

Summary
Cis-regulatory changes play a central role in morphological divergence, yet the regulatory principles underlying emergence of human traits remain poorly understood. Here we use epigenomic profiling from human and chimpanzee cranial neural crest cells to systematically and quantitatively annotate divergence of craniofacial cis-regulatory landscapes. Epigenomic divergence is attributable to genetic variation within TF motifs at orthologous enhancers, with a novel motif being most predictive of activity biases. We explore properties of this cis-regulatory change, revealing the role of particular retroelements, uncovering broad clusters of species-biased enhancers near genes associated with human facial variation, and demonstrating that cis-regulatory divergence is linked to quantitative expression differences of crucial neural crest regulators. Our work provides a wealth of candidates for future evolutionary studies and demonstrates the value of ‘cellular anthropology’, a strategy of using in vitro-derived embryonic cell types to elucidate both fundamental and evolving mechanisms underlying morphological variation in higher primates.
Keywords: cis-regulatory, evolution, enhancers, neural crest, chimpanzee, craniofacial
Graphical Abstract
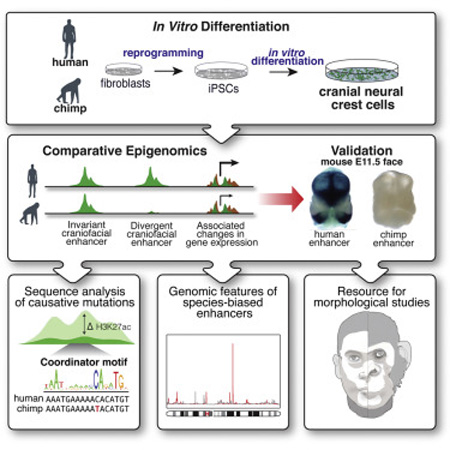
Introduction
Since the discovery that the protein-coding regions of the genome remain largely conserved between humans and chimpanzees, it has long been postulated that morphological divergence between closely related species is driven principally through quantitative and spatiotemporal changes in gene expression, mediated by alterations in cis-regulatory elements (Carroll, 2008; King and Wilson, 1975; Wray, 2007). A number of excellent case studies has validated these early predictions and demonstrated that mutations or deletions affecting distal regulatory elements called enhancers can alter ecologically-relevant traits (Gompel et al., 2005; Shapiro et al., 2004; Attanasio et al., 2013). Recent successes in full genome sequencing and epigenomic strategies have enabled the first genome-wide comparisons of transcription factor (TF) binding and regulatory landscapes in closely related species, demonstrating the value of comparative epigenomics in the context of high genome orthology for understanding principles of cis-regulatory evolution (Bradley et al., 2010; He et al., 2011; Stefflova et al., 2013). Nonetheless, despite the availability of human and chimpanzee genomes, our knowledge of cis-regulatory divergence between humans and our closest evolutionary relatives remains fairly speculative. Previous efforts have relied heavily on computational approaches to pinpoint conserved non-coding elements that were either deleted or had undergone accelerated change specifically in the human lineage (McLean et al., 2011; Pollard et al., 2006; Prabhakar et al., 2006). Functional epigenomic comparisons between humans and other primates have been largely limited to lymphoblastoid cell lines (Cain et al., 2011; Shibata et al., 2012; Zhou et al., 2014;) or to profiling whole organs from more distantly related species (Cotney et al., 2013; Villar et al., 2015).
Recently, iPSCs were made available from our nearest living evolutionary relative the chimpanzee (Marchetto et al., 2013), offering an opportunity to derive developmentally-relevant and previously-inaccessible tissue types in vitro. This allows aspects of species-specific development to be recapitulated in a dish, facilitating ‘cellular anthropology’ through the discovery of cell type-specific regulatory changes that occurred during recent human evolution. Here we focus on the neural crest (NC), one of the embryonic cell populations most relevant to emergence of uniquely human traits. In vivo, NC cells (NCCs) arise during weeks ~3–5 of human gestation from the dorsal part of the neural tube ectoderm, and migrate into the branchial arches and what will later become the embryonic face, consequently establishing the central plan of facial morphology (Bronner and LeDouarin, 2012; Cordero et al., 2011; Jheon and Schneider, 2009). Within our recent evolutionary history, the modern human craniofacial complex has undergone dramatic changes in shape and sensory organ function, which helped to build a recognizably human face and were required to accommodate the transition to bipedal posture, enlargement of the brain, extension of the larynx for speech, and compensatory rotations of the orbits, olfactory bulb and nasomaxillary complex. (Bilsborough and Wood, 1988; Lieberman, 1998; Spoor et al., 1994).
