

abstract
The oncogenic mutation of BRAFV600E has been found in approximately 8% of all human cancers, including more than 60% of melanoma and 10% of colorectal cancers. The clinical proof of concept in treating BRAFV600E-driving melanoma patients with the BRAF inhibitors has been well established. We have sought to identify and develop novel BRAFV600E inhibitors with more favorable profiles. Our chemistry effort has led to the discovery of EBI-907 as a novel BRAFV600E inhibitor with potent anti-tumor activity in vitro and in vivo. In a LanthaScreen BRAFV600E kinase assay, EBI-907 showed an IC50 of 4.8 nM, which is >10 -fold more potent than Vemurafenib (IC50 = 58.5 nM). In addition, EBI-907 showed a broader kinase selectivity profile, with potent activity against a number of important oncogenic kinases including FGFR1-3, RET, c-Kit, and PDGFRb. Concomitant with such properties, EBI-907 exhibits potent and selective cytotoxicity against a broader range of BRAFV600E-dependent cell lines including certain colorectal cancer cell lines with innate resistance to Vemurafenib. In BRAFV600E-dependent human Colo-205 and A375 tumor xenograft mouse models, EBI-907 caused a marked tumor regression in a dose-dependent manner, with superior efficacy to Vemurafenib. Our results also showed that combination with EGFR or MEK inhibitor enhanced the potency of EBI-907 in cell lines with innate or acquired resistance to BRAF inhibition alone. Our findings present EBI-907 as a potent and promising BRAF inhibitor, which might be useful in broader indications.
KEYWORDS: BRAF, BRAFV600E, EBI-907, inhibitors, kinase, MAPK, RAF
Abbreviations
- BRAF
v-raf murine sarcoma viral oncogene homolog b1
- FGFR
fibroblast growth factor receptor
- RET
Rearranged during transfection
- PDGFRb
platelet-derived growth factor receptor B
- MAP
mitogen-activated protein
- ERK
extracellular signal-regulated kinase
- MEK
MAP/ ERK kinase
- TK
Tyrosine Kinase
Introduction
The RAS/RAF/MEK/ERK pathway acts as a signal transducer to sense and send extracellular signals such as hormones, cytokines, and various growth factors into cell nucleus, directing a range of biochemical and physiological processes including cell differentiation, proliferation, growth, and apoptosis.1 However, the pathway is frequently found to be constitutively activated in many human cancers. In many cases, some critical components within this pathway become mutational “driving forces” to fuel the unchecked growth of malignant cells.2 BRAF is a member of the Raf kinase family of serine/threonine-specific protein kinases, which function through phosphorylation and activation of a downstream kinase, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK). Gain-of-function mutations of BRAF have been found in approximate 7-8% of all human cancers, including >50 % of malignant melanomas, ˜45% of papillary thyroid cancer, ˜10% of colorectal cancer, ˜10% of ovarian cancer, and small percentage of many other human cancers.3 Recently it was reported that almost all hairy-cell leukemia patients carry the BRAFV600E mutation and inhibition of the enzyme caused significant remission of the disease.4 These findings have greatly fueled drug discovery efforts in searching for small molecule inhibitors targeting the kinase activity of BRAF mutants (especially the most common form of BRAFV600E 5,6). Recently, BRAF-specific inhibitors such as Vemurafenib (Zelboraf, Roche) and Dabrafenib (GSK2118436, GSK) have been approved by FDA and EMEA for the treatment of BRAF-mutated metastatic melanoma, and many other BRAF inhibitors are under different stages of development.7
Despite recent therapeutic advances, there are still unmet medical needs for BRAF mutant cancers due to not only the innate and acquired drug resistance but also a drug-induced paradoxical activation of downstream effectors MEK and ERK, leading to the onset of secondary skin cancers. We have sought to identify and develop novel BRAFV600E inhibitors with more favorable pharmacology and safety profiles. Through a structure-guided approach, we have identified a novel BRAFV600E inhibitor, EBI-907, with potent anti-tumor activity in vitro and in vivo. More importantly, our results showed that EBI-907 has a broader kinase selectivity profile than existing BRAF inhibitors, suggesting a possibility to develop next generation targeted therapies for BRAF mutated cancers.
Results
Identification of EBI-907 as a potent BRAFV600E inhibitor
Through an extensive structure-guided chemistry approach, we have synthesized a series of novel aminoisoquinolines as candidates for BRAFV600E inhibitors (WO2014/043296). After screening the compound pools against the full-length BRAFV600E and several rounds of activity-guided structural optimization, one compound called EBI-907 (Fig. 1A) was identified as a potent BRAFV600E inhibitor with an IC50 of 4.9 nM in the LanthaScreen kinase assay against the full-length BRAFV600E, which was 12-fold more potent than Vemurafenib (Fig. 1B). The kinase selectivity profile of EBI-907 was evaluated by measuring its inhibitory activities against a panel of over 350 unique kinases. As shown in Fig. 1C, EBI-907 showed the highest activity against all RAF kinases and a feature of broad kinase selectivity, exhibiting potent inhibition (with IC50 less than 100 nM) in many kinases within the branches of Tyrosine Kinase-like (TKL) and Tyrosine Kinase (TK). The top kinases hit by EBI-907 are listed in Fig. 1D.
