

Abstract
The residual presence of integrated transgenes following the derivation of induced pluripotent stem (iPS) cells is highly undesirable. Here we demonstrate efficient derivation of iPS cells free of exogenous reprogramming transgenes using an excisable polycistronic lentiviral vector. A novel version of this vector containing a reporter fluorochrome allows direct visualization of vector excision in living iPS cells in real time. We find that removal of the reprogramming vector markedly improves the developmental potential of iPS cells and significantly augments their capacity to undergo directed differentiation in vitro. We further propose that methods to efficiently excise reprogramming transgenes with minimal culture passaging, such as those demonstrated here, are critical since we find that iPS cells may acquire chromosomal abnormalities, such as trisomy of chromosome 8, similar to ESC after expansion in culture. Our findings illustrate an efficient method for the generation of transgene-free iPS cells and emphasize the potential beneficial effects that may result from elimination of integrated reprogramming factors. In addition, our results underscore the consequences of long-term culture that will need to be taken into account for the clinical application of iPS cells.
Keywords: induced pluripotent stem (iPS) cells, reprogramming, excisable single lentiviral vector, stem cell cassette, endoderm
INTRODUCTION
Reprogramming of somatic cells to a pluripotent state has been achieved by the introduction of four transcription factors, Oct4, Klf4, Sox2 and cMyc using four independent retroviral vectors [1–6]. It is now widely accepted that iPS cells share many of the characteristics of embryonic stem cells (ESC), including gene expression profiles, epigenetic signatures and pluripotency [3, 7–10].
Since iPS cells can be generated from mature mouse and human somatic cells, such as skin fibroblasts [11–13], enabling the derivation of patient-specific cells and autologous tissues, it is often predicted that iPS cells will become a powerful tool for biological research as well as a potent source for regenerative medicine. In order for this technology to become clinically relevant, however, methods need to be developed that improve the safety profile of the technology while increasing the overall efficiency of the production of the cells. Several studies have demonstrated that reactivation or sustained expression of reprogramming transgenes can result in deleterious outcomes such as tumor formation [2] or the disruption of pluripotency [14, 15]. Moreover, the study of the biology of reprogramming and the ability to evaluate how closely iPS cells and ESC functionally resemble each other will greatly benefit from the use of homogeneous populations devoid of any residual transgene expression.
Recently, a series of studies have demonstrated proof of principle in the generation of murine iPS cells without viral integrations [16, 17]. It should be noted, however, that the efficiency of reprogramming in these studies was low and their application for deriving iPS cells from diverse, easily-accessible target somatic cells appears to be limited. Similarly, transposon-mediated reprogramming, albeit at low efficiency, was able to generate murine iPS cells free of exogenous factors [18]. The potential toxicity associated with the use of the transposon/transposase system in iPS cells, however, remains to be studied [19, 20]. Most recently, Jaenisch and colleagues demonstrated Cre-mediated excision of multiple integrated reprogramming lentiviral vectors in human iPS cells following the completion of reprogramming [21]. This important advance demonstrated that prior to excision the transcriptome of iPS cells differs slightly from control ESC. To date it remains unclear whether these subtle differences in global gene expression between iPS cells and ESC are accompanied by significant functional differences. Moreover, if indeed removal of transgenes is necessary for proper functioning of iPS cells, methods that are simple, efficient, and involve minimal screening strategies for deriving `transgene-free' iPS cells represent important advances for the clinical translation of this technology.
We have recently described the use of a single lentiviral `stem cell cassette' (STEMCCA) for the efficient generation of iPS cells from post-natal fibroblasts [22]. Importantly, the use of a single polycistronic vector, expressing Oct4, Klf4, Sox2, and cMyc, allowed us to obtain iPS cell clones with a single integration. Here we adapt the STEMCCA vector to derive iPS cells free of exogenous reprogramming transgenes. Using a single integrated copy of this polycistronic vector, encoding either 3 or 4 reprogramming factors flanked by loxP sites, we accomplish efficient reprogramming of post-natal fibroblasts, followed by highly efficient Cre-mediated excision of the vector. A direct comparison of iPS cell clones before and after excision reveals that removal of the reprogramming vector markedly improves the developmental potential and differentiation capacity of iPS cells.
MATERIALS AND METHODS
Construction of pHAGE-STEMCCA-LoxP vectors
The EF1α-STEMCCA lentiviral vector allows for constitutive expression of the four proteins Oct4, Klf4, Sox2 and cMyc from a single polycistronic transcript [22]. In order to obtain a floxed version of STEMCCA after viral integration, that would enable Cre-mediated excision of the vector following reprogramming, a 34-bp loxP site was inserted in the 3' dU3 LTR region of EF1α-STEMCCA. During reverse transcription, the loxP sequence is copied to the 5' LTR resulting in a vector flanked by two loxP sites. Detailed methods for primers and construct design are included as supplemental data.
Cell culture
Tail tip fibroblasts from newborn Sox2-GFP knock-in mice were cultured using standard procedures in fibroblast growth media as described [22]. TTFs were infected at passage 3 for generation of iPS cells.
Lentivirus production and infection
Lentiviruses were produced in 293T packaging cells as previously described [23]. iPS colonies were mechanically isolated 15 to 20 days post-infection with STEMCCA-loxP or 25–30 days post-infection with STEMCCA-loxP-RedLight based on morphology and expanded by plating on Mitomycin C treated MEFs in ES cell media. Copy numbers of each STEMCCA vector integrated in the genome of reprogrammed cells was assessed by Shouthern blot analysis as previously described [22].
