

Abstract
Emerging evidence indicates that methylglyoxal (MG) can inhibit tumorigenesis. Glyoxalase I (GLOI), a MG degradation enzyme, is implicated in the progression of human malignancies. However, little is known about the roles of MG and GLOI in breast cancer. Our purpose was to investigate the anticancer effects of MG and inhibition of GLOI on breast cancer cells and the underlying mechanisms of these effects. Our findings demonstrate that cell viability, migration, invasion, colony formation, and tubule formation were significantly restrained by addition of MG or inhibition of GLOI, while apoptosis was significantly increased. Furthermore, the expression of p-JNK, p-ERK, and p-p38 was markedly upregulated by addition of MG or inhibition of GLOI, whereas MMP-9 and Bcl-2 expression levels were dramatically decreased. These effects were augmented by combined treatment with MG and inhibition of GLOI. Collectively, these data indicate that MG or inhibition of GLOI induces anticancer effects in breast cancer cells and that these effects are potentiated by combination of the 2. These effects were modulated by activation of the MAPK family and downregulation of Bcl-2 and MMP-9. These findings may provide a new approach for the treatment of breast cancer.
KEYWORDS: Bcl-2, breast cancer cell line, glyoxalase I, MAPKs, Methylglyoxal, MMP-9
Abbreviations
- Bcl-2
B-cell lymphoma protein-2
- DMEM
Dulbecco Modified Eagle medium
- ER
estrogen receptor
- ERK
extracellular signal-regulated kinases
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- GLOI
glyoxalase I
- GLOII
glyoxalase II
- JNK
c-Jun N-terminal kinases
- MAPKs
mitogen-activated protein kinases
- MG
methylglyoxal
- MMP9
matrix metallopeptidase 9
- NF-κB
nuclear factor-κB
- p38
p38 mitogen-activated protein kinase
- PBS
phosphate-buffered saline solution
- PI
propidium iodide
- PKC
protein kinase C
- RT-PCR
reverse-transcriptase polymerase chain reaction
- TNF
tumor necrosis factor.
Introduction
Despite advances in early diagnosis and treatment, breast cancer is the most prevalent cancer and the second most common cause of cancer death in women in the US, where breast cancer accounts for 29% of all newly diagnosed cancers and 15% of all deaths from cancer in 2014.1 Although the incidence of breast cancer in China used to be low, it has increased dramatically in recent years and continues to rise.2,3 The high mortality rate is due mainly to tumor recurrence.4 The recurrent tumors are often more aggressive than the primary tumor, possess an intrinsic resistance to therapy, and have a high metastatic potential, all of which contribute to a poor prognosis. Therefore, the need for novel and effective therapeutic approaches that suppress the mechanisms for breast cancer recurrence is particularly urgent.
Methylglyoxal (MG), a highly reactive dicarbonyl compound, is formed spontaneously during glycolysis.5 Accumulating evidence indicates that MG exhibits antitumor activity in various cancer cells, partially by a strong cytotoxic effect.6 A great deal of evidence indicates that MG promotes the activation of protein kinase C (PKC), c-Jun N-terminal kinases (JNK), and p38 mitogen–activated protein kinase (MAPK) signaling, thus inducing cell apoptosis and necrosis.7-9 Talukdar et al. reported that MG prevents tumor growth by inhibiting mitochondrial respiration and glycolysis of malignant cells.10 Nevertheless, the role of MG in breast cancer is largely unknown.
The glyoxalase system is a ubiquitous detoxification pathway consisting of glyoxalase I (GLOI) and glyoxalase II (GLOII), which act in concert to convert the spontaneously formed hemithioacetal adduct between glutathione and MG into D-lactate and glutathione.9 It is known that cancer cells have lower MG and higher GLOI levels and activity than normal cells.5,6,9,11 Abnormal expression and activity of GLOI and MG levels have been demonstrated in breast cancer cells as well.12,13 Increased expression and activity of GLOI are believed to be associated with the increased proliferation, progression, and drug resistance of tumor cells.14
Matrix metallopeptidase 9 (MMP9) is one of a class of zinc-dependent proteinases that degrade extracellular matrix components and is therefore important in tumorigenesis and cancer metastasis.15 Production and secretion of MMP-9 in tumor cells is a critical element in promoting metastasis.16
B-cell lymphoma protein-2 (Bcl-2), the founding member of the Bcl-2 family, which includes both anti-apoptotic and pro-apoptotic proteins, is known to modulate the cell cycle. Bcl-2 is a key regulator of the intrinsic apoptosis pathway by interfering with the release of cytochrome C from the mitochondria or its binding to Apaf-1 through interaction with Bax.17 An earlier study showed that overexpression and/or activation of Bcl-2 was correlated with cancer pathogenesis and chemoresistance in several apoptosis-resistant cell lines and tumor specimens.18 Hence, Bcl-2 is a prime target for novel specific anticancer therapeutics.
It has been shown that the MAPK family, which includes p38 kinases, extracellular signal–regulated kinases (ERK), and JNK, all play important roles in cell death, differentiation, and survival.19 JNK and p38 are activated in response to several stress signals, including tumor necrosis factor (TNF) and hyperosmotic conditions, and this activation is associated with induction of apoptosis.20 ERK activation is induced by growth factors and oxidant injury and plays a major role in regulating cell growth and differentiation.21
Emerging evidence suggests that MG and the glyoxalase system are involved in tumorigenesis. Yet, the role of MG, especially in relation to the glyoxalase pathway in breast cancer and the mechanisms underlying that role have not been elucidated. In this study, we investigated the anticancer effects of MG and inhibition of GLOI and their potential molecular mechanisms in breast cancer cells.
Results
GLOI knockdown suppressed GLOI mRNA, protein, and enzyme activity in human breast cancer cell lines
After transfection with short hairpin GLOI (shGLOI), the expression of GLOI mRNA (Fig. 1A) and protein (Fig. 1B) were significantly suppressed in MCF-7 and T47D (estrogen receptor [ER] positive) and MDA-MB-231 (ER negative) breast cancer cells compared to cells transfected with short hairpin control (shNC; p < 0.01 to 0.001). Similarly, GLOI enzyme activity was also significantly decreased in the breast cancer cells transfected with shGLOI compared to those transfected with shNC (p < 0.01; Fig. 1C).
Figure 1.