Figure 1.

Molecular structure and in vitro kinase activities of EBI-907. (A) chemical structure of EBI-907. (B) Inhibitory activity of EBI-907 and Vemurafenib in BRAFV600E kinase assay. (C) Selectivity profile of EBI-907 against human protein kinase superfamily as shown by a KinomeScan dendogram. The figure was generated by using an online Kinome Render program (http://bcb.med.usherbrooke.ca/kinomerenderLig.php). (D) IC50 values of EBI-907 against a panel of top kinases hit by the compound.
EBI-907 showed potent and selective cytotoxicity to BRAFV600E-dependent cell lines
The effect of BRAF inhibitors on cellular proliferation was assessed using a melanoma cell line A375 and a colorectal cancer cell line Colo205, both of which harbor a BRAFV600E mutation. As shown in Fig. 2, EBI-907 potently inhibited proliferation of both cell lines in a concentration-dependent manner (GI50 = 13.3 and 13.8 nM, respectively), exhibiting >10 -fold potency than Vemurafenib) (Fig. 2C). In contrast, EBI-907 did not show meaningful anti-proliferative effects on the growth of Calu-6 cells carrying a wild-type BRAF, displaying at least 40-fold selectivity toward BRAFV600E-dependent cells lines (Fig. 2C).
Figure 2.
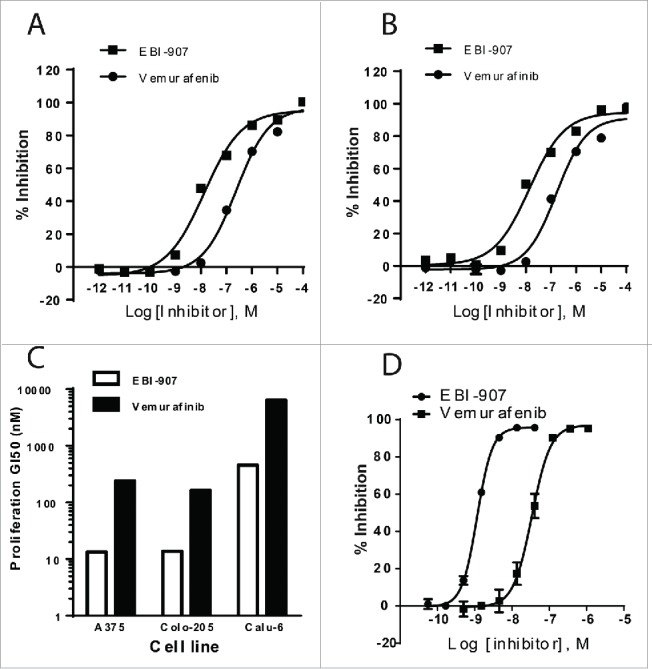
Effect of EBI-907 and Vemurafenib on cell proliferation and ERK phosphorylation in different cell lines. (A) inhibition of A375 cell proliferation by EBI-907 and Vemurafenib. (B) inhibition of Colo205 cell proliferation by EBI-907 and Vemurafenib. (C) the GI50 values of EBI-907 and Vemurafenib in different cell lines. D, inhibition of cellular ERK phosphorylation in A375 cells by EBI-907 and Vemurafenib. All experiments were performed in triplicate and the values in inhibition curves are mean ± SD.
The phosphorylation of extracellular signal-regulated kinases (ERK) 1 and 2 is a critical step in driving the survival and proliferation of BRAFV600E dependent cells. In order to further evaluate the target engagement in a cellular model, we next examined the effect of EBI-907 on ERK1/2 phosphorylation in the A375 cell line. The assay is performed by using an AlphaScreen®SureFire™phospho-ERK Kit (Perkin Elmer) quantitatively detecting the phosphorylation of ERK1/2 in cell lysates. As shown in Fig. 2D, EBI-907 inhibited the phosphorylation of ERK1/2 in a dose-dependent fashion in A375 cells, with an IC50 of 1.2 nM. Consistent with the inhibition of growth in BRAF-mutant cells, EBI-907 also showed a greater potency than Vemurafenib (IC50 = 34 nM) in inhibiting ERK1/2 phosphorylation.
Given that EBI-907 showed a broader kinase selectivity profile, with potent activity against many important oncogenic kinases including FGFRs and RET, we further tested the anti-proliferation activity of EBI-907 in NCI-H1581 (driven by FGFR1) and TT (driven by RET) cells. EBI-907 showed moderate potency in inhibiting the growth of NCI-H1581 (GI50 = 0.82 µM) and TT cells (GI50 = 0.89 µM).