PCR and RT-PCR analysis
Detection of the proviral DNA by PCR was carried out using 50 ng of genomic DNA and the primers endo-MycS (5'-ACGAGCACAAGCTCACCTCT-3') and A-WPRE (5'-TCAGCAAACACAGTGCACACC-3'). PCR reactions consisted of 30 cycles of 95°C for 30 seconds, 65°C for 45 seconds and 72°C for 45 seconds. RT-PCR was performed as previously described [22].
Infection of iPS cells with Adeno-Cre
Excision of STEMCCA was performed by infecting iPS cells with a defective adenoviral vector expressing Cre-recombinase (Adeno-Cre), a kind gift of Jeng-Shin Lee and Richard C. Mulligan from the Harvard Gene Therapy Initiative. The recombinant adenovirus was propagated in 293 cells, purified by CsCl gradient centrifugation and desalted on a Sephadex G-50 column (GE Healthcare UK Limited, Little Chalfont, Buckinghamshire, U.K., http://www.gehealthcare.com) in PBS/3% glycerol. For Adeno-Cre infection, 100,000 iPS cells in 100 μl of ESC media were mixed with 3 μl of Adeno-Cre in a microfuge tube and incubated for 6 hours at 37°C. Cells were then washed with PBS, seeded on Mitomycin C treated MEFs and cultured in ESC media until colonies appeared. For each iPS clone infected with Adeno-Cre, several subclones were isolated and expanded as described above. Finally, the efficiency of Cre-recombinase activity was assessed by PCR and Southern Blot or by mCherry red fluorescence, as indicated in the text.
Alkaline phosphatase staining and immunofluorescence
Alkaline phosphatase staining was performed with the Vector Red Substrate Kit (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com) according to the manufacturer's protocol. For immunofluorescence, cells were fixed in 4% paraformaldehyde, incubated with mouse anti-SSEA-1 (Santa Cruz Biotechnology, Santa Cruz, CA, http://www.scbt.com) followed by secondary antibody, Alexa Fluor 568 conjugated goat anti-mouse IgM (Invitrogen, Carlsbad, CA, http://www.invitrogen.com).
ES and iPS cell differentiation
Prior to differentiation all ESC and iPS cells were adapted to serum-free maintenance media [24] by culture expansion on mitomycin C-inactivated mouse embryonic fibroblasts (MEFs). Maintenance media consisted of 50% Neurobasal medium (Invitrogen) and 50% Dulbecco's Modified Eagle Medium/F12 medium (Invitrogen); with N2 and B27 supplements (Invitrogen), 1% penicillin/streptomycin, 0.05% bovine serum albumin, LIF (1000 U/ml; ESGRO; Chemicon; Millipore, Bedford, MA, http://www.millipore.com), 10 ng/ml human BMP-4 (R&D Systems, Minneapolis, MN, www.rndsystems.com), and 1.5 10–4 M monothioglycerol (MTG) (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com). Mouse ESC (129/Ola; containing GFP targeted to the brachyury locus and hCD4 targeted to the Foxa2 locus) were the generous gift of Dr. Gordon Keller, Mount Sinai Medical Center, New York, NY [24, 25]. Differentiation into primitive streak- and endoderm-like phenotypes was performed in serum free media, as previously described by Keller and colleagues [24, 25]. Briefly, embryoid bodies were formed in suspension culture by plating ESC or iPS cells in non-adherent culture plates for 2 days in the absence of LIF. On day 2 embryoid bodies were dispersed by trypsinization followed by replating for 3 more days in serum-free media with activin A (50ng/ml; R&D systems, 338-AC). On day 5 embryoid bodies were dissociated with trypsin/EDTA (2 min, 37°C) and harvested for RNA extraction. Differentiation towards mesoderm and ectoderm was performed as previously described [26, 27]. In brief, mesoderm differentiation was quantified as the number of cells expressing the early hemangioblast marker, flk1, by FACS analysis of EBs cultured for 4 days in serum-free media supplemented with 10 ng/ml BMP4. Ectoderm differentiation was quantified as the number of cell colonies positively immunostained for expression of the neuronal marker Tuj1, after EB formation for 5 days in the presence of 100 nM retinoic acid and 1 μM Smoothened agonist 1.3, followed by 2 days incubation in poly-D-lysine/laminin plates with 10 ng/mL of the neurotrophic factors GDNF, BDNF, and CNTF.
Quantitative RT-PCR
qRT-PCR was carried out in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com) using Taqman custom primers and probe specific to the genes of interest as described by the manufacturer. Amplification of the viral transcript was performed with a Taqman assay designed to amplify a cMyc to WPRE fragment present in the STEMCCA vector. Reactions were performed in triplicate using 1/20 of the cDNA obtained as described above. Gene expression levels were normalized to β-actin and relative quantification of expression was estimated using the comparative Ct method. For qRT-PCR analyses of changes in gene expression in response to activin stimulation, total RNA was purified with RNeasy Mini kit (Qiagen, Chatsworth, CA, http://www.qiagen.com) and treated with RNase-Free DNase I (Qiagen). One microgram of RNA was reverse-transcribed using TaqMan Reverse Transcription Reagents kit (Applied Biosystems) according to the manufacturer's instructions. qPCR analyses of cDNAs was performed in an Applied Biosystems Sequence Detection System 7300 using the following Taqman inventoried primers and probes (Applied Biosystems): Nanog (Mm02384862_g1), Rex1 (Mm01194089_g1), Brachyury (Mm00436877_m1), FoxA2 (Mm00839704_mH), Sox17 (Mm00488363_m1), Gata4 (Mm00484689_m1), and Gata6 (Mm00802636_m1). Reactions were performed in duplicate using 1/20 diluted cDNA obtained as described above. Gene expression levels were normalized to 18S rRNA (4319413E) and relative expression of each gene compared to undifferentiated ESC was quantified using the 2−[delta][delta]Ct method.