To overcome the inability to obtain cranial NCCs (CNCCs) directly from higher primate embryos, we here employ a pluripotent stem cell-based in vitro differentiation model in which specification, migration and maturation of human and chimpanzee CNCCs are recapitulated in the dish (Bajpai et al., 2010; Rada-Iglesias et al., 2012, this study). We compared TF and coactivator binding, histone modifications and chromatin accessibility genome-wide to annotate the divergent regulatory element repertoire of human and chimpanzee CNCCs. This information allowed us to explore, with unprecedented comprehensiveness and resolution, the mechanisms of tissue-specific enhancer landscape evolution within a developmentally-relevant tissue type in humans and our nearest evolutionary relative.
Results
Derivation of human and chimpanzee CNCCs
Given the similarities in hominid gestational environment, we hypothesized that non-human primate CNCCs could be derived from pluripotent cells using the same cell culture conditions that we have previously applied to human embryonic stem cells (ESCs)/iPSCs (Bajpai et al., 2010; Rada-Iglesias et al., 2012). Chimp iPSCs have recently become available and can be maintained in vitro under identical conditions as human ESCs/iPSCs (Marchetto et al., 2013). Upon differentiation of our chimp iPSCs, we observed formation of highly-mobile stellate cells that were morphologically indistinguishable from human CNCCs, expressed a broad range of migratory NC markers at levels equivalent to those seen in human cells, and had very low level of HOX gene expression, a profile consistent with CNCC identity (Figures 1A–C, S1A; Figures 1B, 1C). To characterize staging and homogeneity of our human and chimp CNCC populations, we identified a panel of five cluster of differentiation (CD) markers, whose expression is sensitive to the developmental progression of CNCC (see Methods, Figure S1B). These markers provided a platform for us to monitor and optimize our cell culture protocol for derivation and maintenance of primate CNCCs achieving metrics of homogeneity greater than 90% regardless of the genetic background, initial cell source (e.g. iPSC vs ESC), or species (human vs chimp); see Figure S1C and Methods. Cultured primate CNCCs show a high correlation of expression signatures and epigenomic profiles with CNCCs isolated from chick embryos, reinforcing the NC identity of these in vitro-derived cells (Figures S2A, S2B). Importantly, derived human and chimp CNCCs are both capable of prolonged maintenance (for up to 18 passages) and sustained differentiation capacity into both mesenchymal and non-mesenchymal lineages (Figure S2C). Furthermore, xenotransplantation of cultured human and chimp CNCCs into the dorsal neural tube of early chick embryos demonstrates their ability to engraft and then follow endogenous migration cues into the distal branchial arches (Figures S2D, S2E).
Figure 1. Derivation of human and chimpanzee CNCCs and epigenomic annotation of craniofacial enhancers.