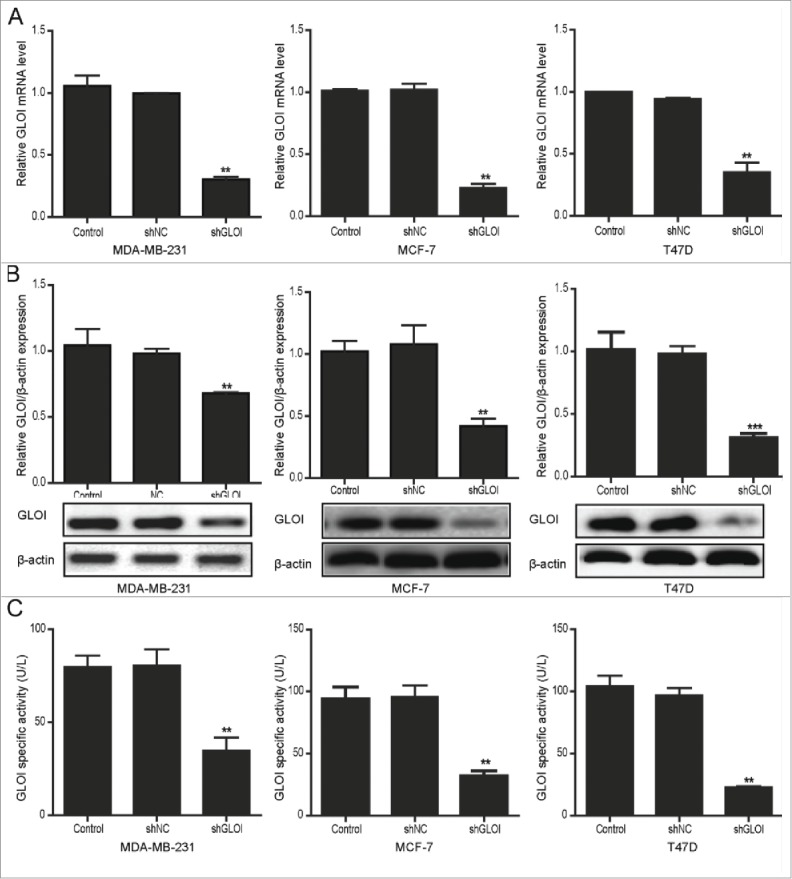
Transfection with shGLOI reduced GLOI mRNA and protein levels and enzyme activity in breast cancer cells. The expression of GLOI mRNA (A) GLOI protein (B) and GLOI enzyme activity (C) was reduced after transfection with shGLOI. **p < 0.01, ***p < 0.001 vs. cells transfected with shNC.
MG and inhibition of GLOI reduced breast cancer cell viability, colony formation, and migration
Cell viability was inhibited in a dose- and time-dependent manner in breast cancer cells (Fig. 2A). Incubation with MG (0.4 or 0.8 mM) or inhibition of GLOI for 12 h (Fig. 2B) or 24 h (Fig. 2C) significantly reduced cell viability (p < 0.05 to 0.01). The combination of MG with inhibition of GLOI augmented these effects.
Figure 2.
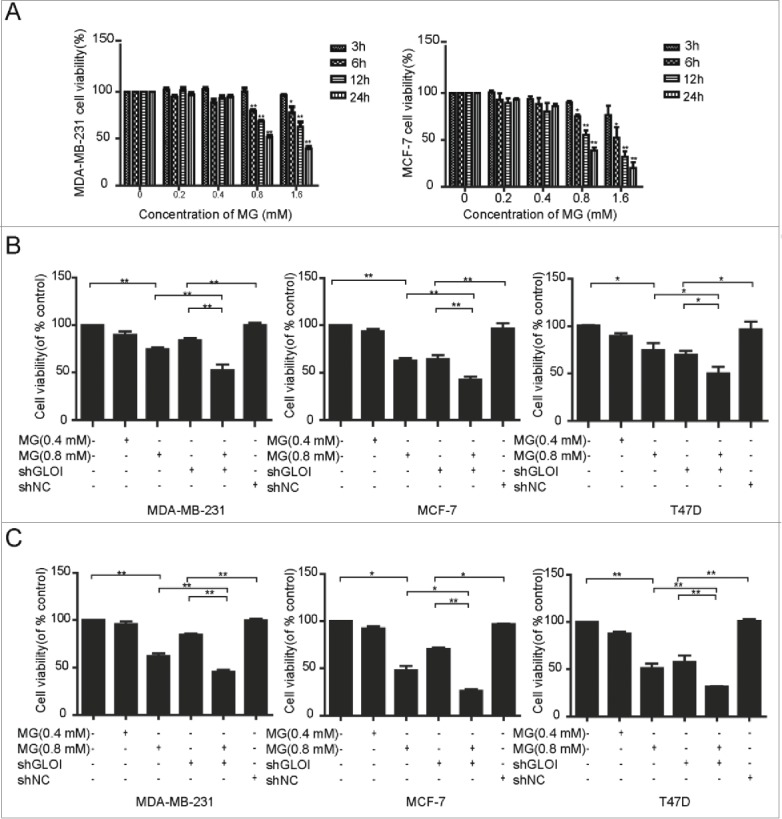
Treatment with MG and/or inhibition of GLOI reduced breast cancer cell viability. The viability of breast cancer cells was inhibited (A) after incubation with MG at various concentrations and different time points, (B) by MG and/or inhibition of GLOI for 12 h, and (C) by MG and/or inhibition of GLOI for 24 h. *p < 0.05, **p < 0.01.
The number of colonies formed by breast cancer cells was reduced significantly by incubation with 0.1 mM MG (p < 0.05 to 0.01 compared to controls) and to an even greater degree by 0.2 mM MG (p < 0.01) (Fig. 3). Inhibition of GLOI by transfection of shGLOI had a significant inhibitory effect on colony formation by breast cancer cells (p < 0.01). Furthermore, the combination of MG and GLOI inhibition showed a much greater growth-suppressive effect than either treatment alone.
Figure 3.
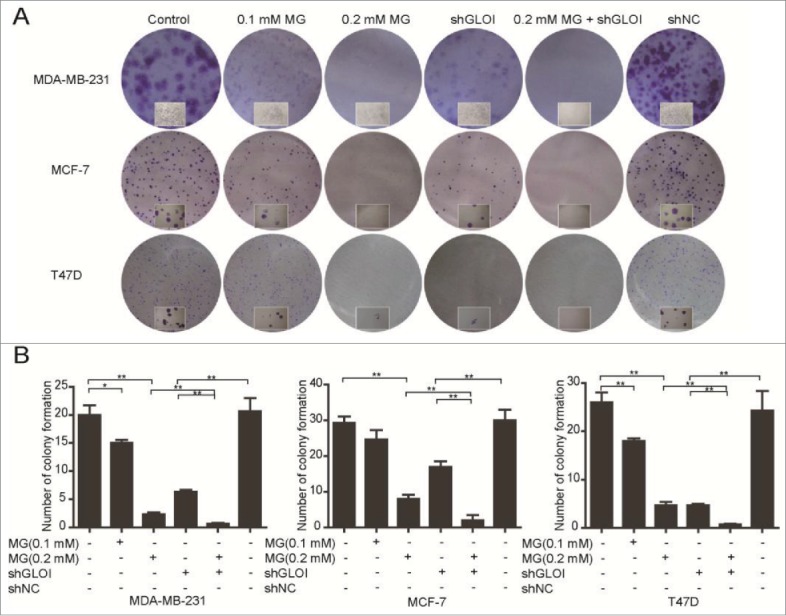
Treatment with MG and/or inhibition of GLOI reduced colony formation by breast cancer cells. (A) Colony formation by breast cancer cells was significantly suppressed by MG and/or inhibition of GLOI. Representative plates are shown. (B) Colonies formed by breast cancer cells were counted under a microscope. *p < 0.05, **p < 0.01.
Treatment with MG (0.4 or 0.8 mM) or inhibition of GLOI significantly reduced the number of breast cancer cells that migrated through a transwell insert membrane compared to the control cells or shNC-transfected cells, respectively. Moreover, co-treatment with MG and inhibition of GLOI decreased breast cancer cell migration to a dramatically greater extent than either treatment alone (p < 0.05 to 0.01) (Fig. 4).