EBI-907 elicited a dichotomous effect on MAPK signaling, which was effectively blocked by a combined inhibition of MEK
It has been reported that many historical RAF inhibitors including Vemurafenib induced MAPK activation in cells with wild-type BRAF, as opposed to a potent inhibition of MAPK signaling in BRAFV600E mutant cells.8 We have tested the effects of EBI-907 on MAPK signaling in Calu-6 cells with wild-type BRAF and mutant KRAS. As shown in Fig. 3A, both Vemurafenib and EBI-907 induced MEK and ERK phosphorylation in a dose-dependent manner in the Calu-6 cells. However, in comparison to a strong induction by Vemurafenib at high concentrations, EBI-907 started to elicit MEK and ERK phosphorylation at very low concentration range, and exhibited complete inhibition on MEK and ERK phosphorylation at high concentrations (Fig. 3A). In addition, combined inhibition of MEK1 kinase activity by a specific MEK1 inhibitor EBI-1051 completely blocked the ERK phosphorylation (Fig. 3B).
Figure 3.
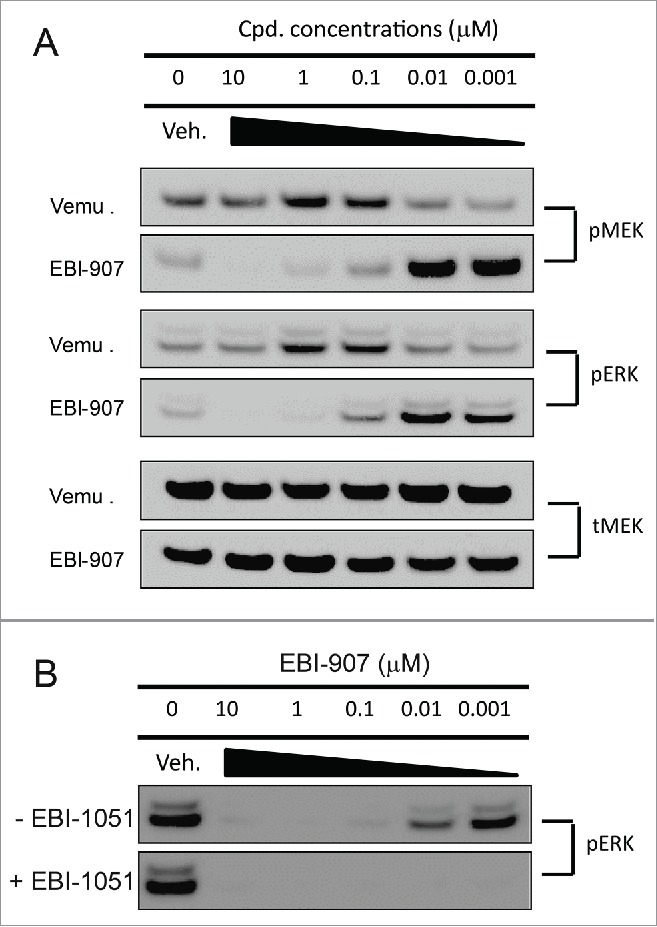
Effect of BRAF and MEK inhibitors on MEK and ERK phoshphorylation in Calu-6 cells. (A) Comparison of Vemurafenib (Vemu) and EBI-907 on MEK and ERK phosphorylation. (B) Effect of EBI-907 on ERK phosphorylation with or without MEK inhibitor EBI-1051. The cells were treated with increasing concentrations of the indicated BRAF inhibitors and/or MEK inhibitor for 2 hrs and the levels of p-MEK and p-ERK in cell lysates were measured by Western blot analysis. Total MEK protein level was also monitored and served as loading control.
EBI-907 displayed favorable pharmacokinetic profiles in preclinical species
We have performed single-dose pharmacokinetic studies in rats and dogs for EBI-907. As shown in Fig. 4A–B, after a single dose of i.v. administration, the plasma concentrations of EBI-907 displayed a typical first-order elimination kinetics in both species, with half-life of 3.42 (rats) and 7.77 (dogs) hours, respectively. After a single oral administration, EBI-907 reached the maximal plasma concentration at around 2-4 hours, followed by the first order clearance, in both rats and dogs, with half-life of 3.35 and 11.3 hours, respectively. A summary of pharmacokinetic parameters is shown in Table 1. Overall EBI-907 has a decent in vivo exposure and favorable PK profiles in both species. Notably, the oral bioavailability of EBI-907 in dogs is favorably higher than rats (44.9% vs 19.7%).
Figure 4.
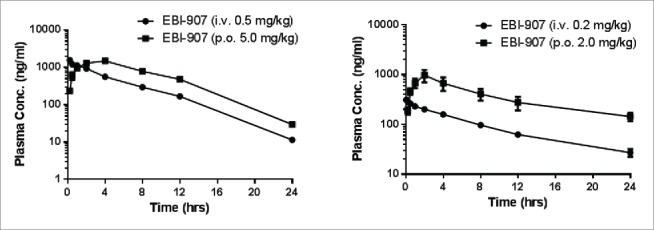
The plasma concentration-time curves of EBI-907 in rats (A) and dogs (B). Data were expressed as mean ± SD (n = 4).