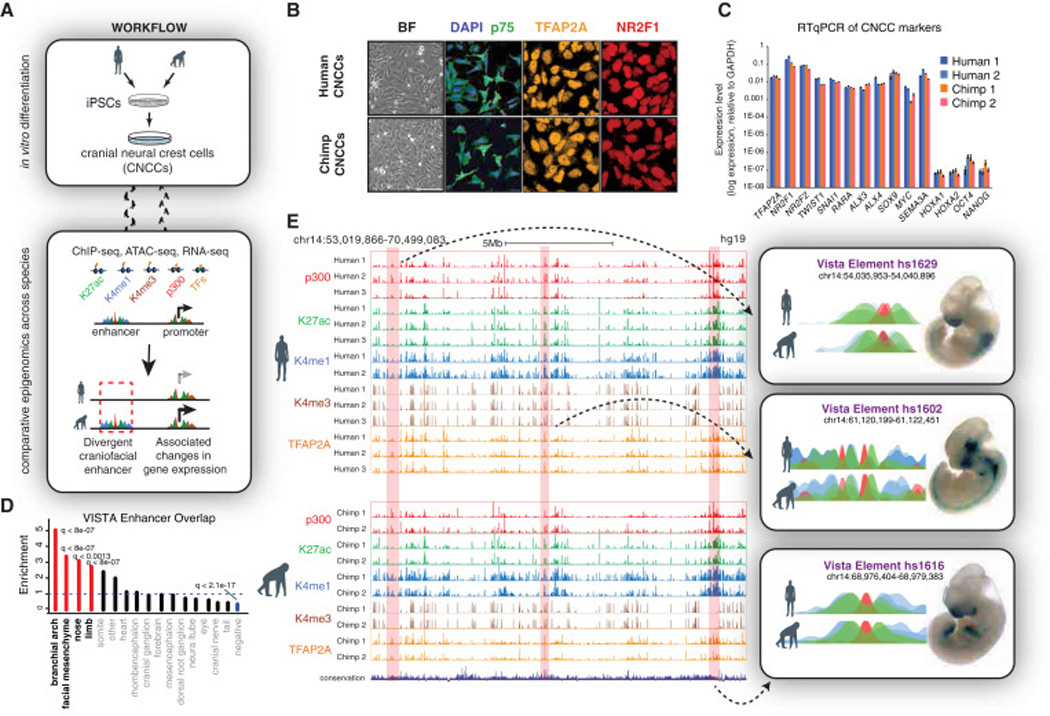
(A) Workflow of comparative epigenomic strategy.
(B) Confocal immunofluorescence detection of NC markers p75, TFAP2A, and NR2F1 in human and chimp CNCCs at passage 4.
(C) RT-qPCR of NC markers, HOXs and pluripotency markers OCT4 and NANOG in derived human and chimp CNCCs from 2 genetic backgrounds of each species.
(D) Enrichment of annotated expression domain categories from overlap of top 15,000 enhancer calls with regions in the VISTA enhancer database. P-values were calculated with Fisher’s exact test and corrected for pFDR. Categories with q-value < 0.05 are indicated in red (enrichment) or blue (depletion).
(E) Representative UCSC Genome Browser tracks showing ChIP-seq profiles for p300 (red), H3K27ac (green), H3K4me1 (blue), H3K4me3 (brown), and TFAP2A (orange) from both species aligned to hg19 reference genome. Representative elements tested through the VISTA enhancer database (Visel et al., 2007) displayed on the right next to the reported lacZ expression domains.
Epigenomic profiling of human and chimpanzee CNCCs
For epigenomic profiling, we derived CNCCs from H9 hESCs and from iPSCs from two humans and two chimpanzees (Marchetto et al., 2013). We subsequently performed chromatin immunoprecipitation and sequencing (ChIP-seq) using antibodies against CNCC TFs (TFAP2A and NR2F1), a general coactivator (p300) and histone modifications associated with active regulatory elements (H3K4me1, H3K4me3, and H3K27ac) (Figures 1A, 1E). In parallel, we mapped genome-wide chromatin accessibility using an assay for transposase-accessible chromatin (ATAC-seq) (Buenrostro et al., 2013).
One crucial advantage of performing comparative epigenomics between human and chimpanzee, as opposed to a more distant primate relative, is the large similarity between genomes which permits reciprocal mapping of sequencing reads to the reference genomes of both species. This allows for quantification of read enrichments from each species in the context of both reference genomes, removing otherwise difficult-to-control-for biases due to mappability, ambiguous liftOver and other technical caveats. Importantly, we could unambiguously assign one-to-one orthology between genomes for over 95% of all enhancer candidates from either species, with the remaining 4–5% representing enhancers that fall within putative species-specific structural variants. We found that enrichments for all ChIP-ed factors and for chromatin accessibility were largely independent of the chosen reference genome, and excluded all candidate elements for whom enrichment divergence was dependent upon the reference (<0.1%) or that did not map uniquely in both genomes (see Experimental Procedures). Globally, the observed epigenomic patterns at candidate regions were highly correlated for human and chimp CNCCs (Figure 1E, Figure S4A).