Figure 4.
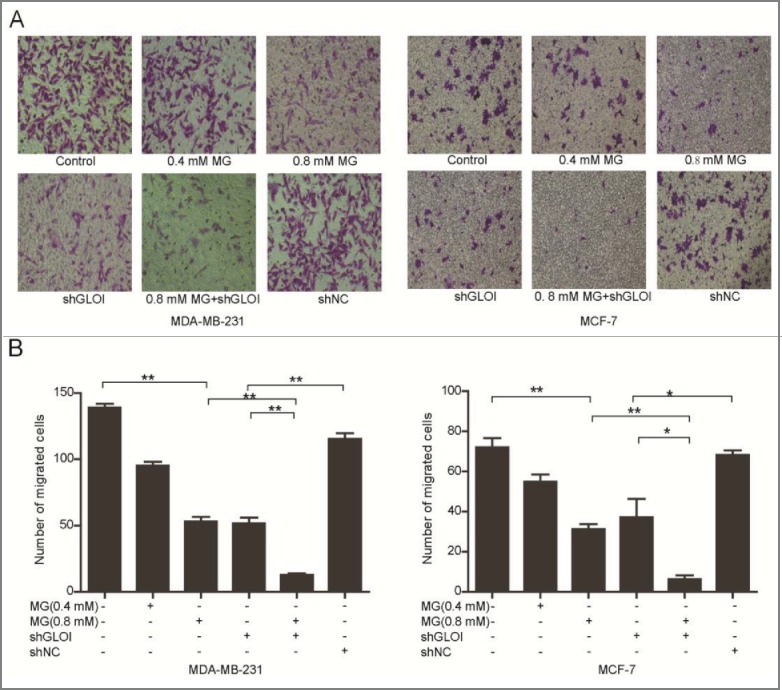
Treatment with MG and/or inhibition of GLOI reduced breast cancer cell migration. (A) Migration of treated breast cancer cells was evaluated by penetration of a transwell insert membrane. Representative membranes are shown. (B) Cells that penetrated the insert membrane were counted under a microscope. *p < 0.05, **p < 0.01.
MG and inhibition of GLOI reduced invasion and MMP-9 protein expression in
MDA-MB-231 cells
Treatment with MG (0.4 or 0.8 mM) or inhibition of GLOI significantly reduced MDA-MB-231 cell invasion compared to the controls or shNC-transfected cells, respectively (p < 0.01; Figs. 5A & B). The combination of MG treatment and inhibition of GLOI greatly increased this effect (p < 0.01; Figs. 5A & B). We also examined the effect of MG and inhibition of GLOI on MMP-9 protein expression in these cells. As shown in Figures 5C and 5D, either treatment alone markedly suppressed MMP-9 protein expression (p < 0.05 to 0.01), and combination of the 2 treatments augmented this inhibitory effect (p < 0.01).
Figure 5.
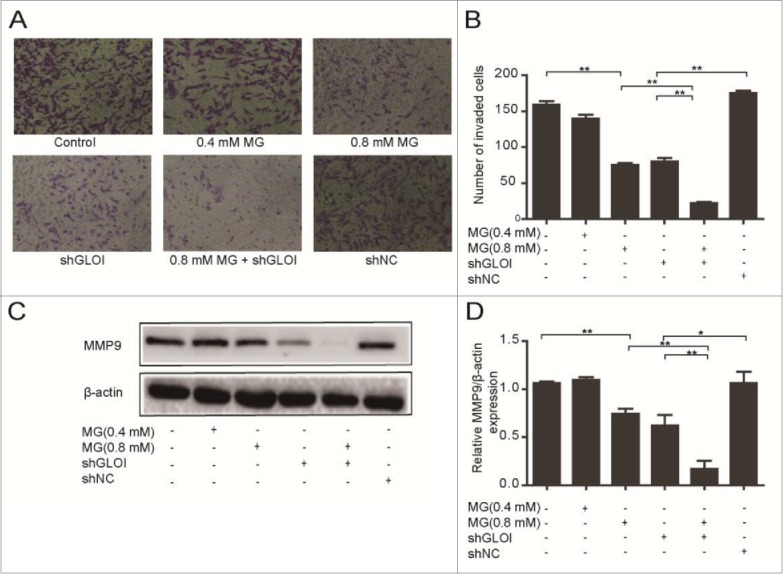
Treatment with MG and/or inhibition of GLOI reduced MDA-MB-231 breast cancer cell invasion and expression of MMP-9 protein. (A) Invasiveness of treated MDA-MB-231 cells was assessed by penetration of a transwell insert membrane. Representative membranes are shown. (B) Cells that penetrated the insert membrane were counted under a microscope. (C) The expression of MMP-9 in MDA-MB-231 cells was attenuated by MG, inhibition of GLOI, or both. (D) MMP-9 protein expression in these cells was determined by densitometric analysis. *p < 0.05, **p < 0.01.
MG and inhibition of GLOI increased apoptosis of MDA-MB-231 cells
Apoptosis was significantly increased after MG treatment or inhibition of GLOI in MDA-MB-231 cells (p < 0.01; Figs. 6A & B). The combination of MG (0.8 mM) with GLOI inhibition increased cell apoptosis to a greater degree than either treatment alone (p < 0.05 to 0.01).
Figure 6.
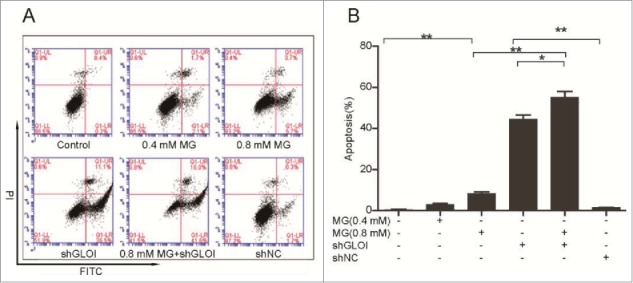
Treatment with MG and/or inhibition of GLOI increased breast cancer cell apoptosis. (A) Apoptosis of treated MDA-MB-231 breast cancer cells was detected by annexin V–propidium iodine (PtdIns) flow cytometry. (B) Quantification of apoptotic cells shows that MG and/or inhibition of GLOI promoted apoptosis. *p < 0.05, **p < 0.01.
MG and inhibition of GLOI reduced tubule formation by breast cancer cells
In MDA-MB-231 cells, tubule formation was reduced markedly after treatment with MG (0.4 or 0.8 mM) or inhibition of GLOI inhibition alone and to an even greater degree in cells subjected to the combination of MG (0.4 mM) and GLOI inhibition (p < 0.01). In MCF-7 cells, tubule formation was significantly inhibited by MG treatment (p < 0.01), but GLOI inhibition had no detectable effect (Figs. 7A & B).
Figure 7.