Teratoma formation
Two million iPS cells were injected subcutaneously into each flank of recipient C.B-17 scid mice (The Jackson Laboratory, Bar Harbor, ME, http://www.jax.org). Three weeks after injection, tumors were dissected and fixed in 4% paraformaldehyde. Finally, paraffin-embedded sections of the teratoma specimens were stained with hematoxylin and eosin, and two independent investigators each assessed 3 individual sections per teratoma according to previously described morphological criteria [28]. All animal experiments were performed in accordance with Boston University Institutional Animal Care and Use Committee (IACUC).
Generation of chimeric animals
iPS cells were injected into C57BL/6J-Tyrc-2J (Jackson laboratory) mouse blastocysts and implanted into pseudopregnant foster mothers using routine methods. Pregnant mice were sacrificed at embryonic day E11.5 and whole embryos were photographed with an inverted fluorescence microscope.
Karyotyping and SKY analysis
Metaphase spreads were prepared according to standard protocols [29]. Spectral karyotyping was performed with a mouse SKY paint kit (Applied Spectral Imaging Inc., Vista, CA, http://www.spectral-imaging.com) according to the manufacturer's instructions. Analysis was performed with the HiSKY and ScanView softwares (Applied Spectral Imaging).
RESULTS
Cre-mediated excision of a loxP-containing polycistronic reprogramming vector allows the derivation of `transgene-free' iPS cells
In order to develop a simplified method for the derivation of transgene-free iPS cells, we sought to utilize a vector system that would result in efficient reprogramming with a single reagent, without the need for concurrent additional vectors, transgenes, or chemical exposures. Hence, in contrast to other studies in which an inducible system was used [10, 21, 30], we chose to use constitutively expressed versions of the lentiviral STEMCCA vector under regulatory control of a human EF1α promoter [22]. This single reagent accomplishes efficient and reliable reprogramming of post-natal cells by expressing four factors (Oct4, Klf4, Sox2, and cMyc) and obviates the need for additional genetic modification since the transactivator (i.e. rtTA) is not required to induce expression of the reprogramming cassette. Because previous studies have shown that iPS cells can be derived without the presence of exogenous cMyc [31], we also developed a modified 3 factor STEMCCA vector by substituting cMyc with the coding sequence of the red fluorochrome mCherry. This modified vector, hereafter named STEMCCA-RedLight, constitutively expresses mCherry as well as the 3 reprogramming factors, Oct4, Klf4 and Sox2 from a single polycistronic mRNA, thus allowing monitoring of STEMCCA gene expression in living cells.
In order to allow for excision of the 3 factor or 4 factor STEMCCA vectors, we first introduced a loxP site in the deleted U3 (dU3) region of each lentiviral vector's 3'LTR [22] (Fig. 1A). During the normal reverse transcription cycle of the virus before integration, the U3 region is copied to the 5' LTR of the proviral genome, creating a loxP-flanked or `floxed' version of the STEMCCA vector that integrates into the host chromosome. These floxed STEMCCA vectors (hereafter STEMCCA-loxP and STEMCCA-loxP-RedLight) were used to generate iPS cells from tail tip fibroblasts (TTFs) of Sox2-GFP knock in mice as previously described [22]. The introduction of loxP sites did not affect viral titers (data not shown), and the STEMCCA-loxP vector was able to generate iPS cell colonies with the same kinetics and the same reprogramming efficiency (~0.5%) as demonstrated previously using STEMCCA [22]. Initial selection of iPS colonies generated with this vector was based solely on morphological criteria and 20 out of 24 (83%) picked colonies selected on this basis resulted in Sox2-GFP expressing cell lines after expansion. As expected 3 factor reprogramming using STEMCCA-loxP-RedLight was slower and less efficient than 4 factor reprogramming. Sox2-GFP+ colonies appeared only 25–30 days after transduction with STEMCCA-loxP-RedLight (compared to 15–20 days when using the four factors vector), and overall reprogramming efficiency was 0.01%, or 50 fold lower than that observed with STEMCCA-loxP. Importantly, persistent expression of the polycistronic STEMCCA-loxP-RedLight vector, driven by the constitutively active EF1α promoter could be readily visualized by red fluorescence microscopy during reprogramming and was maintained in picked Sox2-GFP+ iPS clones after the completion of reprogramming (Fig. 1B).
Figure 1.
