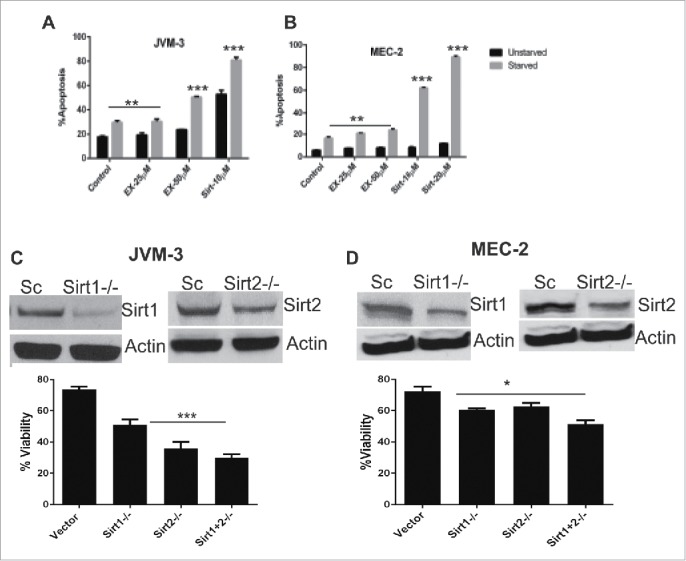
Starvation enhances the effect of SIRT inhibitors and knock down of sirtuins reduces cell viability. (A) JVM-3 and (B) MEC-2 cells were exposed to EX-527 or sirtinol in normal and nutrient deprived medium for 24 hr. Apoptosis was measured by Annexin V/PtdIns staining followed by flow cytometry. Statistically significant differences (by Student t test) between un-starved and starved cells is indicated by an asterisk (*, P< 0.01, **, P < 0.001; ***, P < 0.0001). (C, D) JVM-3 and MEC-2 were transiently transduced with lentivirus containing short hairpin RNA for SIRT1 or SIRT2. Cell viability was measured using MTT assay following 48 hr. of transduction. The error bars represent SDs of mean and statistically significant differences (by Student t test) between cells tranduced with scrambeled and/or SIRT-shRNA are indicated by an asterisk (*, P < 0.01, **, P < 0.001; ***, P < 0.0001).
shRNA knock down of SIRT1 and SIRT2 impairs cell growth
Our results demonstrate that sirtuin activity is compromised in CLL cells exposed to SIRT inhibitors. To confirm whether sirtuins are essential to CLL cell growth, we knocked down SIRT1, SIRT2 or both in JVM-3 and MEC-2 cell lines and followed cell viability for 48 hr. Fig. 7C and D show that transcriptional silencing of SIRT1 or SIRT2 impaired the viability of both JVM-3 and MEC-2 cells at 48 hr. Furthermore, cells lacking both SIRT1 and SIRT2 show further reduction in cell viability.
Discussion
Sirtuins (SIRT) are class III histone deacetylases, which are uniquely dependent on NAD+ for deacetylase activity. The mammalian SIRT1 is the direct homolog of the yeast SIRT2 and has a wide range of substrates and cellular functions. SIRT1 can induce chromatin silencing through de-acetylation of histones H1, H3, and H419 and regulates a wide variety of biological processes and cellular functions by de-acetylating a large number of non-histone proteins, including transcription factor p53, FOXO1, FOXO3a, NF-kB, c-MYC and E2F1,20-25 by regulating cell cycle progression and survival; PGC1α, PPARγ25 and LXR26 and by regulating metabolism. The roles and functions of SIRT1 in cancer development have become increasingly complex and are still not well understood. Although SIRT1 can act as either a tumor promoter or a tumor suppressor,27 the knock down of SIRT1 reduces the growth of various tumor cells.28,29 In the present study, we have explored the role of sirtuin expression in the proliferation and survival of CLL cells and in PLL cell lines (a surrogate since no CLL cells lines are extant) JVM-3 and MEC-2 using 2 SIRT inhibitors, EX-527 and sirtinol.