Table 1.
The PK parameters of EBI-907 in rats and dogs after an oral dose (mean ± SD, n = 4).
| Rats | Dogs | |
|---|---|---|
| Dose (mg/kg) | 5 | 2 |
| Tmax (h) | 3.33 ± 1.15 | 2.0 ± 0.01 |
| Cmax (ng/ml) | 1498 ± 474 | 972 ± 465 |
| AUCall(h*ng/ml) | 14186 ± 3717 | 8906 ± 4119 |
| AUCinf (h*ng/ml) | 14330 ± 3744 | 11239 ± 4657 |
| MRT (h) | 6.71 ± 0.22 | 15.2 ± 2.3 |
| t1/2 (h) | 3.35 ± 0.28 | 11.3 ± 1.8 |
| CL (ml/min/kg) | 6.12 ± 1.77 | 3.45 ± 1.77 |
| Vdss (ml/kg) | 1787 ± 612 | 15.2 ± 2.3 |
EBI-907 potently inhibited the tumor growth in the Colo-205 xenograft mice model
Having observed favorable in vitro activities and decent oral exposure in vivo, we next investigated the in vivo efficacy of EBI-907 in the Colo-205 xenograft mice model. To this end, EBI-907 at two doses (20 and 60 mg/kg) was administered twice daily for 14 d to mice bearing Colo205 tumor xenografts, along with the reference compound PLX-4720 (an analog of Vemurafenib) (60 mg/kg, bid). The volume of tumors was measured twice weekly by calipers to monitor the anti-tumor effects of testing compounds. As shown in Fig. 5A, EBI-907 significantly inhibited tumor growth for both doses studied. Upon completion of the experiment, the growth of established Colo-205 xenografts was reduced by 75% and 95% in mice treated with EBI-907 at 20 and 60 mg/kg bid, respectively. Importantly, EBI-907 at 60 mg/kg induced a near complete remission in tumor growth and showed a superior efficacy to PLX-4720 (Fig. 5B). All treatments were well tolerated with no mortality and meaningful changes in body weight (Fig. 5C).
Figure 5.

Effect of BRAF inhibitors on tumor growth in the Colo-205 xenograft mice model. (A) tumor growth curve in mice bearing colo-205 xenograft treated with daily dosing of compounds (p.o, twice a day) for 14 d (B) The relative volume of tumor xenografts at the end of study. (C) The body weight of mice during the course of treatment. Data were expressed as mean ± SE (n = 9 to 12) ***P<0.001 vs. Vehicle; #P<0.05 vs. 0.05.
EBI-907 potently inhibited the tumor growth in the A375 xenograft mice model
EBI-907 at two doses (15 and 50 mg/kg) was administered twice daily for 15 d to mice bearing A375 tumor xenografts, along with Vemurafenib (PLX-4032) at 50 mg/kg, bid. EBI-907 showed a marked inhibition on A375 tumor growth at both doses (Fig. 6A). The relative tumor volume was reduced by ˜75% after 10 d of treatment with EBI-907 at 15 or 50 mg/kg, whereas PLX4032 (Vemurafenib) at 50 mg/kg reduced the tumor burden by 40% (Fig. 6B). Consistent with the Colo-205 xenograft study, all treatments were well tolerated with no meaningful changes in body weight (Fig. 6C).
Figure 6.

Effect of BRAF inhibitors on tumor growth in the A-375 xenograft mice model. (A) tumor growth curve in mice bearing A-375 xenograft treated with daily dosing of compounds (p.o, twice a day) for 15 d (B) The relative volume of tumor xenografts on day 10 after treatment. (C) The body weight of mice during the course of treatment. Data were expressed as mean ± SE (n = 7 to 8) **P<0.01, ***P<0.001 vs. Vehicle; ##P<0.05.
Combination with appropriate targeted therapies is an effective approach to overcome resistance to BRAF inhibitors
Although historical BRAF inhibitors such as Vemurafenib have proven effective in treating BRAF-mutant melanoma, they have shown very limited efficacy in treating colorectal cancers harboring BRAF mutations.6,9 Several lines of evidence showed that EGFR-mediated MAPK pathway reactivation might be responsible for such an innate resistance to BRAF inhibitors, and combined inhibition of RAF and EGFR or other important cell growth pathways improved the anti-proliferative efficacy of BRAF inhibitors.6,9 Our data showed that, despite of a slight attenuation in maximal inhibition, EBI-907 exerted potent activities in inhibiting the cell growth of BRAFV600E-bearing HT-29 and WiDr colorectal cancer cell lines with high expression of EGFR (Fig. 7A). Combination with a specific EGFR inhibitor SHR1258 further enhanced the specific cytotoxicity of EBI-907 against the WiDr cells (Fig. 7B). A calculation of combination index (CI) values of 0.2 strongly supports this synergism.
Figure 7.
