

Abstract
Temporal lobe epilepsy (TLE) is the most common form of acquired epilepsy that can be caused by several inciting events including viral infections. However, one-third of TLE patients are pharmacoresistant to current antiepileptic drugs and therefore, there is an urgent need to develop antiepileptogenic therapies that prevent the development of the disease. Oxidative stress and redox alterations have recently been recognized as important etiological factors contributing to seizure-induced neuronal damage. The goal of this study was to determine if oxidative stress occurs in the TMEV (Theiler’s murine encephalomyelitis virus) model of temporal lobe epilepsy (TLE). C57Bl/6 mice were injected with TMEV or with PBS intracortically and observed for acute seizures. At various time points after TMEV injection, hippocampi were analyzed for levels of reduced glutathione (GSH), oxidized glutathione (GSSG) and 3-nitrotyrosine (3NT). Mice infected with TMEV displayed behavioral seizures between days 3 and 7 days post-infection (dpi). The intensity of seizures increased over time with most of the seizures being a stage 4 or 5 on the Racine scale at 6 days p.i. Mice exhibiting at least one seizure during the observation period were utilized for the biochemical analyses. The levels of GSH were significantly depleted in TMEV infected mice at 3, 4 and 14 days p.i. with a concomitant increase in GSSH levels as well as an impairment of the redox status. Additionally, there was a substantial increase in 3NT levels in TMEV infected mice at these time points. These redox changes correlated with the occurrence of acute seizures in this model. Interestingly, we did not see changes in any of the indices in the cerebellum of TMEV-infected mice at 3 dpi indicating that these alterations are localized to the hippocampus and perhaps other limbic regions. This is the first study to demonstrate the occurrence of oxidative stress in the TMEV model of infection-induced TLE. The redox alterations were observed at time points coinciding with the appearance of acute behavioral seizures suggesting that these changes might be a consequence of seizure activity. Our results support the hypothesis that redox changes correlate with seizure activity in acquired epilepsies, regardless of the inciting insults, and suggest oxidative stress as a potential therapeutic target for their treatment.
Keywords: Theiler’s murine encephalomyelitis virus, Temporal lobe epilepsy, Oxidative stress, Nitrosative stress, Redox status, Seizures
Graphical Abstract
Introduction
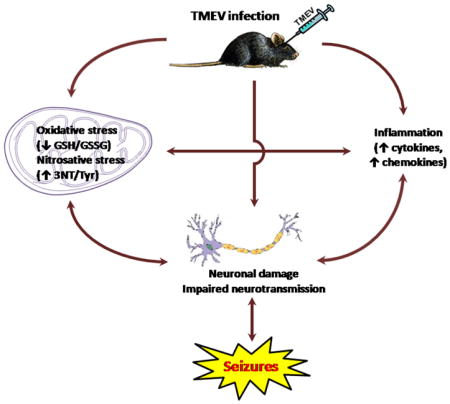
Temporal lobe epilepsy or TLE, the most common form of acquired epilepsy is initiated by a variety of insults including traumatic brain injury, stroke, status epilepticus and infections, which can cause early seizures, and following a latent period, lead to the development of spontaneous seizures or epilepsy. The cascade of biochemical, molecular and structural alterations following a precipitating injury and culminating in the development of epilepsy, i.e. epileptogenesis, is thought to involve processes such as neuronal loss, gliosis, axonal sprouting, neurogenesis and inflammation (Dudek & Staley, 2011; Sharma et al., 2007). A recent study indicates that patients that exhibit seizures during viral encephalitis are 22 times more likely to develop epilepsy than the control population (Misra et al., 2008). Thus patients with encephalitis are at high risk for developing epilepsy. A novel mouse model of infection-induced TLE which recapitulates clinical observations has been recently developed which offers a unique opportunity to study the molecular mechanism(s) underlying epileptogenesis and to identify novel therapeutic strategies (Libbey et al., 2008). Theiler’s murine encephalomyelitis virus (TMEV) infected C57BL/6J mice show acute behavioral seizures between 3 and 7 days post-infection (dpi), exhibit neurodegeneration and glial activation in the hippocampus. A significant proportion of mice surviving the infection develop epilepsy after a latent period (Kirkman et al., 2010; Stewart et al., 2010a,b). In addition, the brains of TMEV infected mice show increased expression of mRNA for proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), during the acute seizure period (Kirkman et al., 2010). Given that TNF-α receptor 1 and IL-6 knockout mice have a significantly reduced incidence of seizures during the acute TMEV infection period, inflammation seems to play an important role in the induction of seizures in this animal model (Kirkman et al., 2010).