We observed an increase in SIRT1 mRNA expression in CLL by data mining of an independent microarray dataset in the Oncomine database,16 with a total of 2022 leukemia samples and 74 normal controls. SIRT1 was significantly upregulated in CLL compared with normal PBMC as well as other leukemia types. Overexpression of SIRT1 has been reported in various malignancies30 and high SIRT1 expression is associated with poor prognosis in diffuse large B cell lymphomas,31 ovarian and breast cancers.32,33 Analysis of SIRT2 expression from the same data set did not show any significant difference between CLL samples and other leukemia types or normal PBMC. These observations suggested that SIRT1 might play a role in CLL.
In our studies, Western blot analysis of 9 CLL patient samples and 2 PLL cell lines revealed that both SIRT1 and SIRT2 are overexpressed in CLL cells as well as in the PLL cell lines. SIRT1 expression was significantly higher than SIRT2 in all the CLL samples tested while the difference in SIRT1 and SIRT2 expression in the cell lines was minimal. Moreover, SIRT1 expression was 4-fold higher in CLL patient samples compared with the cell lines while cell lines had higher expression of SIRT2 compared with CLL samples. These observations demonstrate that cell line data are not always consistent with the data in primary cancer cells and that both need to be investigated when possible to explain the sometimes differential biology in vivo and in vitro. Interestingly, single-agent treatment with the specific SIRT1-inhibitor EX527 at low concentration (<25 µM) had minimal effect on leukemic cell proliferation or apoptosis, but treatment with higher concentrations (>50 µM) of EX-527 or the SIRT1/2 inhibitor, sirtinol induced significant apoptosis. These data indicate redundant functions of SIRT1 and SIRT2. Furthermore, both compounds potently inhibited SIRT1 protein expression, while no change in SIRT2 expression was observed in either CLL cells or cell lines. Interestingly, both inhibitors induced α-tubulin acetylation in the cell lines and in CLL cells. Tubulin can be de-acetylated by 2 enzymes, the NAD-independent histone deacetylase HDAC6 34 and SIRT2.35 We observed an increase in tubulin acetylation following treatment with EX-527 as well as sirtinol. While EX-527 is a more specific SIRT1 inhibitor at lower concentrations and both sirtinol and EX-527 display a higher degree of inhibitory activity toward SIRT1 than SIRT2,36 higher concentration of these inhibitors has significant effect on SIRT2 activity as well. We believe that the concentrations of EX-527 and sirtinol that we used for our studies are high enough to inhibit SIRT2 activity, resulting in increased tubulin acetylation. Furthermore, we observed that increase in tubulin acetylation in cell lines was much higher with EX-527 compared with sirtinol. Sirtinol, however induces more apoptosis than EX-527. It is possible that sirtinol has some other non-specific targets, while EX-527 is specific to SIRT1 and SIRT2.
Mammalian sirtuins have diverse cellular locations, target multiple substrates, and affect a broad range of cellular functions, such as regulation of oxidative stress, DNA damage and metabolism.37 We also demonstrated that sirtinol, and EX- 527, pharmacologically distinct inhibitors of SIRT1/SIRT2, induced apoptosis in CLL cells and PLL cell lines, which included a decrease in mitochondrial transmembrane potential (ΔΨm) and enhanced mitochondrial ROS followed by caspase and PARP activation. Using shRNA to knock down SIRT1 and SIRT2 we demonstrated that both SIRT1 and SIRT2 are important for cell survival. Additionally the nutrient deprivation data described in Fig. 7 confirms the role of SIRT1 and SIRT2 in metabolism in CLL. Many cancer cells display a greater sensitivity to nutrient deprivation. Caloric restriction and oxidative stress induce autophagy and SIRT1 expression in tumor cell lines.38 In our studies apoptosis induced by sirtuin inhibition was enhanced in nutrient-deprived conditions. These results support a protective role of sirtuins in tumor cells in which blood supply, nutrients and oxygen are limited.
Using shRNA to knock down SIRT1 and SIRT2 we demonstrated that SIRT1 and SIRT2 expression is crucial for the survival of CLL cells.