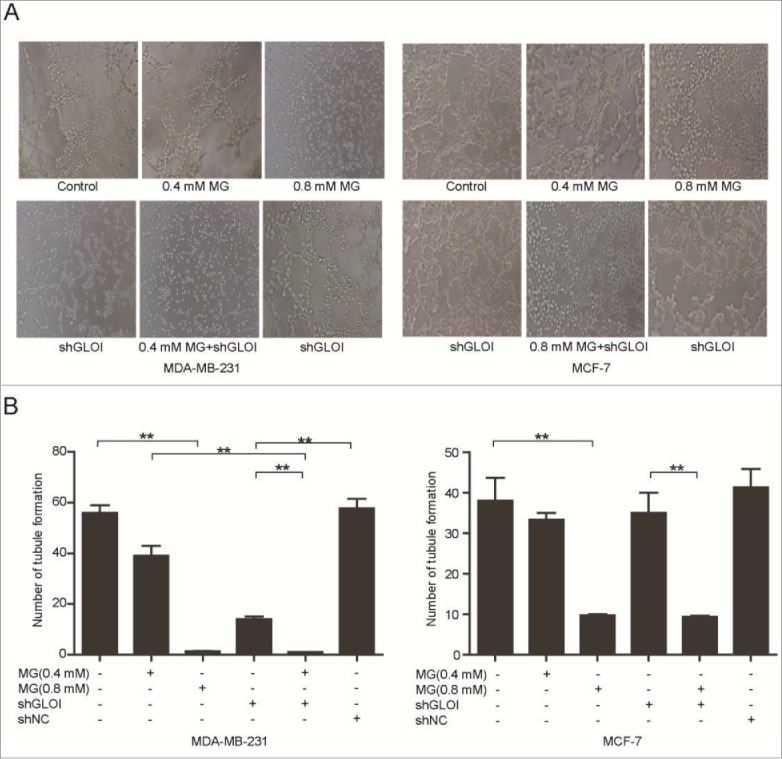
Treatment with MG and/or inhibition of GLOI attenuated tubule formation by breast cancer cells. (A) Tubule formation was reduced in treated breast cancer cells. Representative samples are shown. (B) Tubules formed by breast cancer cells were counted under a microscope. *p < 0.05, **p < 0.01.
MG and inhibition of GLOI increased MAPK phosphorylation in breast cancer cells
Treatment with MG or inhibition of GLOI markedly increased the phosphorylation of JNK (p-JNK; p < 0.05 to 0.01; Fig. 8A) and ERK (p-ERK; p < 0.05 to 0.01; Fig. 8B) in MDA-MB-231 and MCF-7 cells. A similar effect on phosphorylation of p38 (p-p38; p < 0.05 to 0.01) was observed in MCF-7 cells but not in MDA-MB-231 cells (Fig. 8C). Even stronger effects were observed in cells treated with combination of MG and inhibition of GLOI (p < 0.05 to 0.01).
Figure 8.
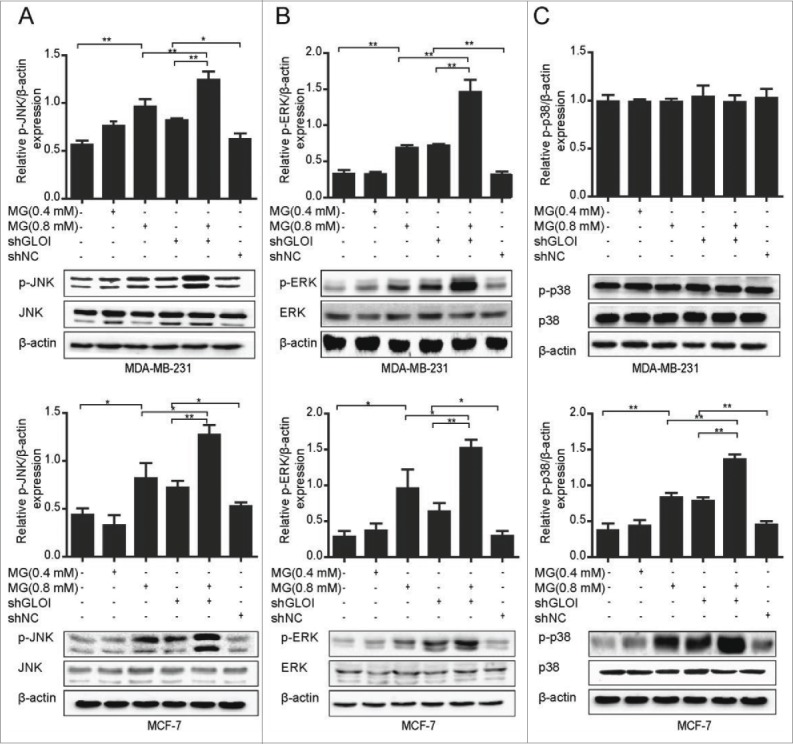
Treatment with MG and/or inhibition of GLOI increased MAPK activation in breast cancer cells. The expression of p-JNK (A), p-ERK (B), and p-p38 (C) in breast cancer cells after MG treatment and/or inhibition of GLOI is shown. *p < 0.05, **p < 0.01.
MG and inhibition of GLOI downregulated expression of Bcl-2 protein in breast cancer cells
As shown in Fig. 9, expression of the Bcl-2 protein was markedly downregulated in MDA-MB-231 cells treated with MG or transfected with shGLOI (p < 0.01). Even more potent Bcl-2 inhibitory effects were observed in MCF-7 cells (p < 0.001). These effects were clearly augmented by combination of MG treatment with GLOI inhibition (p < 0.05).
Figure 9.
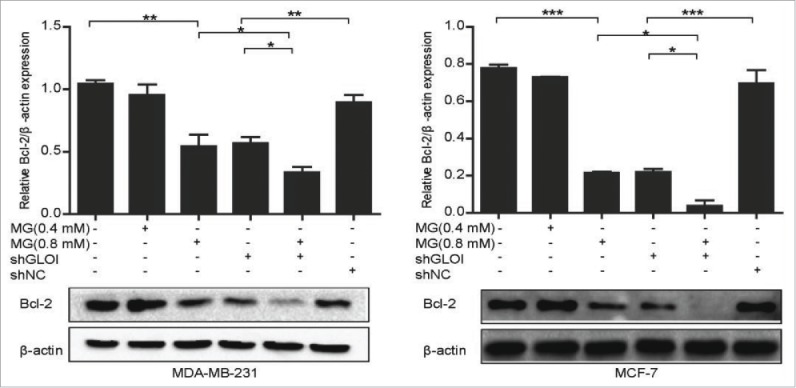
Treatment with MG and/or inhibition of GLOI suppressed Bcl-2 protein expression in breast cancer cells. Bcl-2 protein expression in MDA-MB-231 or MCF-7 cells treated with MG and/or GLOI inhibition is shown. *p < 0.05, ** p < 0.01, ***p < 0.001.
Discussion
Our findings demonstrate that treatment with MG or inhibition of GLOI suppressed proliferation, migration, and invasion, and induced apoptosis, in ER-positive MCF-7 and T47D cells and ER-negative MDA-MB-231 breast cancer cells. These effects appeared to be modulated by the activation of MAPK family kinases and downregulation of Bcl-2 and MMP-9, proteins known to promote cancer development and progression.