Oxidative stress is an important mechanism known to occur following brain injuries, sufficient to cause epilepsy (Liang et al., 2000). Endogenous antioxidants can overcome normal production of reactive oxygen and nitrogen species (ROS and RNS). However, their excessive production can overwhelm the natural antioxidant defenses and shift the redox state to a more oxidized environment which can lead to oxidative damage of various cellular targets. In fact, both mitochondrial and extracellular ROS play a role in mediating seizure-induced neuronal death (Liang et al., 2000; Patel et al., 2005). Additionally, oxidative stress has been shown to occur throughout epilepsy development in chemoconvulsant models of TLE (Waldbaum & Patel 2010; Patel 2004; Liang & Patel 2006). Whether oxidative stress is a common mechanism underlying diverse epileptogenic injuries is unclear. We hypothesized that oxidative stress occurs in the Theiler’s virus infection model of TLE for the following reasons. (1) Viral infections often cause increased formation of ROS and RNS either due to direct effects of the virus on the cells or as a consequence of host inflammatory responses to the infections (Schwarz, 1996; Valyi-Nagy et al., 2000). In response to viral infections, increased levels of cytokines, chemokines and other inflammatory mediators can directly damage mitochondria, resulting in oxidative stress. Herpes simplex virus (HSV) and Japanese encephalitis virus (JEV) are among the most common viruses which cause encephalitis and are both associated with acute seizures in patients (Theodore, 2014). Acute and chronic HSV-1 infection in mice results in inflammation and oxidative damage to the neurons and non-neuronal cells in the brain (Valyi-Nagy & Dermody, 2005). JEV infection has also been shown to stimulate the formation of oxidative stress in rat cultured cortical glial cells and in an acute JEV rat model (Liao et al., 2002; Srivastava et al., 2009). (2) Oxidative stress and mitochondrial dysfunction have the potential to lower seizure threshold by a variety of mechanisms including impaired ATP production (Jamme et al., 1995) and altered expression of transporters and enzymes crucial in the homeostasis of synaptic levels of neurotransmitter and intracellular calcium levels, thus tilting the balance of synaptic neurotransmission towards hyperexcitation (Waldbaum & Patel, 2010). Oxidative stress can further damage neurons by directly inducing apoptosis or necrosis and such aberrant neuronal loss can facilitate seizure generation (Kannan & Jain, 2000). Therefore, both inflammation and oxidative stress following viral infection may contribute to the development of acute seizures in the TMEV model.
While inflammation has been well documented in the TMEV model of infection-induced epilepsy, it is currently unknown if oxidative stress is observed during the acute seizure stage in TMEV infected mice. Therefore, the goal of the present study was to investigate the time course of oxidative stress in the TMEV-infection mouse model of TLE. We report here that TMEV-infected animals have a significant depletion of reduced glutathione (GSH), increase in oxidized glutathione (GSSG) levels, as well as an increase in 3-nitrotyrosine/tyrosine (3NT/Tyr) ratio. This data suggests that oxidative stress occurs in the TMEV model of CNS infection-induced epilepsy coincident with inflammation and acute seizure activity.
Methods
Animals
Male C57BL/6 mice aged between 4–5 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME, USA). After arrival, mice were allowed to acclimatize for 3 days prior to the experiment. Mice were provided food and water ad libitum and kept in a facility providing 12 hours of light and lark cycle starting at 6 am. All the procedures performed were in accordance with the guidelines provided and approved by the Institutional Animal Care and Use Committee of the University of Utah.