Taken together, these findings suggest that CLL cells are characterized by increased expression and function of SIRT1 and SIRT2, both directly inhibited by SIRT inhibitors. SIRT1 and SIRT2 inhibition using specific inhibitors could be a novel therapeutic approach for the treatment of CLL and other SIRT expressing hematologic malignancies.
Materials and methods
Primary chronic lymphocytic leukemia (CLL) cells
After approval by the Northwestern University Institutional Review Board (IRB) and written informed consent in accordance with the declaration of Helsinki, peripheral blood was drawn from patients with CLL. Malignant cells were purified by diluting the blood 1:1 with PBS (Ca2+ and Mg2+ free) and layered on top of Ficoll-Paque Plus (Sigma-Aldrich). Samples were then centrifuged at 150g for 20 minutes at room temperature; the buffy coat layer was removed and washed with PBS twice and subsequently placed in culture with RPMI medium containing 10% heat-inactivated FBS and 200 U of penicillin/streptomycin (Mediatech) under 5% CO2 and 37°C.
Cell lines
B cell-prolymphocytic leukemia (PLL) cell lines JVM-3 (ACC-18) and MEC-2 (ACC-500) were obtained from DSMZ (German collection of microorganisms and cell cultures). JVM-3 and MEC-2 cell lines were grown in RPMI-1640 and IMDM (Life technologies) respectively, consisting of 10% heat-inactivated FBS and 200 U of penicillin/streptomycin (Mediatech) under 5% CO2 and 37°C.
JVM-3 was established from the peripheral blood of a 73-year-old man with PLL and is characterized by Trisomy 12. The cell line was established by EBV-transformation during treatment with phorbol ester TPA; cells express mRNA of the proto-oncogenes BCL2 and BCL3.39 The MEC-2 cell line was established from a patient with CLL in the prolymphocytic phase and expresses the same light (κ) and heavy chains (μ, δ) as the fresh parental B-CLL cells at the same high intensity. MEC-2 is characterized by expression of mature B cell markers (CD19, CD20, CD21, CD22), CD11a, CD18, CD44, CD49d, CD54 and high levels of both CD80 and CD86.40
Antibodies and reagents
Antibodies used were anti-SIRT1 (Upstate-Millipore Biotechnology), anti-p53, anti–acetylated-Lys382-p53, anti-caspase-3, anti-PARP1, anti-Mcl1, anti-Bim, anti-p21, anti-β-Actin, anti-mouse IgG-HRP conjugated and anti-rabbit HRP-conjugated (all from Cell Signaling Technologies). EX-527 and Sirtinol are from Sigma-Aldrich.
Establishment of SIRT1 and SIRT2 knock down cell lines
The phoenix packaging cell line was transfected with SIRT1 or SIRT2 shRNA in GIPZ lentivirus (Open Biosystem) using Mirus 2020 (Mirus Bio LLC) transfection reagent. After 48 hr., the medium containing lentivirus was collected. Lentivirus was incubated with 8 µg/ml polybrene for 10 min and added to JVM-3 and MEC-2 cell lines followed by spin infection at 2200 rpm and 25°C for 2 hr. Following transduction, cells were selected in puromycin-containing media for 14 d Stably transduced cells that were viable after knocking down SIRT1 and SIRT2 proteins were selected for further analysis
WST-1 cell viability assay
Cells were plated at a concentration of 1 × 104 cells per well in 96-well plates and cells were incubated with several different concentrations of EX-527 or Sirtinol. After 24, 48 or 72 h, 10 μL of WST-1 reagent (Roche, Mannheim, Germany) was added into each well and the absorbance was measured at 450 nm after 1 h of incubation using a Model Synergy HT Microplate Reader (Bio-Tek).