Genome-wide annotation of human and chimpanzee CNCC regulatory elements uncovers enhancers with craniofacial activity
To annotate enhancers genome-wide, we promiscuously identified candidate cis-regulatory regions by the presence of TF or p300 enrichment, and/or increased chromatin accessibility. We then restricted our analysis primarily to enhancers by assessing the ratio of H3K4me1/H3K4me3 enrichment at these candidate sites, which distinguishes distal enhancers from promoters (Heintzman et al., 2007), and further using H3K27ac enrichment to differentiate active from inactive elements (Creyghton et al., 2010; Rada-Iglesias et al., 2011). The resulting enhancer candidates had enriched conservation signatures compared to surrounding genomic regions and were near genes annotated with craniofacial ontologies – consistent with bona fide NC enhancer status (Figures S3A–C). Furthermore, cross-referencing our enhancer list with the VISTA Enhancer Browser database (Visel et al., 2007) identified 247 regions overlapping CNCC enhancers that were functionally tested for activity in mouse embryos. Of those 247 regions: (i) 208 were active at E11.5 (odds ratio 6.33 and p < 5×10−32) and, (ii) these 208 active enhancers were significantly enriched for activity in NC-derived head tissues (branchial arches and facial mesenchyme; Figure 1D, examples are shown in Figures 1E (right), S3D). Thus, our analysis captures regulatory regions relevant for distinct spatial identities in the developing face in vivo (Figure 1D). Taken together, our epigenomic approach thus comprehensively annotates putative human and chimp NC enhancers, at least a subset of which is active in facial structures during embryogenesis.
Quantitative analysis of H3K27ac enrichments predicts species-biased enhancers
We hypothesized that in closely related species, quantitative modulation of activity at orthologous regions is a major form of enhancer divergence. To identify such divergence, we used H3K27ac enrichment data in biological quadruplicate (i.e., independent CNCC derivations from each individual) to quantitatively approximate activity at all annotated CNCC enhancers detected for either species. Global comparisons of H3K27ac enrichments between individuals of the same species revealed high concordance of signals with some minor variation due to either differences in genetic background or experimental variability (Figure 2A, highlighted in red, Figure S4A). Human and chimpanzee CNCC H3K27ac enrichment was also highly correlated when mapped to the same reference genome, and human and chimpanzee CNCC H3K27ac profiles clustered together distinctly from 49 other human cell types (Figures S4A–B). Despite this high conservation of profiles, a substantial subset of elements demonstrated a significant species bias (Figure 2A, FDR<0.01 highlighted in blue), which we thereafter considered to be our species-biased enhancer candidates. H3K27ac ChIP-qPCR at select candidate enhancers from independent CNCC derivations recapitulated this species-bias (Figure S4C).
Figure 2. Identification of species-biased enhancers using H3K27ac enrichments at orthologous loci.