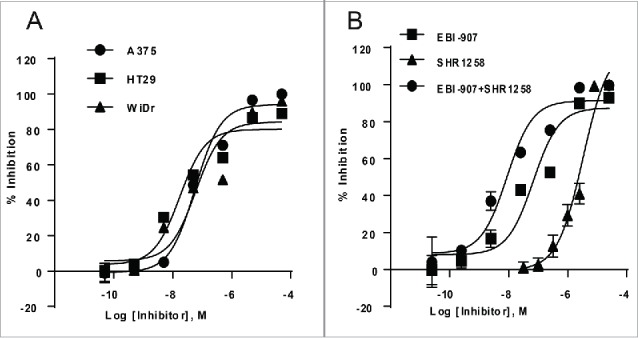
(A) Effect of EBI-907 on proliferation of colorectal cancer cell lines with innate resistance to Vemurafenib. (B) combination with EGFR inhibitor (SHR1258) enhanced the potency of BRAF inhibitor EBI-907 in inhibiting the proliferation of colorectal cancer cell lines. Data were expressed as mean ± SD (n = 3).
Similar to other targeted therapies, acquired drug resistance also develops after initial responses in patients treated with historical BRAF inhibitors such as Vemurafenib. We have generated Vemurafenib-resistant A375 cell lines by chronic exposure to Vemurafenib (GI50 value greater than 10 uM, Fig. 8A). Vemurafenib-resistant cells also showed certain level of resistance to EBI-907 as the GI50 was dramatically elevated (Fig. 8B). Multiple mechanisms for Vemurafenib-induced resistance have been proposed, including MAPK reactivation and activation of alternative survival pathways.10 To study the mechanisms of acquired resistance in our cell model, we examined the MAPK and PI3K/AKT signaling, and found that the level of phosphorylation for both AKT and ERK was increased in Vemurafenib-resistant cells compared with parental A375 cells (Fig.8C). Furthermore, our data showed that EBI-907 potently inhibited the induced ERK phosphorylation in a dose-dependent manner (Fig. 8D), possibly contributing to a residual sensitivity of Vemurafenib-resistant cells to EBI-907.
Figure 8.
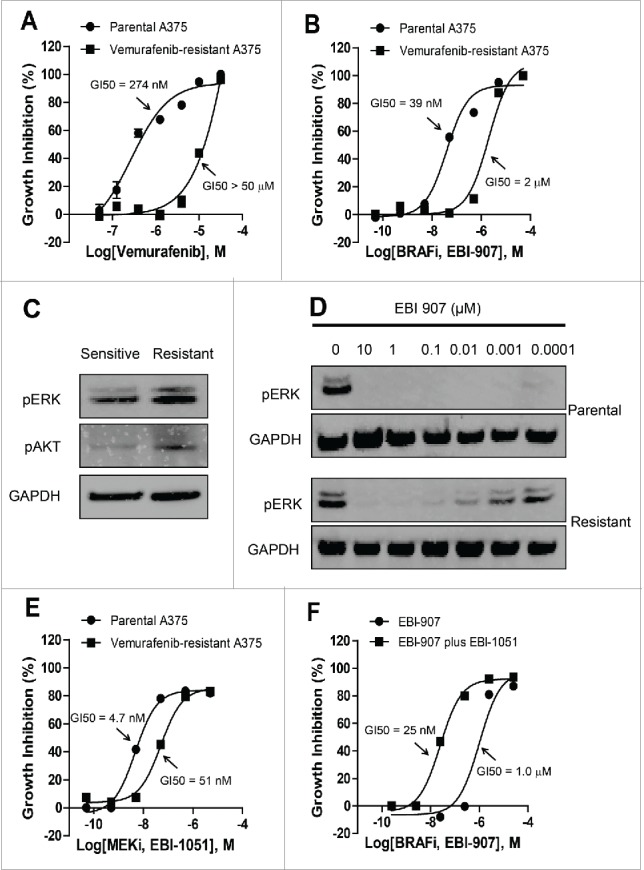
(A) acquired resistance of isolated A375 cells to Vemurafenib. (B) resistance of vemurafenib-resistant A375 cell line to EBI-907. (C) Western blot analysis of ERK and AKT phosphorylation in vemurafenib-sensitive or resistant A375 cells. (D) Western blot analysis of ERK phosphorylation in vemurafenib-sensitive or resistant A375 cells treated with EBI-907. GAPDH was used to as a loading control in panel C and D. (E) Relative growth of parental and vemurafenib-resistant A375 cells in the presence of MEK inhibitor EBI-1051. (F) MEK inhibitor EBI-1051 enhanced the sensitivity of vemurafenib-resistant A375 cells to BRAF inhibition. Quantitative data were expressed as mean ± SD (n = 3).
MEK inhibition restores the sensitivity to vemurafenib in acquired resistance through efficiently blocking ERK phosphorylation. We tested the effect of MEK1/2 inhibitor alone and in combination with EBI-907 on cell growth inhibition of Vemurafenib-resistant A375 cells. The resistant cells also showed a certain level of resistance to a specific MEK inhibitor EBI-1051 with a marked right-shift of the inhibitory curve (Fig. 8E). The combination of MEK and BRAF inhibitors effectively inhibited the growth of the Vemurafenib-resistant A375 cell line (Fig. 8F), suggesting that combined use of BRAF and MEK inhibitors represents an effective strategy in combating the acquired resistance to BRAF inhibitor alone.