Treatment of mice and seizure monitoring
Mice were anesthetized briefly using a mixture of isoflurane and compressed air. Mice were then injected with 20 μl of either phosphate-buffered saline (PBS, n=30) or 3 × 105pfu (plaque forming units) of Daniels strain of TMEV (n=50) intracortically to a depth of 2 mm in the temporal region of the right hemisphere (posterior and medial of the right eye). Mice were agitated by briefly shaking their cages and monitored for behavioral seizures for 1 hour, twice a day and a minimum of 2 hours apart from 8:00 am to 5:00 pm, until 10 days post-injection (p.i.) as previously described (Libbey et al., 2008). The intensity of the seizure activity was graded using the Racine scale as follows: Stage 1, mouth and facial movements; Stage 2, head nodding; Stage 3, forelimb clonus; Stage 4, rearing; and Stage 5, rearing and falling (Racine, 1972). Mice were sacrificed at 8 hour p.i. (hrpi), and 1, 2, 3, 4, and 14 days pi (dpi). For 3, 4, and 14 dpi, only TMEV-infected mice that had acute behavioral seizures were used, while non-seized mice from the TMEV group were excluded from the studies. The ipsilateral and contralateral portions of the hippocampus were micro-dissected and collected separately. Cerebellum was also collected. All the tissue samples were flash frozen using 2-methylbutane chilled on dry ice and stored at −80 °C. The samples were shipped overnight on dry ice to the laboratory of Dr. Manisha Patel at the University of Colorado, Aurora, CO for the analysis.
HPLC determination of GSH and GSSG
Reduced and oxidized forms of GSH were measured by HPLC with electrochemical detection (HPLC-EC) following minor modifications to previously described methods (Lakritz et al., 1997; Liang & Patel, 2006). GSH and GSSG were detected using a CoulArray system (Model 5600, ESA) on two coulometric array cell modules, each containing eight electrochemical sensors attached in series. Electrochemical detector potentials were 150/300/450/570/690/800/850 mV (vs Pd.). Frozen hippocampi obtained from Dr. Karen Wilcox’s laboratory were sonicated with 0.1N perchloric acid (HClO4) immediately before thawing in a 1:10 weight by volume ratio. The homogenates were centrifuged at 13,000g for 10 minutes at 4°C. Aliquots of the supernatant (20μL) were injected into the HPLC and separated on a 5μM, 250x4.6-mm C-18 ODS-80Tm column (Tosoh Bioscience, Japan). The mobile phase comprised of 100mM sodium phosphate, 1% methanol, pH 2.7 and a flow rate of 0.6 ml/min was maintained. Control values were normalized to one hundred percent and data is represented as percent of control (% control).
HPLC determination of 3-Nitrotyrosine
3-Nitroyrosine (3NT) and tyrosine levels were measured using HPLC method similar to GSH and GSSH measurements and as previously described (Ryan et al., 2014). Frozen hippocampi samples were processed in the same way as described above and 20μl of the supernatant was injected into an ESA 5600 CoulArray HPLC (Chelmsford, MA). Separation was achieved using the same column and mobile phase as mentioned above.
Statistical analyses
GraphPad Prism 6 was used for all statistical analyses performed. Group differences were determined by Analysis of variance (ANOVA) with Sidak’s multiple comparison tests.
Results
Acute behavioral seizures in TMEV-infected mice
Behavioral seizures occur between days 3 and 7 dpi in the TMEV model (Stewart et al., 2010a). Accordingly, seizures were not observed in animals sacrificed prior to day 3. As it was not possible to predict which animals would have gone on to develop seizures, the number of animals was increased (n=8 per group) for the TMEV samples that were obtained at 8 hrpi and at 1 and 2 dpi. The remaining animals (n=26) were then assessed for seizure activity and 81% of those animals were observed to have had at least one seizure prior to sacrifice. However, on any given observation day, approximately only 50% of the animals had a seizure (Fig. 1a). As previously described, seizure severity increased over the course of the observation period, with the majority of seizures observed being either a stage 4 or 5 seizure by day 6 pi (Figs. 1b and c). Only those animals that exhibited at least one seizure during the observation periods were used for harvesting tissue for the remaining time points (3, 4, and 14 dpi; n=5 per group).
Figure 1. Acute behavioral seizures in TMEV-infected mice.