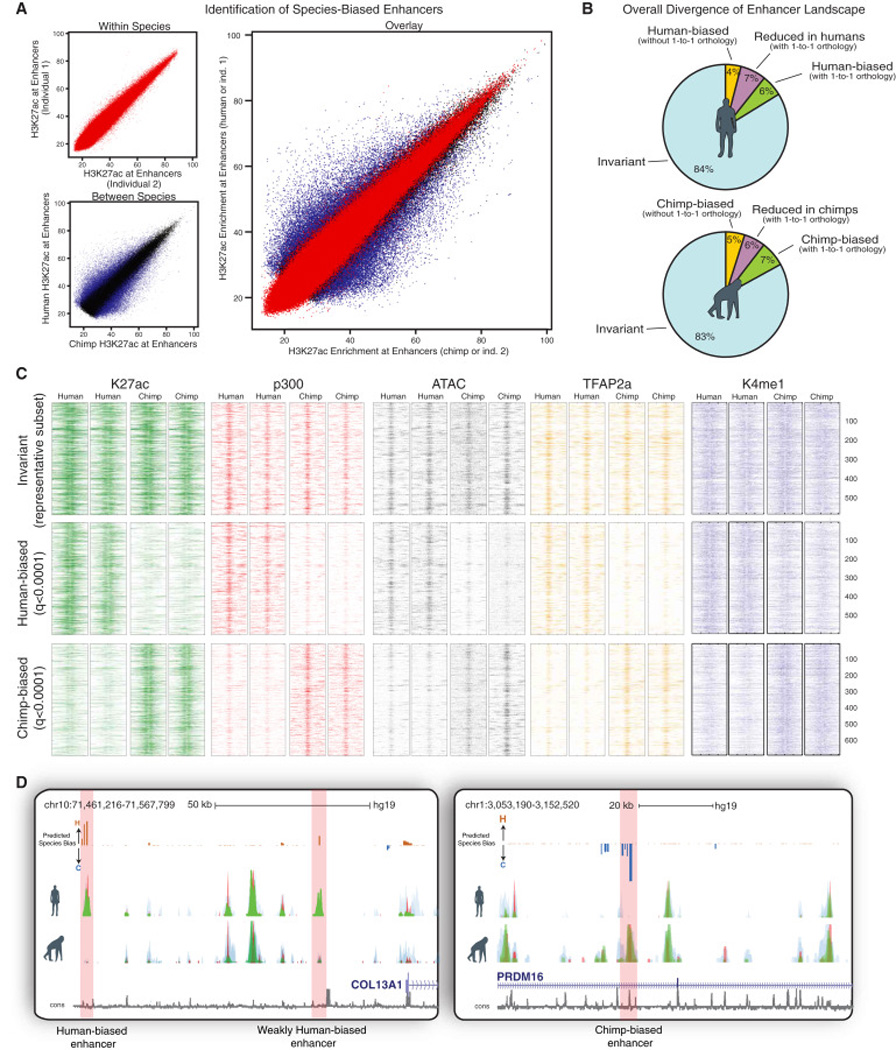
(A) Enrichment of H3K27ac at candidate enhancer elements compared within individuals of the same species (red) or across species (blue/black), with overlay shown on the right. Enhancers with significant inter-species divergence indicated in blue (padj<0.01).
(B) Pie charts showing the percentage of total active CNCC enhancers classified as either species-biased enhancers with gained activity (green), species-biased enhancers with decreased activity (purple), enhancers without clear orthology across genomes (yellow), or invariant enhancers (blue) using a human reference genome (above) or chimp reference genome (below).
(C) Heat map of raw ChIP-seq and ATAC-seq counts across species-biased and invariant CNCC enhancers for two human and two chimp genetic backgrounds. Each row represents a 2kb window (1kb each direction) centered around the middle of human-biased (n=598, q<0.0001), chimp-biased (n= 691, q<0.0001) or invariant (n=584 representative subset, q>0.95) enhancers for H3K27ac (green), p300 (red), TFAP2A (yellow), K4me1 (blue) and ATAC-seq (gray). All reads were aligned to hg19.
(D) Representative browser tracks showing overlaid H3K4me1 (blue), p300 (red) and H3K27ac (green) from human and chimp CNCCs mapped to hg19. Examples of strongly human-biased, weakly human-biased, or strongly chimp-biased enhancers highlighted in pink. Predicted species-bias track shown above for candidate enhancers, the magnitude of the bias track represents -log10 (adjusted p-value of divergence) with negative sign (indigo) representing chimp bias and positive (bronze) human bias.
Importantly, consistent with the premise that H3K27ac is a suitable readout of enhancer activity, the bias in H3K27ac status alone was highly predictive of biases in TF and p300 binding, as well as chromatin accessibility (Figure 2C; examples are shown in Figures 2D, S4D). Furthermore, this approach enabled genome-wide assignment of signed significance scores on a per-enhancer basis, visualizable as a genome browser track (Figure 2D, “Predicted Species Bias” track).
Altogether, of all annotated active human CNCC enhancers (n = 14,606), 84% were invariant, 4% fell at non-orthologous sites, and 6% and 7% demonstrated quantitative increase or decrease respectively (Figure 2B). One limitation is the low number of currently available chimpanzee iPSC lines, especially given the high reported degree of polymorphism among chimps (Kaessmann et al., 1999). To estimate false positive rate for identifying true fixed inter-species differences we applied our strategy to previously published ChIP-seqs from chimp lymphoblastoid cell lines and estimated a conservative FDR of 0.15 when using only two chimp genetic backgrounds. This suggests that the vast majority of identified differences represent functionally fixed differences across species (the rest represent enhancers that are still divergent but remain polymorphic within one of the species). Our observations agree with the emerging notion that quantitative modulation of enhancer activity is the prevalent source of regulatory landscape divergence among closely related species.