Discussion
In the past decade, BRAF mutants have added to the growing list of oncogenic driving kinases that can be therapeutically targeted by a selective inhibitor. The effectiveness of specific BRAF inhibitors on cancers driven by BRAF mutations has been well documented and clinically proved. Following rapid development paths, vemurafenib (PLX-4032) and Dabrafenib (GSK-2118436) were approved for the treatment of BRAF-driven metastatic melanoma in US and other counties. However, despite of the effectiveness of Vemurafenib and Dabrafenib in various clinical settings, they carried certain liabilities (e.g, paradoxical activation of MAPK pathway) and showed only limited efficacy in some patients harboring the same BRAF oncogenic lesion (e.g, colorectal cancer patients). In addition, drug resistance rapidly developed in many patients despite of initial good response.
We have sought to identify and develop potent and selective BRAF (V600E) inhibitors with more favorable pharmacological and toxicological profiles. An extensive SAR optimization campaign led directly to the identification of EBI-907 as a novel BRAF (V600E) inhibitor with potent anti-tumor activity both in vitro and in vivo. More importantly, the compound displayed unique and broad selectivity profile against human kinases, without causing non-specific cytotoxicity in BRAF wild type cells. For example, it showed potent activity against FGFR1-3, RET, c-Kit, and PDGFRb, which is potentially desirable for not only extending its clinical applications, but mitigating the emergence of drug resistance. An up-regulation of PDGFRb was reported in Vemurafenib-resistant melanoma cells, causing the drug resistance.11-13 Therefore, inclusion of the PDGFRb activity in EBI-907 may help decrease the chance of drug resistance development due to PDGFRb activation.
EBI-907 also demonstrated to be orally bioavailable in multiple preclinical species (mice, rats, and dogs) with excellent pharmaceutical profiles. In a BRAF (V600E)-dependent human Colo-205/A375tumor xenograft mouse model, EBI-907 showed potent anti-tumor activity, causing tumor regression in a dose-dependent manner, with superior efficacy to Vemurafenib. A notable feature of BRAF inhibitors is that they selectively suppressed the ERK signaling in BRAF mutant tumors while spared cells carrying wild-type BRAF. Such property of BRAF inhibitors is thought to be the major mechanistic basis for the broad therapeutic window of these compounds. In consistence with this notion, EBI-907 showed a very high safety margin as evidenced by lack of any noticeable toxicity in all dosing groups in the xenograft studies. In contrast, MEK inhibitors inhibit ERK signaling in normal cells, which is thought to account for the substantial toxicities for MEK inhibition in both preclinical and clinic settings.
Despite of the striking anti-tumor effects of BRAF inhibitors in melanoma, their long-term therapeutic benefits appeared to be limited, primarily due to the intrinsic or acquired drug resistance. For example, the vast majority of patients with BRAF-V600E colorectal cancer did not achieve an objective response after treatment with Vemurafenib.14 Similarly, clinical data available so far also suggest that the response in BRAF-mutant lung cancer patients to Dabrafenib is not as impressive as seen in BRAF-mutant melanoma patients.15 The mechanistic basis for such efficacy disparity in different tumor lineages has been an area of active investigation. Multiple groups reported that the alternations in basal RTK signaling such as EGFR-driving signaling is responsible for lack of efficacy of BRAF inhibitors in the BRAFV600E colorectal cancer.6,9 Our results showed that EBI-907 is effective in inhibiting the growth of BRAFV600E-bearing WiDr and HT-29 colorectal cancer cell lines, suggesting a favorable application of EBI-907 in BRAFV600E-bearing colorectal cancer. Furthermore, an enhanced anti-tumor efficacy was achieved by combined inhibition of BRAF (via EBI-907) and EGFR (via our in-house EGFR inhibitor SHR1258). Similar to other targeted therapies, acquired resistance to BRAF inhibitors arises in most melanoma patients in 2 to 12 months despite of an initial substantial response.16 Surprisingly, the gatekeeper mutations, a common mechanism of resistance to other kinase inhibitors, have not been identified in BRAF-V600E in Vemurafenib-treated patients.16 Several mechanisms, including upstream activation of RAS due to mutations or alternative activation of RTK-driving signaling, have been proposed.12,13 Therefore, identification of potent activity in inhibiting multiple RTK (e.g, PDGFRb) by EBI-907 is expected to confer an advantage in delaying or preventing the emergence of drug-resistance in clinical settings.