Mice infected with TMEV show acute behavioral seizures between 3 and 7 days post-infection. (A) While 81% of the TMEV infected animals exhibited at least one seizure during the observation period, only approximately 50% of the mice were observed to have behavioral seizures on any given day between 3–7 dpi. (B) Distribution of mice which experienced acute seizures following TMEV infection based on the seizure intensity graded according to the Racine scale. For a mouse which had more than one seizure on any given day, the highest scale of seizure intensity was considered for the plot. Decreasing numbers of animals over time reflect the fact that animals were being sacrificed at different time points throughout the study. (C) Consistent with previous studies (Stewart et al., 2010b; Umpierre et al., 2014), the total numbers of seizures based on the Racine scale on each day during seizure monitoring show that seizure intensity increases over the course of observation period. (dpi = days post-injection).
Impaired glutathione redox status in TMEV model
To determine if oxidative stress occurs in TMEV infected mice, we measured the tissue redox status in the ipsi- and contralateral hippocampus of PBS and TMEV injected mice. GSH is the most abundant non-protein thiol as well as an extremely important non-enzymatic antioxidant in the body. The ratio of GSH to its oxidized form GSSH, a disulfide redox partner, is an excellent indicator of the overall tissue redox status and can be measured by HPLC analysis (Schafer & Buettner, 2001; Liang & Patel, 2006). A higher GSH/GSSG ratio indicates a reduced environment whereas a decrease in this ratio denotes a relatively oxidized tissue environment. Decreased GSH/GSSG correlates with structural damage to cellular membranes, DNA damage, post-translational modifications to proteins and inactivation of essential enzymes, which can all affect neuronal excitability (Andersen, 2004; Waldbaum & Patel, 2010). Whole hippocampal (ipsilateral) GSH (Fig. 2a) decreased significantly in TMEV infected mice at 72hrs (mean ± S.E.M. = 74.712 ± 3.6%), 96hrs (73.494 ± 4.236%), and 14 days (83.91 ± 5.010%) post-infection with a concomitant increase in GSSH (Fig. 2b) levels (203.422 ± 45.697%, 238.291 ± 14.27%, 231.849 ± 41.642% for 72hrs, 96hrs and 14 days respectively), compared to PBS injected control mice. The ratio of GSH/GSSG (Fig. 2c) was also significantly decreased at these time points. The contralateral hippocampi displayed similar levels as PBS injected mice. This correlates with the onset of acute behavioral seizures indicating that tissue redox status is decreased as a consequence of either seizure activity and/or inflammation in the TMEV model, which is consistent with previous findings in the lithium-pilocarpine and kainate chemoconvulsant models of TLE (Waldbaum et al., 2010; Ryan et al., 2014; Jarrett et al., 2008).
Figure 2. Impaired GSH redox status in TMEV-infected mice.

Hippocampal GSH redox status is impaired in mice infected with TMEV. Levels of GSH (A), GSSG (B) and the ratio of GSH/GSSG (C) were measured in the mouse ipsilateral hippocampus using HPLC with electrochemical detection. Mice were injected with PBS or TMEV and sacrificed at indicated times (n≥5 mice per group). Values were normalized to percent of control values and presented as values ± S.E.M. The dotted line indicates 100%. Statistics: *=p<0.05, **=p<0.01, ***=p<0.001 and ****=p<0.0001 versus respective PBS controls; two-way ANOVA with Sidak’s Multiple Comparison test.
Increased 3-nitrotyrosine levels in TMEV-infected mice
3-nitrotyrosine (3NT) is a marker for protein nitration, a post-translational modification that can lead to protein dysfunction or turnover. A major source of 3NT is peroxynitrite (ONOO−), produced from the reaction between nitric oxide (NO) and superoxide (O2·−), two highly reactive free radical species. ONOO− attacks tyrosine residues of proteins specifically, to form 3NT (Sawa et al., 2000). The levels of 3NT and free tyrosine (Tyr) can be measured utilizing HPLC and a higher ratio of 3NT/Tyr is an indicator of oxidative and/or nitrosative stress in tissues. The ratio of 3NT/Tyr in the ipsilateral hippocampus was significantly elevated at 72hrs, 96hrs and 14 days (means ± S.E.M. of 3NT/Tyr × 1000 = 4.707 ± 0.566, 6.297 ± 0.801 and 4.44 ± 0.428 respectively) post-infection compared to their respective controls (Fig. 3). Thus, this data demonstrates that acute seizure activity in mice infected with TMEV leads to an increase in protein nitration suggestive of increased production of reactive oxygen and nitrogen species.