These findings indicate that MG and GLOI exert important biological effects on cell activity and colony formation in breast cancer cells. Bair et al. showed that MG can exert anti-proliferative activity in human malignant melanoma.22 MG fully inhibited PC-3 cell growth, leading ultimately to cell death.23 MGBCP formed by MG also resulted in a marked growth inhibition of T-47D cells.24 GLOI is overexpressed in murine fibrosarcoma, hepatocellular carcinoma, and breast cancer cells, while inhibition of GLOI expression significantly suppresses the proliferation of these tumor cells.25,26,27 Successful GLOI inhibition was confirmed by reduced level of GLOI mRNA, protein and enzyme activity in our study. These findings are consistent with our results. In contrast, recent studies suggest that MG did not reduce proliferation of human prostate cancer cells9 and even promoted the proliferation of vascular smooth muscle cells,28 suggesting that the MG effect is cell line or tumor specific; the mechanisms underlying these effects remain an open question.
Our finding of marked increase of apoptosis with downregulation of Bcl-2 after MG treatment or GLOI inhibition in MDA-MB-231 or MCF-7 cells are in line with a previous report that MG increased apoptosis through suppression of Bcl-2 protein expression in Neuro-2A neuroblastoma cells.29 Hiroya et al. showed that GLOI inhibition or MG enhanced TNF-related apoptosis-inducing ligand–induced apoptosis via downregulation of Bcl-2 expression mediated by nuclear factor kappa B (NF-κB) inhibition.30 GLOI inhibition also resulted in a activation of mitochondrial apoptotic pathway via Hsp27, p53 and NF-κB in irradiated MCF-7 cell apoptosis.31 Our finding that MG treatment or GLOI inhibition markedly reduced Bcl-2 expression suggested that their antitumor effects in MDA-MB-231 and MCF-7 breast cancer cells were partially mediated by apoptosis through downregulation of the Bcl-2 pathway.
The marked reduction of MDA-MB-231 cell invasion by MG administration or downregulation of GLOI was associated with downregulation of MMP-9, an important indicator of breast cancer prognosis that is significantly associated with higher grade breast tumors (grades II and III).32,33 Direct targeting of MMP-9 expression in invasive breast cancer cells has been shown to significantly inhibit cell invasion and metastasis in vitro and in vivo.16,34 These findings suggest that both MG and GLOI inhibition play suppressive roles against breast cancer cell migration and invasion. This is supported by published reports of similar roles of MG and GLOI in the migration and metastasis of gastric cancer35 and liver cancer cells.36
Tumoral angiogenesis is considered an important step in the metastatic cascade of tumors and tumor growth, supplying nutrients and oxygen and disposing of catabolic products.37 Thus, inhibition of angiogenesis by tumor cells is associated with a significant delay in tumor growth and is a promising therapeutic strategy for prevention of cancer metastasis. Tubule formation was suppressed by MG treatment or GLOI inhibition in MDA-MB-231 cells but only by MG treatment and not by GLOI inhibition in MCF-7 cells. We speculate that the ER or other unknown biological properties of these cells could be responsible for the difference. However, the underlying mechanisms of these effects are not fully understood and further investigation is warranted.
The MAPKs, a group of evolutionarily conserved Ser/Thr protein kinases, play a dominant role in various cellular activities, such as mitosis, differentiation, proliferation, and cell survival/apoptosis.38,39 Pal et al. showed that MG activated inducible nitric oxide synthase by a p38/NF-κB–induced pathway and contributed to reactive oxygen species generation in macrophages by ERK and JNK activation in a sarcoma-180 mouse model.40 Furthermore, p38 activation was suggested to be a key signaling intermediate of MG-induced apoptosis in kidney cells41 and Schwann cells,42 while the JNK pathway appears to be important for MG-induced apoptosis in human osteoblasts.8 These findings are consistent with our results demonstrating the activation of MAPKs following MG treatment and GLOI inhibition in breast cancer cells, though p38 was not affected in ER-negative MDA-MB-231 cells. We speculated that the activation of p38 may be related to cell characteristics, such as the expression of ER, degree of malignancy, and susceptibility to MG and GLOI inhibition of the 2 cell lines, which need to be further studied. Our results suggest that MAPK pathway activation could be involved in the anti-proliferative and pro-apoptotic effects of MG and silencing of GLOI in breast cancer cells. Nevertheless, the exact molecular mechanisms as how these pathways are regulated and their relationships with each other need further investigation.
Not surprisingly, all these effects were intensified by combination of MG with inhibition of GLOI. Downregulation of GLOI in tumor cells led to marked accumulation of MG and increased its cytotoxicity, enhancing its antitumor effect.5,6,9 For this study, we selected 3 breast cancer cell lines representing different expression of ER and different degrees of malignancy. Our findings in these cells are preliminary. Further studies in animal models are needed to better define these anticancer effects and their molecular mechanisms.
In summary, our data clearly demonstrate that MG treatment or GLOI inhibition can restrain proliferation, migration, and invasion and induce apoptosis in human breast cancer cells. These effects may be modulated by activation of the MAPK signaling pathway and downregulation of Bcl-2 and MMP-9. Our findings suggest a promising therapeutic approach against breast cancer.
Materials and Methods
Cell culture and reagents
ER-positive human breast cancer cell lines MCF-7 and T47D were cultured in RPMI-1640 medium (Gibco, 11875093). ER-negative human breast cancer cell line MDA-MB-231 was cultured in high-glucose Dulbecco modified Eagle medium (DMEM) (Gibco, 11995065). All media were supplemented with 10% fetal calf serum (FCS) (Bioind, 040011A) and 100 U/mL penicillin-streptomycin (Solarbio, P1400). Cells were cultured at 37°C in a 5% CO2 atmosphere. All three cell types were used in all experiments unless otherwise noted.
MG was purchased from Sigma (M0252). The following reagents were used in this study: Annexin V-FITC Apoptosis Detection Kit (KeyGEN Biotech, KGA107); p-JNK antibody (9251), JNK antibody (9252), p-p38 antibody (4511), p-ERK antibody (4370), Bcl-2 antibody (2870S), and MMP-9 antibody (13667; all, Cell Signaling Technology); p38 antibody (Abcam, 32142); ERK antibody (Bioss, bs0022r); GLOI antibody (Epitomics, 67781); β-actin antibody (Beyotime, AA128); goat anti-rabbit IgG (Bioworld, BS13271); and the Quantichrom Glyoxalase I Assay Kit (Hayward, DGLO-100).
Cell transfection
The shRNA sequences targeting GLOI (shGLOI) and empty vector (shNC) were synthesized by GenePharma. The sequences of shGLOI were GGATTCGGTCATATTGGAATT; GGGAGTCAAATTTGTGAAGAA; GGCA
TTTATTCAAGATCCTGA; and GAAGAACTGGGAGTCAAATTT. MDA-MB-
231, MCF-7, and T47D cells were grown to 80%-90% confluence and transfected with plasmids using Lipofectamine 2000 (DNA/Lipofectamine 2000 ratio, 1:2) (Invitrogen, 11668-019) according to the manufacturer's instructions. After transfection for 24 h, cells were examined under a fluorescence microscope for green fluorescence to determine transfection efficiency. Transfected cells with transfection efficiency ≥ 50% were grown in culture medium containing 1000 μg/mL G418 (Sigma, 11811031) for 15 days to build stable cell lines. The clones were verified by western blot and real-time quantitative polymerase chain reaction (RT-PCR). The stably transfected clones were pooled and used for further investigations.