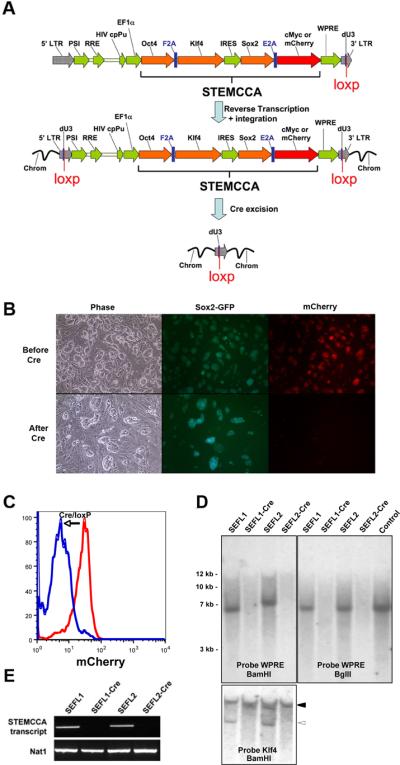
An excisable single lentiviral vector for the generation of iPS cells free of exogenous transgenes. (A) Schematic representation of the STEMCCA-loxP or STEMCCA-loxP-RedLight lentiviral vector. Two versions of the constitutive EF1α-STEMCCA vector [22] were engineered, expressing either 4 reprogramming factors (STEMCCA-loxP) or 3 factors plus mCherry (STEMCCA-loxP-RedLight). A loxP site was introduced within the U3 region of the 3' LTR. Upon formation of the provirus a floxed version of each STEMCCA vector is produced. Following exposure to Cre, the entire cassette is excised. LTR: long terminal repeat; PSI: packaging signal; RRE: rev responsive element; cpPu: central polypurine tract; WPRE: Woodchuck hepatitis virus post-transcriptional regulatory element; dU3: deleted U3. (B) Representative images of iPS cells created using the STEMCCA-loxP-RedLight vector, before and after Cre excision. In the top panel (Before Cre), mCherry expression is evident in all iPS colonies, while in the bottom panel (After Cre), red fluorescence expression is no longer detected, illustrating efficient excision of the STEMCCA-loxp-RedLight cassette. (C) Same cells as in (B) were analyzed using flow cytometry to detect mCherry fluorescence. Cells before (red) and after (blue) Cre treatment are shown. (D) Southern blot analysis of genomic DNA (gDNA) purified from 2 representative iPS clones produced with the STEMCCA-loxP vector, before and after Cre-mediated excision. gDNA was digested with BamHI to expose each individual viral integration. Both, SEFL1 and SEFL2 clones displayed a single integration that is not detected after exposure to Cre (SEFL1-Cre and SEFL2-Cre). Similar results were obtained when probing for Klf4. In this case, the endogenous gene was evident in all clones (closed arrowhead) while the STEMCCA encoded Klf4 is not present following Cre excision (open arrowhead). gDNA was digested with BglII to obtain a band of 6.7Kb that confirms appropriate viral transmission. For a control, STEMCCA-loxP plasmid DNA representing 2.5 copies of the insert was digested with BglII. A single band corresponding to an integration of the correct size was observed in SEFL1 and SEFL2 clones and was not present following Cre treatment. (E) Expression of the STEMCCA transcript was analyzed by RT-PCR to confirm excision. As expected, clones SEFL1-Cre and SEFL2-Cre showed no detectable STEMCCA transcript. Nat1 is a constitutively expressed gene and serves as a control for loading.
Next, we screened Sox2-GFP expressing clones by Southern blot to determine the number of viral integrations, as a first step to pursue vector excision. Three of 9 screened clones generated with STEMCCA-loxP and 3 of 7 clones generated with STEMCCA-loxP-RedLight showed single copy integration (Supplemental online Fig. 1). In order to excise the single integrated copy of each floxed vector, the clones were exposed to an adenoviral vector (Adeno-Cre) to achieve transient expression of Cre recombinase. We employed Adeno-Cre mediated recombination rather than electroporation of Cre-expressing plasmids based on screening studies revealing superior transfection efficiencies of ESC or iPS cells using adenoviral vectors vs. plasmid electroporation (90–100% transfection efficiency vs. 0.5–1%, respectively, data not shown). Adeno-Cre infection of all single integrant iPS cell lines (n=3) resulted in successful Cre-mediated excision of STEMCCA-loxP in 5 out of 5 subclones of each cell line, as evidenced by PCR of gDNA (Supplemental online Fig. 2). In addition, Southern blot analysis confirmed the absence of integrated vector using probes against the WPRE sequence of the STEMCCA vector (Fig. 1D) as well as against the individual reprogramming genes (Fig. 1D and data not shown). PCR screening using oligos specific to Cre was used to confirm that, as expected, no integration of the Adeno-Cre vector had occurred (data not shown). As expected, following excision of the reprogramming cassette and culture expansion of excised iPS cell subclones, the STEMCCA transcript was undetectable as evidenced by RT-PCR (Fig. 1E) and qRT-PCR (Supplemental online Fig. 3), in contrast to the pre-excision parental clones. In iPS cells generated with STEMCCA-loxPRedLight, disappearance of the mCherry reporter was used to precisely monitor and quantify STEMCCA excision efficiency by fluorescence microscopy (Fig.1B) and FACS (Fig. 1C). These iPS cells co-expressed mCherry and Sox2-GFP, however, after Adeno-Cre infection, 96 out of 100 colonies expressed only GFP but not mCherry, suggesting an excision efficiency of almost 100%.
iPS cells display stable growth characteristics and stem cell marker gene expression after excision of reprogramming transgenes
For further study of iPS cells following excision of the reprogramming transgenes, two subclones generated after excision of STEMCCA-loxP were selected and named SEFL1-Cre and SEFL2-Cre, based on their origin from the parental SEFL1 and SEFL2 clones, respectively (Fig. 1D). As shown in Fig. 2A and 2B, after STEMCCA excision iPS cells maintained expression of alkaline phosphatase (AP), SSEA1, Sox2-GFP and a variety of stem cell markers as evidenced by RT-PCR. In addition, the methylation status of the Oct4 and Nanog proximal promoter regions was analyzed by bisulphite sequencing (Fig. 2C). Both before and after STEMCCA-loxP excision, established iPS cell clones exhibited unmethylated CpG islands at these key loci, in contrast to parental fibroblasts. Importantly, iPS cell expansion appeared to be stable following excision of the STEMCCA-loxP as evidenced by the ability of the cells to be maintained in the undifferentiated state in culture for at least 20 passages.
Figure 2.