Figure 3. Increased levels of 3NT in TMEV-infected mice.

Hippocampal 3NT levels were elevated in TMEV-infected mice. Mice were injected with PBS or TMEV and 3NT levels were measured at the indicated times with HPLC-EC (n=4–5 mice per group). Data are represented as the ratio of 3-nitrotyrosine to tyrosine (3NT/Tyrosine × 1000) as values ± S.E.M. Statistics: *=p<0.05 and ****=p<0.0001 versus respective PBS controls; two-way ANOVA with Sidak’s Multiple Comparison test.
No change in oxidative stress indices in the cerebellum of TMEV-infected mice
In order to determine if the alterations in oxidative and nitrosative stress are localized to any regions other than the hippocampus, we measured GSH, GSSG and 3NT levels in the cerebellum of mice injected with TMEV and PBS at 3 dpi. There were no significant differences between PBS and TMEV-infected mice in any of the oxidative stress indices measured (Fig. 4) indicating that the redox changes are specific to the hippocampus and perhaps other limbic areas in this model.
Figure 4. No oxidative stress in cerebellum of TMEV mice at 3 dpi.

Mice were injected with PBS or TMEV and GSH, GSSG, 3NT and Tyr levels were measured at 3dpi with HPLC-EC (n=5 mice per group). GSH and GSSG levels are presented as nmols/gram tissue and as the ratio of GSH/GSSG. 3NT levels are presented as the ratio of 3NT to tyrosine (3NT/Tyr × 1000). All data is represented as values ± S.E.M. Statistical analysis used here was an unpaired t-test.
Discussion
This study demonstrates for the first time, that acute seizures resulting from TMEV infection in mice leads to the occurrence of oxidative and nitrosative stress which along with inflammation, might be contributing to the development of epilepsy in this model. We have shown that (1) the glutathione redox status is significantly impaired and (2) 3-nitrotyrosine levels are significantly increased following acute seizures in the TMEV model of infection-induced epilepsy. These results are consistent with previous findings in the lithium-pilocarpine and kainate chemoconvulsant models of TLE and verify that ROS and RNS are elevated as a consequence of acute seizure activity regardless of the initiating injury and point to the fact that oxidation and nitration induced post-translation modifications might play an important role in epileptogenesis and associated pathologies.
The TMEV mouse model of epilepsy was generated by the injection of the Daniel’s strain of TMEV into the right cortex of C57Bl/6J mice whereby the mice develop short-term encephalitic seizures within 3 to 7 dpi. Previously published research has demonstrated similar results as well as confirmed the development of chronic, spontaneous seizures starting at around 2 months pi by video-EEG recordings (Stewart et al., 2010a). The development of epilepsy was also associated with hippocampal sclerosis and astrogliosis in these animals, hallmarks of mesial TLE (Stewart et al., 2010a). Viral infection-induced models of epilepsy, such as the West Nile (WNV), measles and herpes simplex viruses (HSV), have been difficult to address experimentally due to a variety of reasons including fatality from acute viral encephalitis, or lack of spontaneous seizures or the persistence of the virus throughout the animal’s life. The TMEV infected mice not only survive the infection and develop spontaneous seizures, but also clear the virus by day 14 post-infection (Kirkman et al., 2010). Therefore, TMEV infected B6J mice present us with a potential model to study the mechanisms underlying the development of acute seizures, epileptogenesis and consequent epilepsy following a viral infection of the CNS (Stewart et al., 2010). In addition, this model may be useful in identifying novel disease-modifying therapies for the prevention of epilepsy.
One distinguishing feature of the TMEV model is the occurrence of inflammatory changes in the brains of the infected mice which is believed to contribute to the development of acute seizures. Specifically, activation of the innate but not the adaptive immune system in response to the infection was implicated which included substantial increases in TNF-α and IL-6 mRNA levels, along with microglial infiltration and astrogliosis (Kirkman et al., 2010). Viral infections of the CNS have also been associated with the production of ROS and oxidative stress. Oxidative damage is an important component of acute encephalitis caused by HSV-1, HIV and measles virus. Increased ROS and RNS production can be a direct effect of the virus and a result of the inflammatory response of the host (Valyi-Nagy & Dermody, 2005). Oxidative stress and tissue redox status in turn can stimulate the production of pro-inflammatory cytokines (Iyer et al., 2009). Here, we show that ROS and RNS are increased acutely, as measured by the tissue GSH redox status and 3NT to tyrosine ratios, resulting from seizure activity that arises due to induction of innate immune responses, highlighting the relationship between oxidative stress and inflammation in this model.