Quantification of GLOI mRNA
Total RNA was extracted from breast cancer cells transfected with shGLOI or shNC by using TRIzol (Invitrogen, 3101-100) according to the manufacturer's instructions. Total RNA (500 ng) was reverse-transcribed to cDNA with random primer (TaKaRa, RRO37A). Gene expression was measured by RT-PCR using an Applied Biosystems 7500 Fast Sequence Detection System and SYBR Green PCR Kit (QIAGEN, 20454) under the following conditions: denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 10 sec and annealing and extension at 60°C for 30 sec. The GLOI mRNA expression was normalized to that of β-actin.
Quantification of GLOI enzymatic activity
Cells transfected with shGLOI or shNC were harvested by trypsinization and collected by centrifugation at 600 g for 5 min. After two washes with phosphate-buffered saline solution (PBS), cells were resuspended in 0.1 M K-phosphate buffer (pH 7.0) containing protease inhibitors. Cell suspensions were then frozen-thawed 3 times in liquid nitrogen. The cells were subjected to sonication (100 W, 20 s at 4°C) and centrifugation at 12,000g for 15 min at 4°C. The resulting cell extracts were assayed for GLOI activity by using the Quantichrom Glyoxalase I Assay Kit.
Cell viability assay
Cell viability was assayed by using the CCK-8 Kit (Dojindo, CK04) as we described previously.43 In brief, cells were seeded in 96-well plates at a density of 1×104 cells/well and incubated for 24 h. Cells with or without GLOI knockdown were then treated with various concentrations of MG or control vehicle for 12 h or 24 h. CCK-8 assay reagent (10 μL) was then added to each well and the plates incubated at 37°C for 1 h. Absorbance was measured at 450 nm using an electroluminescence immunosorbent assay reader (Thermo Scientific).
Colony formation assay
The plate colony formation assay was performed to detect cell growth.44 Cells were seeded at 1×103 cells/well in 6-well plates and treated with MG and/or inhibition of GLOI. After 15 days, colonies were visible with the unaided eye. The colonies were fixed with 4% paraformaldehyde for 15 min and stained with crystal violet (Beyotime, C0121) for 15 min at ambient temperature. After washing with PBS, the colonies from 5 randomly selected fields were counted under a microscope at ×100 magnification.
Cell migration assay
Cells were seeded in 6-well plates and incubated with 0.4 or 0.8 mM MG and/or with inhibition of GLOI for 24 h. Cells were collected and washed twice with PBS. Cells were then resuspended with serum-free DMEM or 1640 medium and seeded (MDA-MB-231, 2×104 cells; MCF-7, 1×105 cells) into 24-well transwell plates with an 8-μm pore membrane insert (Corning, 3422) in each transwell. DMEM supplemented with 10% FCS or 1640 medium supplemented with 20% FCS was placed in the lower chamber as a chemoattractant. MDA-MB-231 cells and MCF-7 cells were incubated for 24 h and 48 h, respectively. Cells that penetrated the membrane were fixed with 4% paraformaldehyde for 20 min and stained with crystal violet for 20 min at ambient temperature. The cells attached to the lower surface in 5 randomly chosen fields were counted under a microscope at ×100 magnification.
Cell invasion assay
Because of the low malignancy and invasion capacity of MCF-7 and T47D, only MDA-MB-231 cells were chosen for this experiment. The cell invasion assay was similar to the migration assay except that the transwells were coated with matrigel solution (50 μL per transwell; matrigel:serum-free medium ratio 1:10). After 24 h, MDA-MB-231 cells (4×104 cells/well) were seeded into the upper chambers of the transwells and incubated with MG or inhibition of GLOI, whereas DMEM (600 μL) with 10% FCS was added to the lower chamber. After 24 h, the cells that had penetrated the matrigel and moved to the lower surface of the membrane were fixed with 4% paraformaldehyde and stained with crystal violet. Cells adhering to the upper surface of the membrane were removed with a cotton swab. The cells attached to the lower surface in 5 randomly chosen fields were counted under a microscope at ×100 magnification.
Apoptosis assay
Because of the biological characteristics of cell adhesion, MCF-7 and T47D cells are prone to breakage in the process of digestion and blowing, resulting in serious mechanical damage, only MDA-MB-231 cells were chosen for this experiment. MDA-MB-231 cells were seeded in 12-well plates (5×105 cells/well) and treated with various concentrations of MG (0.4 mM or 0.8 mM) and/or inhibition of GLOI for 24 h. Cells were collected and washed twice with PBS. The cell suspensions were stained with annexin V-enhanced green fluorescent protein (FITC) and propidium iodide (PtdIns) from the Annexin V-FITC Apoptosis Detection Kit for 15 min at room temperature in the dark. Apoptotic cells double-stained with annexin V and PI were isolated immediately by flow cytometry. The percentage of apoptotic cells was determined.
Tubule formation assay
Undiluted matrigel (50 μL) was layered in 96-well plates for 40 min. MDA-MB-231 and MCF-7 cells were added to the precoated 96-well plates at a density of 1×105 cells/well and treated with MG and/or Inhibition of GLOI. The plate was then incubated at 37°C in a humidified CO2 incubator for 6 h. After incubation, the tubules were photographed under an inverted light microscope (Nikon). The tubules formed in 5 randomly selected fields were counted under a microscope at ×100 magnification.
Western blot analysis
Treated cells were harvested and subjected to lysis in the presence of a protease inhibitor cocktail and then subjected to centrifugation at 12,000 g for 15 min at 4°C. The supernatant fraction was collected and the protein concentration was measured using a bicinchoninic acid protein assay kit (Beyotime, P0010). An aliquot of 70 μg of denatured protein from each sample was applied to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Parafilm, PM-996). After blocking with 5% nonfat milk, membranes were incubated at 4°C overnight with the following primary antibodies (1:1000 dilution): anti-GLOI antibody, anti-p-JNK antibody, anti-JNK antibody, anti-p-p38 antibody, anti-p38 antibody, anti-p-ERK antibody, anti-ERK antibody, anti-Bcl-2 antibody, anti-MMP-9 antibody, and anti-β-actin antibody. Membranes were washed with Tris-buffered saline/0.05% Tween-20 solution (TBST) and incubated with horseradish peroxidase–conjugated secondary antibody (1:2500 dilution) for 1 h at ambient temperature. After washing with TBST for 1 h, the membranes were incubated with enhanced chemiluminescence solution for 2 min. The signals were detected and quantified by densitometry using Quantity One software. β-actin was used as an internal control.
Statistical analyses
Statistical analysis was performed with SPSS 17.0 software. All results are expressed as mean ± standard deviation. Each experiment was repeated independently at least 3 times. Differences between groups were analyzed by using analysis of variance (ANOVA) and 2-tailed Student t-test. A p-value < 0.05 was considered statistically significant.]