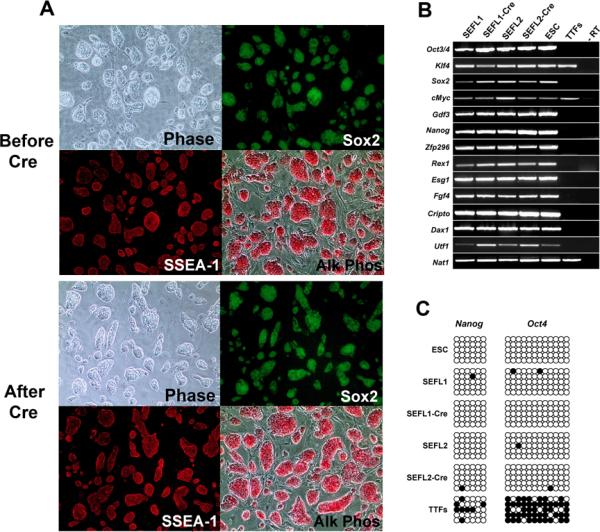
Characterization of iPS clones before and after STEMCCA-loxP excision. (A) Representative images of iPS cells derived using the constitutive STEMCCA-loxP vector before and after Cre-mediated excision show normal colony morphology (Phase), Sox2-GFP reporter gene expression, SSEA1 positive immunostaining, and robust alkaline phosphatase activity (Alk Phos). (B) Expression of ESC `marker' genes detected by RT-PCR in iPS cell clones before (SEFL1 and SEFL2) and after excision (SEFL1-Cre and SEFL2-Cre). Representative samples from murine ESC and unmanipulated tail tip fibroblasts (TTFs) are also shown for comparison. An iPS cell sample prepared without RT was used as negative control (-RT). (C) Promoter regions of Nanog and Oct4 genes were analyzed for methylation status using bisulfite sequencing. Similar to ESC, all iPS cell clones (SEFL1, SEFL1-Cre, SEFL2 and SEFL2-Cre) showed mostly unmethylated CpG motifs (open circles) in sharp contrast to parental TTFs, in which the extracted DNA was mostly methylated (closed circles).
Excision of reprogramming transgenes facilitates the developmental capacity of iPS cells
We have previously reported that iPS cells generated with the constitutive STEMCCA vector were able to differentiate into all three germ layers in teratoma assays, despite the residual presence of exogenous reprogramming genes, even when expressed under regulatory control of constitutively active promoters [22]. Indeed, we confirmed these results using the SEFL1 or SEFL2 clones: regardless of whether these clones were tested before or after excision of the STEMCCA-loxP, SEFL1 and SEFL2 iPS cell lines readily gave rise to differentiated cells with morphologies of all three primary germ layers [28] in teratoma assays (Fig. 3A). We considered the possibility that in vivo silencing or downregulation of STEMCCA expression in vivo in the teratoma assays might account for the unexpected capacity of these iPS cell lines to differentiate in this assay. Indeed, in contrast to the persistence of STEMCCA expression in vitro despite prolonged passaging, qRT-PCR analyses of teratoma tissues (Fig. 3B) indicated that overall levels of STEMCCA expression are markedly lower in teratomas compared to the undifferentiated iPS cell lines pre-transplantation. This lower level of transgene expression possibly explains, in part, the capacity of these cell lines to differentiate in vivo in this assay, which allows a relatively long 3 week period for selection of cells that either are already expressing low STEMCCA levels or undergo subsequent vector silencing in vivo.
Figure 3.

iPS cells generated with the STEMCCA-loxP lentiviral vector are pluripotent and allow for normal in vivo development following the removal of exogenous transgenes. (A) Teratomas derived from iPS cell lines produced with STEMCCA-loxP vector, before and after Cre excision, showing differentiation into cell types of all three germ layers: endoderm (end), mesoderm (mes) and ectoderm (ect). Images are representative of two independent experiments. (B) Representative qRT-PCR data for comparison of the STEMCCA expression levels in iPS cells and teratoma tissues before (SEFL1 and SEFL2) and after (SEFL1-Cre and SEFL2-Cre) excision. (C and D) iPS cells generated with the constitutive STEMCCA vector (no Cre-excision) displayed high levels of embryonic contribution following injection into blastocysts, but also induced gross morphological abnormalities (C). In sharp contrast (D), iPS cells generated using the STEMCCA-loxP vector with subsequent Cre excision, produced a higher percentage of chimeric embryos with normal developmental morphology. Chimerism is evidenced by Sox2-GFP expression in neural crest-derived tissues. (E) Derivation of neonatal chimeric mice from blastocysts injected with iPS cells generated using the STEMCCA-loxp vector with subsequent Cre excision. Chimerism is evidenced by dark coat color.
Constitutive expression of STEMCCA, however, did appear to adversely affect the in vivo developmental potential of iPS cells after transplantation into mouse blastocysts, an assay that affords the cells very short time intervals to undergo precisely regulated differentiation. As shown in Fig. 3C, iPS cells generated with the constitutive STEMCCA vector were injected into 15 blastocysts followed by midgestation (E11.5) harvest. These injections yielded only 5 embryos of which 3 were chimeric. All 3 chimeric embryos, however, displayed severe morphological abnormalities, including exencephaly, neural tube defects, reduced size and defective somite patterning. In addition, in four independent attempts we were unable to obtain live chimeras when using three different iPS cell clones containing the constitutive STEMCCA or STEMCCA-loxP vectors. In marked contrast, following Cre-mediated excision of STEMCCA-loxP, iPS cells injected into 14 blastocysts yielded 11 midgestation embryos of which 7 were chimeric with 5/7 displaying normal developmental morphology (Fig. 3D). Moreover, we were able to obtain live chimeric mice from blastocysts injected with iPS cells after excision of STEMCCA-loxP (Fig. 3E).