MTT assay
In a 96-well flat bottom plate, approximately 104 cells /100 uL were plated and treated for 24, 48 or 72 hours with vehicle or increasing concentrations of EX-527 (25–100 µM) or sirtinol (10–30 µM). After treatment 20 µL of MTS/PMS (Promega Cell Titer 96 Aqueous Non-Radioactive Cell Proloferation assay), solution was added to each well and incubated for 4 hours at 37°C. Plates were then analyzed at 490 nm wavelength. Data are plotted as growth percentage of control. This value was determined by comparing the absorbance reading of each set of control wells in which no drug was added.
Cell apoptosis assays
Apoptosis was measured using the apoptosis detection kit from BD Biosciences using the manufacturer's protocol. In brief, cells were harvested washed and stained with Annexin V-FITC and propidium iodide (PtdIns) for 15 minutes at room temperature followed by flow cytometry using the BD LSR Fortessa instrument. Data were analyzed using FCS Express software. The significance of differences between experimental conditions was determined using the Student's t test.
Western blot analysis
Cells were centrifuged, washed with cold PBS, and lysed on ice for 30 minutes in lysis buffer containing protease and phosphatase inhibitors. Protein concentrations were determined with the Bio-Rad protein assay kit (Bio-Rad, Hercules,CA). Total protein (50μg) was electrophoresed on 12% SDS polyacrylamide gels and transferred to nitrocellulose membranes, blocked for 1 hour with 50 mM Tris buffer, pH7.5 containing 0.15 M NaCl, 0.05% Tween 20 (TBST) and 5 % (wt/vol) nonfat dry milk and probed overnight at 4C with TBST containing primary antibodies. After three 10-minutes washes in TBST, the filters were incubated with horseradish peroxidase-conjugated secondary antibody in the blocking buffer for 1 hour at room temperature. After three 10 minutes washes in TBST, proteins were detected by enhanced chemiluminescence detection reagents (Amersham Biosciences, Buckinghamshire, United Kingdom). Blots were stripped and re-probed with β-actin to use as loading control.
Reactive oxygen species (ROS) measurement
To determine ROS production, JVM-3 and MEC-2 cells were suspended in pre-warmed (37°C) RPMI containing 2.5 µM MitoSOX Red for 30 min followed by washing with PBS and analysis by flow cytometry using the LSRFortessa analyzer (BD, USA). MitoSOX–red is a fluorogenic dye, which specifically targets mitochondria in the live cells.
Measurement of mitochondrial membrane potential (ΔΨm) (MMP)
MMP was measured by flow cytometry using JC-1 staining. JC-1 dye exhibits potential- dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (∼529 nm) to red (∼590 nm). Mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio. Following incubation with EX-527 or sirtinol, cells were further incubated with JC-1 dye (final concentration 2 µM) for 15 minutes. After incubation cells were washed with phosphate buffered saline (PBS) and re-suspended in PBS followed by flow cytometry. Carbonyl cyanide m-cholorophenylhydrazone (CCCP) was used as a positive control.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. David Gius for helpful discussions. Authors also thank the flow cytometry core facility at the Robert H. Lurie Comprehensive Cancer Center. This work was supported in part by the Shannahan Family Foundation and the Bligh Foundation (LIG).
Funding
This work was supported in part by the Shannahan Family Foundation and the Bligh Foundation (LIG).
Author contributions
S.B. designed and performed research, analyzed data, and wrote the paper; L.I.G. designed research, analyzed data, and wrote the paper.