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported in part by grants from the National Natural Science Foundation of China (81170257).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5-29; PMID:25559415; http://dx.doi.org/ 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2. Hong W, Dong E. The past, present and future of breast cancer research in China. Cancer Lett 2014; 351:1-5; PMID:24735750; http://dx.doi.org/ 10.1016/j.canlet.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 3. Linos E, Spanos D, Rosner BA, Linos K, Hesketh T, Qu JD, Gao YT, Zheng W, Colditz G. Effects of reproductive and demographic changes on breast cancer incidence in China: a modeling analysis. J Natl Cancer Inst 2008; 100:1352-60; PMID:18812552; http://dx.doi.org/ 10.1093/jnci/djn305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maughan KL, Lutterbie MA, Ham PS. Treatment of breast cancer. Am Fam Physician 2010; 81:1339-46; PMID:20521754 [PubMed] [Google Scholar]
- 5. Lee DY, Chang GD. Methylglyoxal in cells elicits a negative feedback loop entailing transglutaminase 2 and glyoxalase 1. Redox Biol 2014; 2:196-205; PMID:24494193; http://dx.doi.org/ 10.1016/j.redox.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antognelli C, Mezzasoma L, Fettucciari K, Mearini E, Talesa VN. Role of glyoxalase I in the proliferation and apoptosis control of human LNCaP and PC3 prostate cancer cells. Prostate 2013; 73:121-32; PMID:22653787; http://dx.doi.org/ 10.1002/pros.22547 [DOI] [PubMed] [Google Scholar]
- 7. Godbout JP, Pesavento J, Hartman ME, Manson SR, Freund GG. Methylglyoxal enhances cisplatin-induced cytotoxicity by activating protein kinase Cdelta. J Biol Chem 2002; 277:2554-61; PMID:11707430; http://dx.doi.org/ 10.1074/jbc.M100385200 [DOI] [PubMed] [Google Scholar]
- 8. Chan WH, Wu HJ, Shiao NH. Apoptotic signaling in methylglyoxal-treated human osteoblasts involves oxidative stress, c-Jun N-terminal kinase, caspase-3, and p21-activated kinase 2. J Cell Biochem 2007; 100:1056-69; PMID:17131386; http://dx.doi.org/ 10.1002/jcb.21114 [DOI] [PubMed] [Google Scholar]
- 9. Antognelli C, Mezzasoma L, Fettucciari K, Talesa VN. A novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int J Biochem Cell Biol 2013; 45:836-44; PMID:23333621; http://dx.doi.org/ 10.1016/j.biocel.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 10. Talukdar D, Chaudhuri BS, Ray M, Ray S. Critical evaluation of toxic versus beneficial effects of methylglyoxal. Biochemistry (Mosc) 2009; 74:1059-69; PMID:19916918; http://dx.doi.org/ 10.1016/10.1134/S0006297909100010 [DOI] [PubMed] [Google Scholar]
- 11. Baunacke M, Horn LC, Trettner S, Engel KM, Hemdan NY, Wiechmann V, Stolzenburg JU, Bigl M, Birkenmeier G. Exploring glyoxalase 1 expression in prostate cancer tissues: targeting the enzyme by ethyl pyruvate defangs some malignancy-associated properties. Prostate 2014; 74:48-60; PMID:24105621; http://dx.doi.org/ 10.1002/pros.22728 [DOI] [PubMed] [Google Scholar]
- 12. Rulli A, Carli L, Romani R, Baroni T, Giovannini E, Rosi G, Talesa V. Expression of glyoxalase I and II in normal and breast cancer tissues. Breast Cancer Res Treat 2001; 66:67-72; PMID:11368412; http://dx.doi. org/ 10.1023/A:1010632919129 [DOI] [PubMed] [Google Scholar]
- 13. Fonseca-Sánchez MA, Rodríguez Cuevas S, Mendoza-Hernández G, Bautista-Piña V, Arechaga Ocampo E, Hidalgo Miranda A, Quintanar Jurado V, Marchat LA, Alvarez-Sánchez E, Pérez Plasencia C, et al. Breast cancer proteomics reveals a positive correlation between glyoxalase 1 expression and high tumor grade. Int J Oncol 2012; 41:670-80; PMID:22614840; http://dx.doi.org/ 10.3892/ijo.2012.1478 [DOI] [PubMed] [Google Scholar]
- 14. Dong L, Zhou Q, Zhang Z, Zhu Y, Duan T, Feng Y. Metformin sensitizes endometrial cancer cells to chemotherapy by repressing glyoxalase I expression. J Obstet Gynaecol Res 2012; 38:1077-85; PMID:22540333; http://dx.doi.org/ 10.1111/j.1447-0756.2011.01839.x [DOI] [PubMed] [Google Scholar]
- 15. Akter H, Park M, Kwon OS, Song EJ, Park WS, Kang MJ. Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumour Biol 2015; 36(8):6053-62; PMID:25724188; http://dx.doi.org/ 10.1007/s13277-015-3282-9 [DOI] [PubMed] [Google Scholar]
- 16. Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014; 5:2736-49; PMID:24811362; http://dx.doi.org/ 10.18632/oncotarget.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review). Int J Oncol 2003; 22:15-20; PMID:12469180; http://dx.doi.org/ 10.3892/ijo.22.1.15 [DOI] [PubMed] [Google Scholar]
- 18. Osford SM, Dallman CL, Johnson PW, Ganesan A, Packham G. Current strategies to target the anti-apoptotic Bcl-2 protein in cancer cells. Curr Med Chem 2004; 11:1031-39; PMID:15083809; http://dx.doi.org/ 10.2174/0929867043455486 [DOI] [PubMed] [Google Scholar]
- 19. Yim NH, Kim A, Liang C, Cho WK, Ma JY. Guibitang, a traditional herbal medicine, induces apoptotic death in A431 cells by regulating the activities of mitogen-activated protein kinases. BMC Complement Altern Med 2014; 14:344; PMID:25241226; http://dx.doi.org/ 10.1186/1472-6882-14-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dent P, Grant S. Pharmacologic interruption of the mitogen-activated extracellular-regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin Cancer Res 2001; 7:775-83; PMID:11309321 [PubMed] [Google Scholar]
- 21. Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat 2001; 4:253-67; PMID:11991680; http://dx.doi.org/ 10.1054/drup.2001.0214 [DOI] [PubMed] [Google Scholar]
- 22. Bair WB, 3rd, Cabello CM, Uchida K, Bause AS, Wondrak GT. GLO1 overexpression in human malignant melanoma. Melanoma Res 2010; 20:85-96; PMID:20093988; http://dx.doi.org/ 10.1097/CMR.0b013e3283364903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milanesa DM, Choudhury MS, Mallouh C, Tazaki H, Konno S. Methylglyoxal-induced apoptosis in human prostate carcinoma: potential modality for prostate cancer treatment. Eur Urol 2000; 37:728-34; PMID:10828676; http://dx.doi.org/20226 [DOI] [PubMed] [Google Scholar]