While these results suggested that sustained expression of the reprogramming genes may affect the ability of iPS cells to undergo appropriate embryonic development in chimeric embryos, it is not clear whether the morphological defects in the chimeric embryos resulted directly from failure of the injected iPS cells to differentiate. Indeed the seemingly intact ability of these same iPS clones to undergo tri-lineage differentiation in teratoma assays suggests they retain some capacity to respond to differentiation cues such as soluble growth factors. Hence, in order to more fully evaluate the effects of residual STEMCCA expression on the capacity of iPS cells to differentiate in response to normal developmental cues active in the early embryo, we stimulated iPS cells in culture with activin A (hereafter activin), a protein known to mimic embryonic nodal/TGF-β signaling. Activin has been shown to induce ESC in vitro to differentiate sequentially into primitive streak-like cells followed by definitive endoderm [24, 25, 32].
Following Cre-mediated excision of STEMCCA-loxP, `transgene-free' versions of each iPS cell clone were compared to their parental clones in terms of endodermal potential in vitro in response to activin. In two independent experiments, analyzed by either conventional or quantitative RT-PCR, each constitutive, integrated STEMCCA-loxP clone showed diminished potential to upregulate endodermal markers in response to activin in vitro (Fig. 4A and 4B). Although all clones were able to form embryoid bodies in culture, STEMCCA-loxP containing clones showed little potential to upregulate key endodermal transcription factors, such as brachyury, Foxa2, Sox17, Gata4, and Gata6. Furthermore, these clones showed persistent expression of the pluripotent markers Rex1 and Esg1 after activin stimulation (Fig. 4A). In contrast, excision of STEMCCA-loxP from these clones improved the capacity of each clone to upregulate endodermal transcription factors and downregulate pluripotent loci in response to activin (Fig. 4A and 4B).
Figure 4.

In vitro differentiation of iPS cells toward primitive streak and endoderm is facilitated by excision of reprogramming factors. (A) Representative RT-PCR data of iPS clones subjected to in vitro differentiation toward primitive streak/endoderm using activin A stimulation (Act A). iPS cells before (SEFL1 and SEFL2) and after (SEFL1-Cre and SEFL2-Cre) excision of STEMCCA-loxP are compared to an ESC clone. mRNA extracted on day 0 (−) and day 5 (+) of activin stimulation was used as the template for RT-PCR. Several genes characteristic of ESC, i.e. Rex1, Sox2, Esg1 and Nanog, as well as primitive streak (Brachyury: Bry) and early endoderm (FoxA2) markers are depicted. Note lack of induction of the neuroectoderm marker Pax6. Samples prepared without RT were used as the negative control (-RT). (B) Quantitative RT-PCR data for cells stimulated in vitro as in (A). Data is expressed as fold change normalized to 18S expression. mRNA extracted on day 0 (blue columns) or day 5 (purple columns) of activin stimulation served as the template for qRT-PCR.
In order to determine if the differences in endodermal differentiation potential in vitro are also evident in iPS cells differentiating into other germ layers, iPS clones pre and post excision of the STEMCCA-loxP construct were exposed to defined culture conditions previously described to induce either mesoderm [26] or ectoderm [27] differentiation. As with endoderm formation, the ability of iPS cells to differentiate towards mesodermal flk1-expressing cells or neuroectodermal Tuj1-expressing cells was significantly enhanced in vitro by excision of the reprogramming transgenes (Supplemental online Fig. 4). Taken together these results underscore the importance of obtaining iPS cells free of exogenous transgenes, not only as a means to improve the safety profile of iPS cells but also to enable their appropriate differentiation potential.
iPS cells before and after excision of reprogramming transgenes display frequent trisomy of chromosome 8
We considered the possibility that Cre recombination in iPS cells might cause chromosomal instability, such as translocation events. Hence, we performed karyotyping by light microscopy as well as spectral karyotyping analyses (SKY) on several iPS cell lines before and after Cre-mediated excision of STEMCCA-loxP. Importantly, no translocation events were detected in any of the clones analyzed strongly suggesting that removal of the single integration by Cre-excision has no deleterious effects on the genome of transgene-free iPS cells. However, we noted that two out of eight iPS clones (25%) generated with the STEMCCA-loxP and one out of nine iPS clones generated with the STEMCCA-RedLight-loxP (11%) displayed trisomy of chromosome 8 (Fig. 5A). This chromosomal abnormality appeared to be present prior to Cre-mediated excision of STEMCCA-loxP, but was not present in the parental fibroblast cell line prior to reprogramming (Fig. 5B). In some cells analyzed by SKY (Fig. 5C) an apparently normal number of 40 mouse chromosomes masked the loss of the Y chromosome in combination with trisomy of chromosome 8. These chromosomal abnormalities have been well described in murine ESC with markedly increased frequencies with increased passage number [33, 34]. The appearance of trisomy 8 abnormalities observed in our iPS cell clones was already present by passage 8 and likely favored their outgrowth during culture, as has been described in ESC.
Figure 5.