Cis-sequence changes drive species-biased enhancer activity in vitro and in vivo
To functionally validate our predictions we used a luciferase reporter assay to examine activity of a selected set of orthologous pairs of species-biased human and chimpanzee enhancers. We found that >80% of tested enhancers had correlated species-bias in luciferase expression, which was consistent regardless of whether the reporter assays were performed in human or chimpanzee CNCCs (Figure 3A–B). These results further validate that H3K27ac identifies both enhancer activity and bona fide species-bias; thus, for simplicity we refer to H3K27ac enrichment interchangeably with “activity”. Importantly, these results also demonstrate that enhancer divergence can be largely explained by cis-sequence changes rather than differences in the trans regulatory environments of the human and chimp CNCCs.
Figure 3. In vitro and in vivo validations of species-biased enhancers.
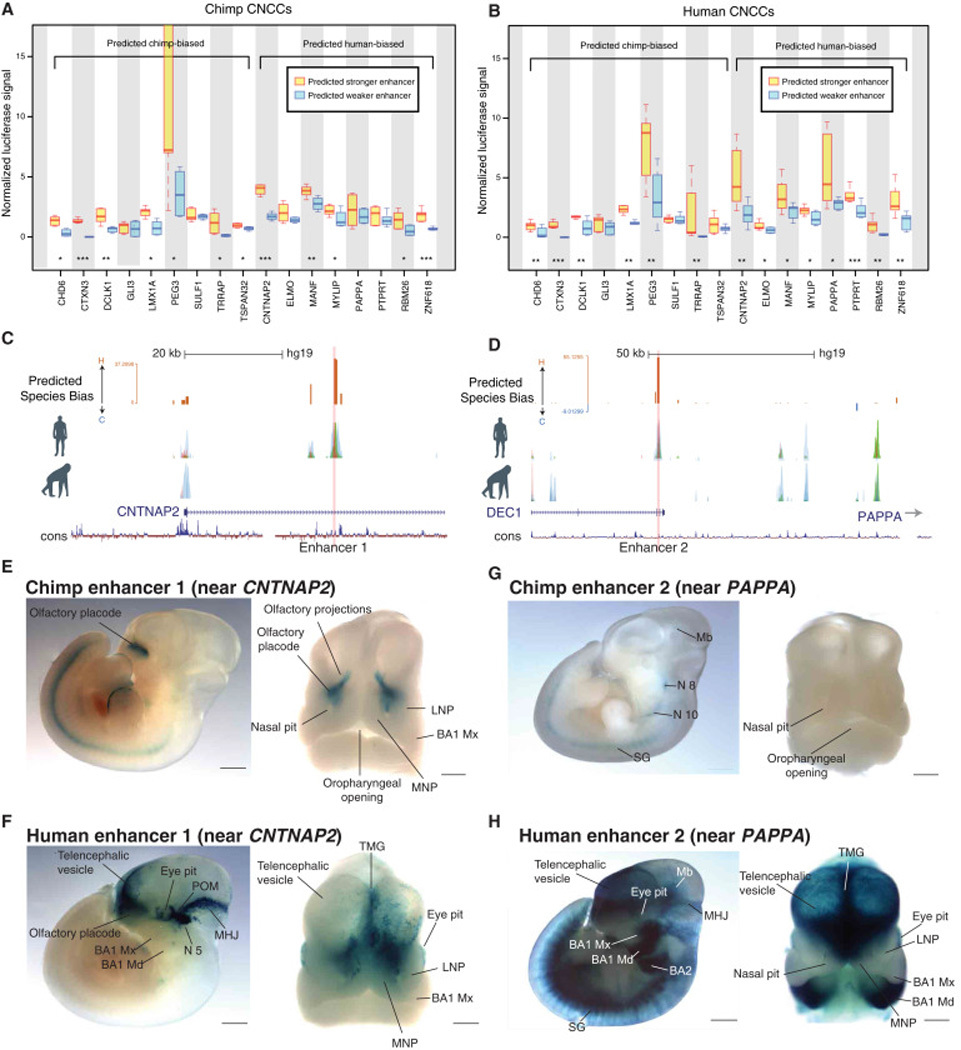
(A) –(B) Luciferase reporter assays performed in chimp CNCCs (A) or human CNCCs (B) for 9 chimp-biased regions (and orthologous weak human enhancers) and 8 human-biased regions (and orthologous weak chimp enhancers). Luciferase signal was normalized to renilla transfection control. Significance tested from three biological replicates from each species with ANOVA followed by residuals testing with Student’s t-test, p-value indicators *<0.05, **<0.01 ***<0.001
(C)–(D) Genome browser tracks showing human-biased Enhancer 1 (near CNTNAP2 gene; C) and Enhancer 2 (near PAPPA gene; D) selected for a lacZ reporter mouse transgenesis assay.