References
- 1.Hallek M. Signaling the end of chronic lymphocytic leukemia: new frontline treatment strategies. Blood 2013; 122:3723-34; PMID:24065239; http://dx.doi.org/ 10.1182/blood-2013-05-498287 [DOI] [PubMed] [Google Scholar]
- 2.Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, Majid A, Dyer M, Siebert R, Taylor AM, et al.. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol 2007; 25:5448-57; PMID:17968022; http://dx.doi.org/ 10.1200/JCO.2007.11.2649 [DOI] [PubMed] [Google Scholar]
- 3.Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, De Paoli L, Spina V, Gattei V, Capello D, et al.. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res 2009; 15:995-1004; PMID:19188171; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1630 [DOI] [PubMed] [Google Scholar]
- 4.Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, Sarno A, Groner S, Mertens D, Busch R, et al.. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114:2589-97; PMID:19643983; http://dx.doi.org/ 10.1182/blood-2009-05-224071 [DOI] [PubMed] [Google Scholar]
- 5.Thompson PA, O'Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, Burger JA, Estrov Z, Jain N, Kantarjian HM, et al.. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 2015; 121:3612-21; PMID:26193999; http://dx.doi.org/ 10.1002/cncr29566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001; 1:194-202; PMID:11902574; http://dx.doi.org/ 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- 7.Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C, Kalavar M, He Z, Burton J, Lichter S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011; 81:325-9; PMID:22237050; http://dx.doi.org/ 10.1159/000334577 [DOI] [PubMed] [Google Scholar]
- 8.Vaquero A. The conserved role of sirtuins in chromatin regulation. Internatl J Dev Biol 2009; 53:303-22; PMID:19378253; http://dx.doi.org/ 10.1387/ijdb.082675av [DOI] [PubMed] [Google Scholar]
- 9.Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. OncoTarget Ther 2013; 6:1399-416; PMID:24133372; http://dx.doi.org/ 10.2147/OTT.S37750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CG, Kurimasa A, et al.. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun 2003; 309:558-66; PMID:12963026; http://dx.doi.org/ 10.1016/j.bbrc.2003.08.029 [DOI] [PubMed] [Google Scholar]
- 11.Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, Shen T, Murphy T, Wickham J, Kanakry C, et al.. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood 2003; 102:652-8; PMID:12649137; http://dx.doi.org/ 10.1182/blood-2002-12-3794 [DOI] [PubMed] [Google Scholar]
- 12.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, et al.. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol 2009; 147:507-14; PMID:19747365; http://dx.doi.org/ 10.1111/j.1365-2141.2009.07881.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Liu S, Sklenar AR, Davis ME, Lucas DM, et al.. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 2005; 105:959-67; PMID:15466934; http://dx.doi.org/ 10.1182/blood-2004-05-1693 [DOI] [PubMed] [Google Scholar]
- 14.Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, Flax EL, Wickham J, Reed JC, Byrd JC, et al.. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia 2004; 18:1207-14; PMID:15116122; http://dx.doi.org/ 10.1038/sj.leu.2403388 [DOI] [PubMed] [Google Scholar]
- 15.Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, Prachand S, Miller R, Gordon LI, Evens AM. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res 2009; 15:3354-65; PMID:19417023; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al.. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007; 9:166-80; PMID:17356713; http://dx.doi.org/ 10.1593/neo.07112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernández JM, Hofmann WK, Mills KI, et al.. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Oncol 2010; 28:2529-37; PMID:20406941; http://dx.doi.org/ 10.1200/JCO.2009.23.4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trend Endocrinol Metabol 2014; 25:138-45; PMID:24388149; http://dx.doi.org/ 10.1016/j.tem.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Molecular cell 2004; 16:93-105; PMID:15469825; http://dx.doi.org/ 10.1016/j.molcel.2004.08.031 [DOI] [PubMed] [Google Scholar]
- 20.Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 2010; 123:4251-8; PMID:21081649; http://dx.doi.org/ 10.1242/jcs.073783 [DOI] [PubMed] [Google Scholar]