- 24. Kaneko H, Hibasami H, Satoh N, Wakabayashi H, Ikeda H, Tsuge N, Yonemaru K, Muraki A, Kawarada Y, Nakashima K. Involvement of apoptosis and cyclin D1 gene repression in growth inhibition of T-47D human breast cancer cells by methylglyoxal bis(cyclopentylamidinohydrazone). Int J Mol Med 1998; 1:931-7; PMID:9852627; http://dx.doi.org/ 10.3892/ijmm.1.6.931 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Kuramitsu Y, Tokuda K, Okada F, Baron B, Akada J, Kitagawa T, Nakamura K. Proteomic analysis indicates that overexpression and nuclear translocation of lactoylglutathione lyase (GLO1) is associated with tumor progression in murine fibrosarcoma. Electrophoresis 2014; 35:2195-202; PMID:24532130; http://dx.doi.org/ 10.1002/elps.201300497 [DOI] [PubMed] [Google Scholar]
- 26. Hu X, Yang X, He Q, Chen Q, Yu L. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol Lett 2014; 36:257-63; PMID:24158671; http://dx.doi.org/ 10.1007/s10529-013-1372-6 [DOI] [PubMed] [Google Scholar]
- 27. Rulli A, Antognelli C, Prezzi E, Baldracchini F, Piva F, Giovannini E, Talesa V. A possible regulatory role of 17 beta-estradiol and tamoxifen on glyoxalase I and glyoxalase II genes expression in MCF7 and BT20 human breast cancer cells. Breast Cancer Res Treat 2006; 96:187-96. PMID:16319983; http://dx.doi.org/ 10.1007/s10549-005-9078-7 [DOI] [PubMed] [Google Scholar]
- 28. Chang T, Wang R, Olson DJ, Mousseau DD, Ross AR, Wu L. Modification of Akt1 by methylglyoxal promotes the proliferation of vascular smooth muscle cells. FASEB J 2011; 25:1746-57; PMID:21321187; http://dx.doi.org/ 10.1096/fj.10-178053 [DOI] [PubMed] [Google Scholar]
- 29. Huang SM, Chuang HC, Wu CH, Yen GC. Cytoprotective effects of phenolic acids on methylglyoxal-induced apoptosis in Neuro-2A cells. Mol Nutr Food Res 2008; 52:940-49; PMID:18481334; http://dx.doi.org/ 10.1002/mnfr.200700360 [DOI] [PubMed] [Google Scholar]
- 30. Taniguchi H, Horinaka M, Yoshida T, Yano K, Goda AE, Yasuda S, Wakada M, Sakai T. Targeting the glyoxalase pathway enhances TRAIL efficacy in cancer cells by downregulating the expression of antiapoptotic molecules. Mol Cancer Ther 2012; 11:2294-300; PMID:22784708; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0031 [DOI] [PubMed] [Google Scholar]
- 31. Antognelli C1, Palumbo I2, Aristei C2, Talesa VN1. Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-κB. Br J Cancer 2014; 111:395-406; PMID:24918814; http://dx.doi.org/ 10.1038/bjc.2014.280 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma VM. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin Cancer Res 2004; 10:7621-28; PMID:15569994; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-1061 [DOI] [PubMed] [Google Scholar]
- 33. Kohrmann A, Kammerer U, Kapp M, Dietl J, Anacker J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: new findings and review of the literature. BMC Cancer 2009; 9:188; PMID:19531263; http://dx.doi.org/ 10.1186/1471-2407-9-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hallett MA, Teng B, Hasegawa H, Schwab LP, Seagroves TN, Pourmotabbed T. Anti-matrix metalloproteinase-9 DNAzyme decreases tumor growth in the MMTV-PyMT mouse model of breast cancer. Breast Cancer Res 2013; 15:R12; PMID:23407024; http://dx.doi.org/ 10.1186/bcr3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng WL, Tsai MM, Tsai CY, Huang YH, Chen CY, Chi HC, Tseng YH, Chao IW, Lin WC, Wu SM, et al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PloS One 2012; 7:e34352; PMID:22479608; http://dx.doi.org/ 10.1371/journal.pone.0034352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loarca L, Sassi-Gaha S, Artlett CM. Two alpha-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a p53-dependent manner. Dig Liver Dis 2013; 45:938-46; PMID:24071451; http://dx.doi.org/ 10.1016/j.dld.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 37. Aozuka Y, Koizumi K, Saitoh Y, Ueda Y, Sakurai H, Saiki I. Anti-tumor angiogenesis effect of aminopeptidase inhibitor bestatin against B16-BL6 melanoma cells orthotopically implanted into syngeneic mice. Cancer Lett 2004; 216:35-42; PMID:15500947; http://dx.doi.org/ 10.1016/j.canlet.2004.06.050 [DOI] [PubMed] [Google Scholar]
- 38. Choi MS, Park HJ, Oh JH, Lee EH, Park SM, Yoon S. Nonylphenol-induced apoptotic cell death in mouse TM4 Sertoli cells via the generation of reactive oxygen species and activation of the ERK signaling pathway. J Appl Toxicol 2014; 34:628-36; PMID:23677851; http://dx.doi.org/ 10.1002/jat.2886 [DOI] [PubMed] [Google Scholar]
- 39. Chye SM, Tiong YL, Yip WK, Koh RY, Len YW, Seow HF, Ng KY, Ranjit A, Chen SC. Apoptosis induced by para-phenylenediamine involves formation of ROS and activation of p38 and JNK in chang liver cells. Environ Toxicol 2014; 29:981-90; PMID:23172806; http://dx.doi.org/ 10.1002/tox.21828 [DOI] [PubMed] [Google Scholar]
- 40. Pal A, Bhattacharya I, Bhattacharya K, Mandal C, Ray M. Methylglyoxal induced activation of murine peritoneal macrophages and surface markers of T lymphocytes in sarcoma-180 bearing mice: involvement of MAP kinase, NF-kappa beta signal transduction pathway. Mol Immunol 2009; 46:2039-44; PMID:19375802; http://dx.doi.org/ 10.1016/j.molimm.2009.03.014 [DOI] [PubMed] [Google Scholar]
- 41. Liu BF, Miyata S, Hirota Y, Higo S, Miyazaki H, Fukunaga M, Hamada Y, Ueyama S, Muramoto O, Uriuhara A, et al. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int 2003; 63:947-57; PMID:12631075; http://dx.doi.org/ 10.1046/j.1523-1755.2003.00829.x [DOI] [PubMed] [Google Scholar]
- 42. Fukunaga M, Miyata S, Higo S, Hamada Y, Ueyama S, Kasuga M. Methylglyoxal induces apoptosis through oxidative stress-mediated activation of p38 mitogen-activated protein kinase in rat Schwann cells. Ann N Y Acad Sci 2005; 1043:151-57; PMID:16037234; http://dx.doi.org/ 10.1196/annals.1333.019 [DOI] [PubMed] [Google Scholar]
- 43. Gan R, Yang Y, Yang X, Zhao L, Lu J, Meng QH. Downregulation of miR-221/222 enhances sensitivity of breast cancer cells to tamoxifen through upregulation of TIMP3. Cancer Gene Ther 2014; 21:290-96; PMID:24924200; http://dx.doi.org/ 10.1038/cgt.2014.29; [DOI] [PubMed] [Google Scholar]
- 44. Ma J, Guo Y, Chen S, Zhong C, Xue Y, Zhang Y, Lai X, Wei Y, Yu S, Zhang J, Liu W. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC Cancer 2014; 14:172; PMID:24612549; http://dx.doi.org/ 10.1186/1471-2407-14-172 [DOI] [PMC free article] [PubMed] [Google Scholar]