Apart from the drug resistance, another confounding issue for the development and clinical application of BRAF inhibitors was a phenomenon called “paradoxical activation:” while the ATP-competitive BRAF inhibitors strongly inhibited ERK signaling in BRAF mutant cells, they enhanced the signaling in cells bearing oncogenic or normally activated RAS.8,17,18 Several regulatory mechanisms, including CRAF activation,19,20 drug-induced dimerization and transactivation,8 and inhibitory autophosphorylation,21 have been proposed and extensively studied to understand and explain this paradox. Similar to other historical BRAF inhibitors such as Vemurafenib, EBI-907 also dose-dependently induced MEK and ERK activation in Calu-6 cells carrying wild-type BRAF and mutant KRAS. However, our data suggest a favorable pattern of EBI-907 in activating MAPK pathway – while the compound elicited MEK and ERK phosphorylation in a range of low concentrations; it completely inhibited ERK signaling with increased concentrations within a safe range. Such a difference in the pattern of MAPK activation between EBI-907 and Vemurafenib might come from a superior potency of EBI-907 against the mutant BRAF (V600E) so that it could offer a complete inhibition on the transactivated RAF dimers at a range of relatively safe concentrations. Given a high therapeutic window of EBI-907, such a property of the compound may provide a possibility to “break” the paradoxical activation or mitigate the risk of developing associated cutaneous malignant lesions in clinic. Indeed, it has been reported that a more potent BRAF inhibitor Dabrafenib had a lower incidence of Cutaneous squamous cell carcinomas (cSCC) compared with Vemurafenib (6-11% vs. 20-26%).22 Furthermore, data generated in the current study and others pointed out that combination with a MEK inhibitor is a very effective means in inhibiting the paradoxical activation and related clinical consequences.23
In summary, we have discovered a structurally novel and orally active BRAF (V600E) inhibitor EBI-907 with favorable pharmaceutical profiles. The compounds also demonstrated to be safe and displayed unique pharmacological properties including broader kinase selectivity. Our data support a further clinical development of EBI-907, which is expected to be added to the growing list of novel targeted therapies for the treatment of BRAF-dependent human cancers.
Materials and methods
BRAFV600E enzymatic activity assay - The BRAFV600E enzymatic assay was performed using a LanthaScreen kinase assay kit purchased from Life Technologies (Grand Island, NY). The assay was conducted according to the procedure provided in the assay kit. In brief, the enzyme reaction was carried out in the kinase reaction buffer containing BRAFV600E (20 ng/mL), ATP (2 µM), Fluorescein-MAP2K1 inactive substrate (0.4 µM), HEPES (50 mM, pH 7.5), 0.01% BRIJ-35, MgCl2 (10mM), and EGTA (1mM) in the presence or absence of the tested compounds at various concentrations in 384-well plate at room temperature (22 ± 1°C) for 60 minutes. The final reaction volume for each reaction was 10 µl. The reaction was started by adding the substrates and stopped by addition of 10 µl of TR-FRET dilution buffer supplemented with kinase quench buffer (10 mM EDTA final) and Tb-anti-pMAP2K1 (2 nM final). The plate was further incubated at room temperature for another 60 minutes, and the fluorescent signals were read on Victor 5 (Perkin Elmer) with excitation at 340 nm and emission at 495 and 520 nm. The assay signal was determined as a ratio of FRET-specific signal measured with emission filter at 520 nm to that of the signal measured with Tb-specific emission filter at 495 nm. IC50 value for compound-mediated enzyme inhibition was calculated using appropriate programs in GraphPad Prism software by plotting the logarithm of the compound concentrations versus percent inhibition.
Cell proliferation assay – A375, Colo-205, Calu-6, and SW-480 cells were purchased from American Type Culture Collection (Manassas, VA). All cells were cultured in the recommended medium with 10% heat-inactivated serum. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2. For cell proliferation assay, cells were seeded in 96-well pates at a density of 1,000 to 5,000 cells per well and cultured overnight at 37°C in medium supplemented with 5-10% FBS. On the next day, the compounds at various concentrations or vehicle control (0.5% DMSO) were added into cell culture. After a 3-day treatment, the growth of cells was assayed by the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). IC50 values were calculated using the GraphPad Prism by plotting the logarithm of the compound concentrations vs. percent inhibition of cell growth as compared with the vehicle control.
Generation of Vemurafenib-resistant A375 cell lines – A375 parental cells were plated in 10-cm dishes, and maintained in DMEM media supplemented with 10% heat-inactivated FBS containing vemurafenib at a series of increasing concentrations. The initial concentration of vemurafenib was set at 1.0 µM and doubled every two weeks. After three months, cell clones that became resistant to high concentration of vemurafenib (32 µM) were selected by limited dilution and amplified for further use.
Erk phosphorylation assay – Cells were cultured in 96-well plates overnight in medium supplemented with 5-10% fetal bovine serum (PBS). After 24 hours, the medium was removed and cells were cultured in serum-free medium at 37°C in the absence or presence of various concentrations of the testing compound for 1 hour. After incubation with the testing compound, phosphorylation of Erk in cells was assessed using an AlphaScreen®SureFire™phospho-ERK Kit (Perkin Elmer) following the manufacturer's instruction. IC50 value was calculated using GraphPad Prism by plotting the logarithm of the compound concentrations versus percent inhibition of Erk phosphorylation.