GSH is an important antioxidant responsible for scavenging harmful reactive species and maintaining a reduced environment in tissues. GSH depletion occurs in several disease states including TLE and the ratio of GSH/GSSG serves as a very important indicator of the tissue redox status and oxidative stress. GSH depletion has been implicated in aging as well as other acute and chronic neuronal disorders (Sims et al., 2004; Liu et al., 2004). Here, we show that the GSH levels are significantly depleted at 3, 4 and 14 dpi concomitant with increases in GSSG levels rendering the overall tissue redox status more oxidized. This is consistent with our previous findings of ROS production and altered redox state in two different chemoconvulsant models of TLE, the kainate and lithium-pilocarpine models, where status-epilepticus is the initiating cause of redox changes (Liang & Patel, 2006; Waldbaum et al., 2010; Ryan et al., 2014). The decrease in the GSH redox status persisted at 14 dpi which represents the latent phase in this model during which the mice do not have any behavioral seizures. Seizures in TMEV-infected mice re-emerge spontaneously 8 weeks p.i (Stewart et al., 2010b) which indicates that the persistent redox changes occurring in the TMEV mice could have a potential role in epilepsy development. However, mechanistic studies are needed to definitively conclude whether these changes in oxidative stress are causal to seizure progression. Nevertheless, our results confirm that decreased GSH redox status is a common phenomenon occurring in animal models of TLE despite the type of precipitating injury, indicating that this could be an important mechanism contributing to spontaneous seizures in TLE.
A variety of mechanisms could lead to the decreased GSH/GSSG ratio in this model. First, it has been previously shown that chemoconvulsant-induced status epilepticus leads to an increase in steady-state mitochondrial O2·− levels resulting in the production of hydrogen peroxide (H2O2) which can diffuse out of the mitochondria into the cellular space and deplete cellular GSH pools (Liang et al., 2000). Second, post-translational inactivation of the iron-sulfur (Fe-S) containing TCA cycle enzyme aconitase mediated by O2·−, which can also pose an additional oxidative burden by producing equimolar amounts of H2O2 per mole of O2·−. Its noteworthy here that the production of O2·− precedes neuronal death in the kainate model and the latter can be prevented by treatment with a broad spectrum antioxidant as well as by overexpressing mitochondrial superoxide dismutase or SOD2 in mice (Liang et al., 2000). Stewart et al., have previously published that TMEV-infected mice have increased neuronal death in the hippocampus at 4–6 dpi (Stewart et al., 2010a). Thus, altered GSH redox state which is observed starting 3 dpi could be a major factor contributing to the brain pathology of TMEV infected mice and the development of epileptic seizures. Therefore, future studies investigating the ability of anti-oxidants to prevent cell death in this model is warranted.
Another important finding of this paper is the increase in the levels of 3NT, a product resulting from nitration of tyrosine residues in proteins by mainly peroxynitrite (ONOO−). ONOO− is the product of the reaction between O2·− and NO, both of which can be generated as a consequence of seizure activity as shown previously in the kainate model (Liang et al., 2000; Ryan et al., 2014). Interestingly, inflammatory cytokines can also lead to the formation of extracellular O2·− and NO by activation of the NADPH oxidases (Nox) and inducible nitric oxide synthase (iNOS), respectively (Dikalov, 2011). Furthermore, our previous work has also illustrated activation of Nox2 in the kainate model (Patel et al., 2005). Therefore, in the TMEV infected mice, 3NT formation could be a consequence of NO production from acute seizures or it could be a direct consequence of the release of pro-inflammatory molecules. The increase in 3NT levels presents with a unique hypothesis where ROS/RNS induced post-translational modifications (PTMs) underlie the damaging subcellular events that promote epileptogenesis. PTMs associated with altered redox status are associated with cysteine (Cys) residues with low pKa or thiol groups in proteins (Giustarini et al., 2004; Jones, 2008). Such modifications can either exert a protection from further oxidation or if the Cys residue is functionally important, PTMs can disrupt the protein’s biological function. As an example, Ryan et al., have recently demonstrated that carbonylation of complex I of the electron transport chain (ETC) is increased in the acute and chronic phases of epileptogenesis in the kainate model, which correlated with decreases in complex I activity. Mass spectrometric analysis of this PTM identified the site as Arg76 within the 75kDa subunit of the complex (Ryan et al., 2012). Moreover, 3NT was shown to accumulate in the hippocampal neurons and not astrocytes in the kainate model implicating RNS as the species contributing to cell death in TLE (Ryan et al., 2014). All this evidence brings forth the notion that PTMs resulting from seizure-induced ROS or RNS production can mediate neuronal death and the development of spontaneous seizures in the TMEV model. Further investigation into the role of ROS/RNS in the TMEV model can be warranted by determining the effect of an antioxidant on cell death and epileptic seizures in this model.