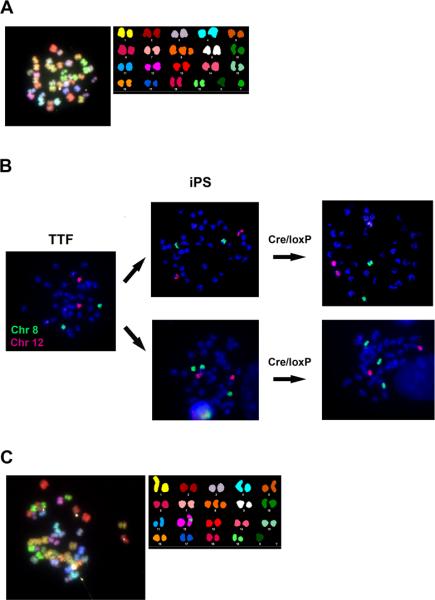
iPS cell clones are free of chromosomal translocations but display trisomy of chromosome 8. (A) Representative metaphase of an iPS clone analyzed by SKY shows no detectable chromosomal translocations and the presence of chromosome 8 trisomy. (B) Chromosome painting demonstrates normal diploid number of chromosome 8 (green probe) in parental tail tip fibroblasts (TTF). After reprogramming, representative iPS clones displaying two (top row) or three (bottom row) copies of chromosome 8 are shown at passage 8 prior to Cre-mediated STEMCCA-loxP excision. No changes in chromosome number were evident after Cre treatment. A red probe for chromosome 12 was used as a control. (C) Representative SKY analysis of an iPS clone displaying an apparently normal number of 40 chromosomes produced by trisomy of chromosome 8 together with the loss of chromosome Y.
DISCUSSION
Our results demonstrate one approach for deriving `transgene-free' iPS cells using a floxed single copy of a polycistronic reprogramming vector. This method allows head-to-head comparisons of the functional capacity of iPS cells before and after excision of reprogramming transgenes. Our findings emphasize the importance of vector excision prior to directed differentiation of iPS cells in culture, if the goal is to adapt differentiation culture conditions originally developed in ESC culture systems. Our findings, however, do not prove that `transgene-free' iPS cells are functionally equivalent to ESC in terms of differentiation capacity. Although our iPS cell lines performed well in endoderm-promoting culture conditions in comparison to the one ESC control line employed in our studies, multiple ESC lines will need to be compared to multiple isogenic `transgene-free' iPS cell lines in culture in order to perform a direct evaluation of the differentiation capacity of ESC vs iPS cells in future studies. Our approach of comparing each transgene-free iPS cell clone to its pre-excision parent, however, allows direct head-to-head comparisons of the effects of transgene excision on clonally-derived cells, while controlling for any potential effects of genetic background, reprogramming methodology or integrant copy number.
To our knowledge this is the first evaluation of the in vitro endodermal potential of iPS cells in serum-free defined culture conditions. In order to generate differentiated precursor cells for modeling or treating human diseases, it is crucial to first show that iPS cells can be directed in culture to recapitulate the sequence of developmental milestones involved in germ layer formation and differentiation. Our results suggest that transgene-free iPS cells upregulate an endodermal differentiation program in response to the same serum-free culture conditions previously employed to derive definitive endoderm from ESC [24]. Although persistent expression of reprogramming factors diminished the response of iPS cells to activin in vitro, the precise mechanism for this effect is not clear. During differentiation stem cells exhibit downregulation of loci encoding pluripotent regulators accompanied by activation of master transcriptional regulators of differentiation. Our results suggest that persistent expression of reprogramming factors interferes with appropriate downregulation of pluripotent loci, such as Rex1 and Esg1 (Fig. 4). Whether persistence of a general pluripotent gene program, or any particular specific reprogramming factor directly resulted in failure to upregulate the endodermal transcriptional regulators Foxa2, Sox17, and Gata4/6 will require further investigation. Recent work suggests that cMyc over-expression during reprogramming leads to downregulation of somatic differentiation gene programs, whereas Oct4, Klf4, and Sox2 over-expression triggers activation of pluripotency [35]. It is conceivable that similar mechanisms account for our findings when reprogrammed cells are then stimulated to differentiate.
The development of methods to derive iPS cells free of exogenous genetic material is particularly important if iPS cells are to be employed for regenerative therapies in human trials. By excising all reprogramming transgenes our approach eliminates the risk of oncogenic transgene reactivation following transplantation of iPS-derived cells. In addition, by excision of only a single vector copy our approach minimizes the risk of chromosomal translocations, an advance over prior methods for Cre-mediated excision of multiple copies of individual reprogramming lentiviral vectors [21]. It should be emphasized, however, that our approach leaves approximately 200 bp of exogenous DNA, primarily viral LTR, in the genome of iPS cells. Although the viral LTR is inactivated in our studies, possessing no intrinsic promoter/enhancer activity [36], it does not completely eliminate the theoretical risk of insertional mutagenesis that may arise from genomically integrated exogenous DNA. While the statistical probability that a single integration event can induce gene disruption and dysregulation is minimal, the simple sequencing of the site of integration would further expose potential dangerous clones. This minimal risk could be further reduced in the future by targeting the STEMCCA into a safe genomic locus. Alternatively, efficient reprogramming methods using small molecules or other methods free of exogenous DNA may still be required in order to further optimize the safety profile of iPS cells.
It should be noted that a number of technical hurdles complicate the use of Cre-mediated excision of DNA sequences from stem cells, potentially limiting the application of these methods to easily generate iPS cells free of floxed transgenes. First, delivery of Cre to ESC or iPS cells has been previously noted to be inefficient; second, screening methods to detect successful Cre-recombination may be cumbersome; and third, clumping of cells after delivery of Cre may result in mosaic colonies containing some cells that have failed to undergo Cre-recombination. Our mCherry-containing STEMCCA-loxP-RedLight vector should be particularly helpful in surmounting these hurdles, since this single reagent accomplishes effective `3 factor reprogramming', and our results demonstrate that Cre-recombination efficiency and excision of reprogramming transgenes from the resulting iPS cells can be readily visualized and monitored in individual living cells and colonies in culture. Indeed, we found adenoviral delivery of Cre as well as Cre recombination to be highly efficient in iPS cells using monitoring of mCherry disappearance to optimize our transgene excision methodology. Furthermore, FACS sorting based on mCherry expression may be easily employed by those investigators wishing to rapidly separate excised from unexcised iPS cells.