Western blot analysis - Following compounds treatment, cells were lysed in lysis buffer (Cell Signaling Technology) supplemented with protease and phosphatase cocktail (Roche Diagnostics). Cell lysates prepared in loading buffer were run on 4-15% gradient polyacrylamide gel (Life Technology), transferred to nitrocellulose membrane, and probed with the following antibodies purchased from the Cell Signaling: anti-phospho-ERK (Thr202/Tyr204), anti-ERK, anti-phospho-MEK (Ser217/221), and anti-MEK.
In vivo pharmacology studies – Xenograft model was developed in athymic mice (nude/nude mouse) with human colon cancer cell line Colo205 and human melanoma cell line A375 purchased from the American Type Culture Collection Company (Manassas, VA). In brief, Colo205 cells (1 × 107) and A375 cells (8 × 106) were implanted s.c. into the hind flank region of each mouse and allowed to grow to the designated size (c.a. 150-200 mm3) before administration of the compounds. Eight to 12 mice were allocated into each treatment group according to the size of tumors. The compounds were given orally at various dose levels twice daily for 14 d (Colo205 xenograft) or 15 d (A375 xenograft). Tumor volume and body weight were measured at least twice a week during the course of the study. All in vivo experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al.. Roles of the MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773:1263–84; PMID:17126426; http://dx.doi.org/12509763 10.1016/j.bbamcr.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3:11–22; PMID:12509763; http://dx.doi.org/ 10.1038/nrc969 [DOI] [PubMed] [Google Scholar]
- 3.Cantwell-Dorris ER, O'Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther 2011; 10:385–94; PMID:21388974; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0799 [DOI] [PubMed] [Google Scholar]
- 4.Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012; 366:2038–40; PMID:22621641; http://dx.doi.org/ 10.1056/NEJMc1202124 [DOI] [PubMed] [Google Scholar]
- 5.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al.. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–54; PMID:12068308; http://dx.doi.org/ 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 6.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al.. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012; 2:227–35; PMID:22448344; http://dx.doi.org/10.1158/2159-8290.CD-11-034123060265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 2012; 11:873–86; PMID:23060265; http://dx.doi.org/ 10.1038/nrd3847 [DOI] [PubMed] [Google Scholar]
- 8.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464:427–30; PMID:20179705; http://dx.doi.org/ 10.1038/nature08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483:100–3; PMID:22281684; http://dx.doi.org/ 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 10.Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, Kolinsky K, Packman K, Kim MJ, Trunzer K, et al.. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res 2012; 72:969–78; PMID:22205714; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1875 [DOI] [PubMed] [Google Scholar]
- 11.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al.. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468:973–7; PMID:21107323; http://dx.doi.org/ 10.1038/nature09626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al.. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014; 4:80–93; PMID:24265155; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res 2011; 71:5067–74; PMID:21803746; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kopetz S DJ, Chan E, Hecht JR, O'Dwyer PJ, Lee RJ, et al.. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol 2010; (suppl) 28:15s (abstract nr 3534); PMID:19933920; http://meetinglibrary.asco.org/content/50595-7419933920 [Google Scholar]
- 15.Planchard D MJ, Riely GJ, Rudin CM, Barlesi F, et al.. Interim results of phase II study BRF113928 of dabrafenib in BRAF V600E mutation–positive non-small cell lung cancer (NSCLC) patients. J Clin Oncol 2013; 31(suppl):(abstr 8009); PMID:23669222; http://meetinglibrary.asco.org/content/112427-13223669222 [Google Scholar]
- 16.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013; 19:1401–9; PMID:24202393; http://dx.doi.org/ 10.1038/nm.3392 [DOI] [PubMed] [Google Scholar]
- 17.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al.. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010; 464:431–5; PMID:20130576; http://dx.doi.org/ 10.1038/nature08833 [DOI] [PubMed] [Google Scholar]
- 18.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al.. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010; 140:209–21; PMID:20141835; http://dx.doi.org/ 10.1016/j.cell.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 2005; 20:963–9; PMID:16364920; http://dx.doi.org/ 10.1016/j.molcel.2005.10.022 [DOI] [PubMed] [Google Scholar]
- 20.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al.. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004; 116:855–67; PMID:15035987; http://dx.doi.org/ 10.1016/S0092-8674(04)00215-6 [DOI] [PubMed] [Google Scholar]
- 21.Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, Tellew J, Hardy S, Hekmat-Nejad M, Stuart DD, et al.. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell 2013; 23:594–602; PMID:23680146; http://dx.doi.org/ 10.1016/j.ccr.2013.03.033 [DOI] [PubMed] [Google Scholar]
- 22.Menzies AM, Kefford RF, Long GV. Paradoxical oncogenesis: are all BRAF inhibitors equal?. Pigment Cell Melanoma Res 2013; 26:611–5; PMID:23795808; http://dx.doi.org/ 10.1111/pcmr.12132 [DOI] [PubMed] [Google Scholar]
- 23.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al.. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367:1694–703; PMID:23020132; http://dx.doi.org/ 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]