To summarize, we have demonstrated that acute seizures, arising from inflammation in the TMEV model of infection-induced epilepsy, lead to the production of ROS and RNS. In our studies we have reported a decrease in the GSH redox status as well as an increase in 3NT levels indicative of oxidative and nitrosative damage respectively. These results agree with previous findings illustrating the occurrence of oxidative and nitrosative stress in SE models of TLE, suggesting that these biochemical changes are common phenomena in animal models of TLE, irrespective of the initiating injury. Furthermore, our results point to the role of ROS and RNS mediated PTMs as well as highlights the interaction between redox and inflammatory processes in the TMEV model, which could be the mechanisms underlying the pathology observed in this model. Finally, this study underscores the relationship between redox mechanisms and seizure activity in the TMEV modeland lays the groundwork for further investigation and development of therapeutic strategies targeting redox processes for the treatment of acquired epilepsies.
Highlights.
The presence of oxidative stress was examined in the TMEV model of infection-induced temporal lobe epilepsy.
Oxidative stress assessed by glutathione redox status and 3-nitrotyrosine levels occurred in the hippocampus of TMEV-infected.
Oxidative stress in TMEV-infected mice occurred concomitantly with acute behavioral seizures.
Oxidative stress may be a potential therapeutic target to treat infection-induced epilepsy.
Acknowledgments
This work was funded by the Skaggs Scholar Award (KSW and MP), R01 NS065434 (KSW) and 1R01NS086423 (M.P.)
Abbreviations
- TLE
temporal lobe epilepsy
- TMEV
Theiler’s murine encephalomyelitis virus
- dpi
days post-infection
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- HSV
Herpes simplex virus
- JEV
Japanese encephalitis virus
- GSH
glutathione
- GSSG
glutathione disulfide
- 3NT
3-nitrotyrosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Staley KJ. The time course of acquired epilepsy: implications for therapeutic intervention to suppress epileptogenesis. Neurosci Lett. 2011;497:240–6. doi: 10.1016/j.neulet.2011.03.071. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox enviroment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Giustarini D, Rossi R, Milzani A, Colombo R, Dalle-Done I. S-glutathionylation: from redox regulation of protein functions to human diseases. J Cell Mol Med. 2004;8:201–12. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Accardi CJ, Ziegler TR, Blanco RA, Ritzenthaler JD, Rojas M, Roman J, Jones DP. Cysteine redox potential determines pro-inflammatory IL-1beta levels. PloS One. 2009;4:e5017. doi: 10.1371/journal.pone.0005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamme I, Petit E, Divoux D, Gerbi A, Maixent JM, Nouvelot A. Modulation of mouse cerebral Na+,K(+)-ATPase activity by oxygen free radicals. Neuroreport. 1995;7:333–337. [PubMed] [Google Scholar]
- Jarrett SG, Liang LP, Hellier JL, Staley KJ, Patel M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol Dis. 2008;30:130–8. doi: 10.1016/j.nbd.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–68. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- Kirkman NJ, Libbey JE, Wilcox KS, White HS, Fujinami RS. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia. 2010;51:454–64. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakritz J, Plopper CG, Buckpitt AR. Validated high-performance liquid chromatography-electrochemical method for determination of glutathione and glutathione disulfide in small tissue samples. Anal Biochem. 1997;247:63–8. doi: 10.1006/abio.1997.2032. [DOI] [PubMed] [Google Scholar]
- Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–70. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med. 2006;40:316–22. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Liao SL, Raung SL, Chen CJ. Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci Lett. 2002;324:133–136. doi: 10.1016/s0304-3940(02)00236-7. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Kirkman NJ, Smith MC, Tanaka T, Wilcox KS, White HS, Fujinami RS. Seizures following picornavirus infection. Epilepsia. 2008;49:1066–1074. doi: 10.1111/j.1528-1167.2008.01535.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang H, Shenvi S, Hagen TM, Liu RM. Glutathione metabolism during aging and in Alzheimer’s disease. Ann N Y Acad Sci. 2004;1019:346–349. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- Misra UK, Tan CT, Kalita J. Viral encephalitis and epilepsy. Epilepsia. 2008;49:13–18. doi: 10.1111/j.1528-1167.2008.01751.x. [DOI] [PubMed] [Google Scholar]
- Patel M, Li QY, Chang LY, Crapo J, Liang LP. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J Neurochem. 2005;92:123–31. doi: 10.1111/j.1471-4159.2004.02838.x. [DOI] [PubMed] [Google Scholar]
- Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med. 2004;37:1951–62. doi: 10.1016/j.freeradbiomed.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ryan K, Backos DS, Reigan P, Patel M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J Neurosci. 2012;32:11250–8. doi: 10.1523/JNEUROSCI.0907-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Liang LP, Rivard C, Patel M. Temporal and spatial increase of reactive nitrogen species in the kainate model of temporal lobe epilepsy. Neurobiol Dis. 2014;64:8–15. doi: 10.1016/j.nbd.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa T, Akaike T, Maeda H. Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J Biol Chem. 2000;275:32467–74. doi: 10.1074/jbc.M910169199. [DOI] [PubMed] [Google Scholar]
- Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med. 1996;21:641–649. doi: 10.1016/0891-5849(96)00131-1. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol. 2007;35:984–99. doi: 10.1080/01926230701748305. [DOI] [PubMed] [Google Scholar]
- Sims NR, Nilsson M, Muyderman H. Mitochondrial glutathione: a modulator of brain cell death. J Bioenerg Biomembr. 2004;36:329–33. doi: 10.1023/B:JOBB.0000041763.63958.e7. [DOI] [PubMed] [Google Scholar]
- Srivastava R, Kalita J, Khan MY, Misra UK. Free radical generation by neurons in rat model of Japanese encephalitis. Neurochem Res. 2009;34:2141–2146. doi: 10.1007/s11064-009-0008-7. [DOI] [PubMed] [Google Scholar]
- Stewart KA, Wilcox KS, Fujinami RS, White HS. Theiler’s virus infection chronically alters seizure susceptibility. Epilepsia. 2010a;51:1418–1428. doi: 10.1111/j.1528-1167.2009.02405.x. [DOI] [PubMed] [Google Scholar]
- Stewart KA, Wilcox KS, Fujinami RS, White HS. Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. J Neuropathol Exp Neurol. 2010b;69:1210–9. doi: 10.1097/NEN.0b013e3181ffc420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore WH. Epilepsy and viral infections. Epilepsy Curr. 2014;14:35–42. doi: 10.5698/1535-7511-14.s2.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umpierre AD, Remigio GJ, Dahle EJ, Bradford K, Alex AB, Smith MD, West PJ, White HS, Wilcox KS. Impaired cognitive ability and anxiety-like behavior following acute seizures in the Theiler’s virus model of temporal lobe epilepsy. Neurobiol Dis. 2014;64:98–106. doi: 10.1016/j.nbd.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyi-Nagy T, Olson SJ, Valyi-Nagy K, Montine TJ, Dermody TS. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology. 2000;278:309–321. doi: 10.1006/viro.2000.0678. [DOI] [PubMed] [Google Scholar]
- Valyi-Nagy T, Dermody TS. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol Histopathol. 2005;20:957–967. doi: 10.14670/HH-20.957. [DOI] [PubMed] [Google Scholar]
- Waldbaum S, Liang LP, Patel M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J Neurochem. 2010;115:1172–82. doi: 10.1111/j.1471-4159.2010.07013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010;88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]