We found trisomy of chromosome 8 to be frequent in our iPS cell lines independent of Cre-mediated excision of reprogramming transgenes. This trisomy is common in ESC but has not been reported previously in iPS cells. While this observation further emphasizes the similarity between iPS cells and ESC, it is not yet known whether the overexpression of reprogramming factors facilitates this chromosomal abnormality. In ESC, frequency of this trisomy increases with passage number, and it is possible that the several passages required to generate stable iPS cells during the gradual reprogramming process allow for more prevalent trisomies in iPS cells than has been previously appreciated. The additional passages required to screen for iPS cell clones with single vector copies, perform vector excision, re-screen for excision, and characterize the transgene-free clones may be particularly problematic in terms of chromosomal instability. For this reason, methods to reprogram cells, maintain chromosomal stability, and excise residual transgenes with the highest efficiency and the lowest possible passage number will be important if iPS cells are to be translated for human therapies. We propose the use of the STEMCCA-loxP-RedLight vector as a useful tool for accomplishing this goal, since excision of the mCherry `RedLight' serves as a simple indicator, requiring minimal screening, for the generation of transgene-free iPS cells.
CONCLUSION
We demonstrate efficient derivation of transgene-free iPS cells using an excisable polycistronic lentiviral vector. A direct comparison of iPS cell clones before and after excision reveals that removal of the reprogramming vector markedly improves the developmental potential of iPS cells and significantly augments their capacity to undergo directed differentiation in vitro. In addition, our results underscore the consequences of long-term culture that will need to be taken into account for the clinical application of iPS cells.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Greg Martin and Katya Ravid from the Transgenic Center at Boston University School of Medicine for technical expertise; Julia Bianco for technical help with karyotyping and SKY; Isabel Dominguez, Matthias Stadtfeld, Konrad Hochedlinger, Justin Ichida, Valerie Gouon-Evans, and Paul Gadue for invaluable advice; Linda Neville and Sarah Ohle for administrative assistance.
Footnotes
Author Contributions C.A.S.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; A.G.S.: conception and design, collection and assembly of data, data analysis and interpretation; T.L., C.C. and D.D.T.: collection and assembly of data; M.G. and F.W.A.: collection and assembly of data; G.J.M: conception and design, data analysis and interpretation; D.N.K and G.M.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
REFERENCES
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 3.Maherali N, Sridharan R, Xie W, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 7.Brambrink T, Foreman R, Welstead GG, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadtfeld M, Maherali N, Breault DT, et al. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 12.Park IH, Arora N, Huo H, et al. Disease-Specific Induced Pluripotent Stem Cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 14.Kopp JL, Ormsbee BD, Desler M, et al. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells. 2008;26:903–911. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 15.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 16.Okita K, Nakagawa M, Hyenjong H, et al. Generation of Mouse Induced Pluripotent Stem Cells Without Viral Vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 17.Stadtfeld M, Nagaya M, Utikal J, et al. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woltjen K, Michael IP, Mohseni P, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geurts AM, Collier LS, Geurts JL, et al. Gene mutations and genomic rearrangements in the mouse as a result of transposon mobilization from chromosomal concatemers. PLoS Genet. 2006;2:e156. doi: 10.1371/journal.pgen.0020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang W, Lin C, Lu D, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:9290–9295. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soldner F, Hockemeyer D, Beard C, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommer CA, Stadtfeld M, Murphy GJ, et al. iPS Cell Generation Using a Single Lentiviral Stem Cell Cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. Epub December 18 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mostoslavsky G, Fabian AJ, Rooney S, et al. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci U S A. 2006;103:16406–16411. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouon-Evans V, Boussemart L, Gadue P, et al. BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat Biotechnol. 2006;24:1402–1411. doi: 10.1038/nbt1258. [DOI] [PubMed] [Google Scholar]
- 25.Gadue P, Huber TL, Nostro MC, et al. Germ layer induction from embryonic stem cells. Exp Hematol. 2005;33:955–964. doi: 10.1016/j.exphem.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Nostro MC, Cheng X, Keller GM, Gadue P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell. 2008;2(1):60–71. doi: 10.1016/j.stem.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Giorgio FP, Carrasco MA, Siao, et al. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat. Neuroscience. 2007;10:608–614. doi: 10.1038/nn1885. Published online 15 April 2007 doi:10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Crussi F. Extragonadal Teratomas. Armed Forces Institute of Pathology; Washington D.C.: 1982. [Google Scholar]
- 29.Franco S, Gostissa M, Zha S, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Wernig M, Lengner CJ, Hanna J, et al. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 32.Kubo A, Shinozaki K, Shannon JM, et al. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131:1651–1662. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 33.Ensenat-Waser R, Santana A, Vicente-Salar N, et al. Isolation and characterization of residual undifferentiated mouse embryonic stem cells from embryoid body cultures by fluorescence tracking. In Vitro Cell Dev Biol Anim. 2006;42:115–123. doi: 10.1290/0509063.1. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Wu H, Loring J, et al. Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev Dyn. 1997;209:85–91. doi: 10.1002/(SICI)1097-0177(199705)209:1<85::AID-AJA8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 35.Sridharan R, Tchieu J, Mason MJ, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.