- 21.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 2009; 185:203-11; PMID:19364925; http://dx.doi.org/ 10.1083/jcb.200809167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JT, Gu W. SIRT1: Regulator of p53 Deacetylation. Gen Cancer 2013; 4:112-7; PMID:24020002; http://dx.doi.org/ 10.1177/1947601913484496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lize M, Pilarski S, Dobbelstein M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Different 2010; 17:452-8; PMID:19960022; http://dx.doi.org/ 10.1038/cdd.2009.188 [DOI] [PubMed] [Google Scholar]
- 24.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al.. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303:2011-5; PMID:14976264; http://dx.doi.org/ 10.1126/science.1094637 [DOI] [PubMed] [Google Scholar]
- 25.Yang T, Fu M, Pestell R, Sauve AA. SIRT1 and endocrine signaling. Trend Endocrinol Metabol 2006; 17:186-91; PMID:16684606; http://dx.doi.org/ 10.1016/j.tem.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 26.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 2007; 28:91-106; PMID:17936707; http://dx.doi.org/ 10.1016/j.molcel.2007.07.032 [DOI] [PubMed] [Google Scholar]
- 27.Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Inter J Biol Sci 2009; 5:147-52; PMID:19173036; http://dx.doi.org/ 10.7150/ijbs.5.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al.. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13:454-63; PMID:18455128; http://dx.doi.org/ 10.1016/j.ccr.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olmos Y, Brosens JJ, Lam EW. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat 2011; 14:35-44; PMID:21195657; http://dx.doi.org/ 10.1016/j.drup.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 30.Ma L, Maruwge W, Strambi A, D'Arcy P, Pellegrini P, Kis L, de Milito A, Lain S, Brodin B. SIRT1 and SIRT2 inhibition impairs pediatric soft tissue sarcoma growth. Cell Death Dis 2014; 5:e1483; PMID:25341037; http://dx.doi.org/ 10.1038/cddis.2014.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang KY, Hwang SH, Kwon KS, Kim KR, Choi HN, Lee NR, Kwak JY, Park BH, Park HS, Chung MJ, et al.. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am J Surg Pathol 2008; 32:1523-31; PMID:18724249; http://dx.doi.org/ 10.1097/PAS.0b013e31816b6478 [DOI] [PubMed] [Google Scholar]
- 32.Jang KY, Kim KS, Hwang SH, Kwon KS, Kim KR, Park HS, Park BH, Chung MJ, Kang MJ, Lee DG, et al.. Expression and prognostic significance of SIRT1 in ovarian epithelial tumours. Pathology 2009; 41:366-71; PMID:19404850; http://dx.doi.org/ 10.1080/00313020902884451 [DOI] [PubMed] [Google Scholar]
- 33.Lee H, Kim KR, Noh SJ, Park HS, Kwon KS, Park BH, Jung SH, Youn HJ, Lee BK, Chung MJ, et al.. Expression of DBC1 and SIRT1 is associated with poor prognosis for breast carcinoma. Hum Pathol 2011; 42:204-13; PMID:21056897; http://dx.doi.org/ 10.1016/j.humpath.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 34.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417:455-8; PMID:12024216; http://dx.doi.org/ 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- 35.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 2003; 11:437-44; PMID:12620231; http://dx.doi.org/ 10.1016/S1097-2765(03)00038-8 [DOI] [PubMed] [Google Scholar]
- 36.Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Therap 2010; 9:844-55; PMID:20371709; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0971 [DOI] [PubMed] [Google Scholar]
- 37.Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Gen Dev 2006; 20:2913-21; PMID:17079682; http://dx.doi.org/ 10.1101/gad.1467506 [DOI] [PubMed] [Google Scholar]
- 38.Ng F, Tang BL. Sirtuins' modulation of autophagy. J Cell Physiol 2013; 228:2262-70; PMID:23696314; http://dx.doi.org/ 10.1002/jcp.24399 [DOI] [PubMed] [Google Scholar]
- 39.Melo JV, Brito-Babapulle V, Foroni L, Robinson DS, Luzzatto L, Catovsky D. Two new cell lines from B-prolymphocytic leukaemia: characterization by morphology, immunological markers, karyotype and Ig gene rearrangement. Int J Cancer 1986; 38:531-8; PMID:3093393; http://dx.doi.org/ 10.1002/ijc.2910380413 [DOI] [PubMed] [Google Scholar]
- 40.Stacchini A, Aragno M, Vallario A, Alfarano A, Circosta P, Gottardi D, Faldella A, Rege-Cambrin G, Thunberg U, Nilsson K, et al.. MEC1 and MEC2: two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leukemia Res 1999; 23:127-36; PMID:10071128; http://dx.doi.org/ 10.1016/S0145-2126(98)00154-4 [DOI] [PubMed] [Google Scholar]