(E)–(F) Analysis of enhancer activity for chimpanzee and human Enhancer 1 in a lacZ reporter transgenic mouse assay. (E) Representative E11.5 transgenic embryo obtained for the chimpanzee Enhancer 1 reporter, shown in lateral view (left) or frontal view (right) of the embryonic head. (F) Representative E11.5 transgenic embryo obtained for the human Enhancer 1 reporter, shown in lateral view (left) or frontal view (right). Midbrain/hindbrain junction (MHJ); periocular mesenchyme (POM); lateral and medial nasal processes (LNP and MNP); maxillary (Mx) and mandibular (Md) processes of branchial arch 1 (BA1) and BA2. Scale bars: 100 µm (left images) and 50 µm (right images).
(G)–(H) Analysis of enhancer activity for chimpanzee and human Enhancer 2 in a lacZ reporter transgenic mouse assay. (G) Lateral view of representative E11.5 transgenic embryo obtained for the chimpanzee Enhancer 2 reporter, shown in lateral view (left) or frontal view (right) of the embryonic head. (H) Representative E11.5 transgenic embryo obtained for the human Enhancer 2 reporter, shown in lateral view (left) or frontal view (right). Midbrain (Mb); cranial nerves VIII and X (N8 and N10 respectively); sympathetic ganglia (SG) telencephalic midline groove (TMG); midbrain/hindbrain junction (MHJ); maxillary (Mx) and mandibular (Md) processes of branchial arch 1 (BA1) and BA2. Scale bars: 100 µm (left images) and 50 µm (right images).
The conservation of trans-environments across species facilitates testing of human and chimp regulatory elements in vivo using a mouse LacZ transgenic reporter assay. We selected two predicted human-biased enhancers near CNTNAP2 (Enhancer 1) and PAPPA (Enhancer 2), respectively (Figure 3C and D). For both predicted human-biased enhancers we observed gains of additional expression domains in head regions, as well as quantitative gains in enhancer strength, as evidenced by the overall higher LacZ staining intensity for the human sequence compared to the chimp ortholog (Figures 3C–H, Figure S5). Notably, to ensure that the negative/weak staining results obtained with the chimp sequences were not a result of undersampling, we performed surplus embryo injections with both chimp enhancer reporters (Figure S5A). Thus, species-biased enhancers identified in our in vitro analysis to drive distinct expression patterns of CNCC-derived tissues in vivo.
Human Accelerated Regions (HARs) overlap with distal CNCC enhancers
Our results suggest that DNA sequence is the predominant driver of enhancer divergence, therefore we began examining sequence properties of species-biased enhancers. Although species-biased enhancers were similar in H3K27ac enrichment levels when compared to invariant enhancers, they showed a distinct reduction of sequence conservation signatures (Figure 4A). Furthermore, we identified 163 ‘Human Accelerated Regions’ (HARs; Hubisz and Pollard, 2014) overlapping active chromatin features in CNCCs, of which 20 showed species-biased activity (at a cutoff of q<0.001; n=48 with a cutoff of q<0.1) (Figure 4B, Figure S6A–D), representing a significant enrichment relative to the whole enhancer set (p<0.025, odds ratio 1.81). It is possible that the HAR-overlapping regions without species-bias in CNCC could manifest divergence in another tissue type, as exemplified by HAR2 (a.k.a. HACNS1), which overlaps an invariant CNCC enhancer (Figure S6D, p-value of species-bias = 0.339) that has a pharyngeal arch activity domain which is conserved in primates, but has human-specific activity in the embryonic limb (Prabhakar et al., 2008).
Figure 4. Global features of species-biased enhancers and correlation of mutations within TF binding motifs with epigenomic divergence.
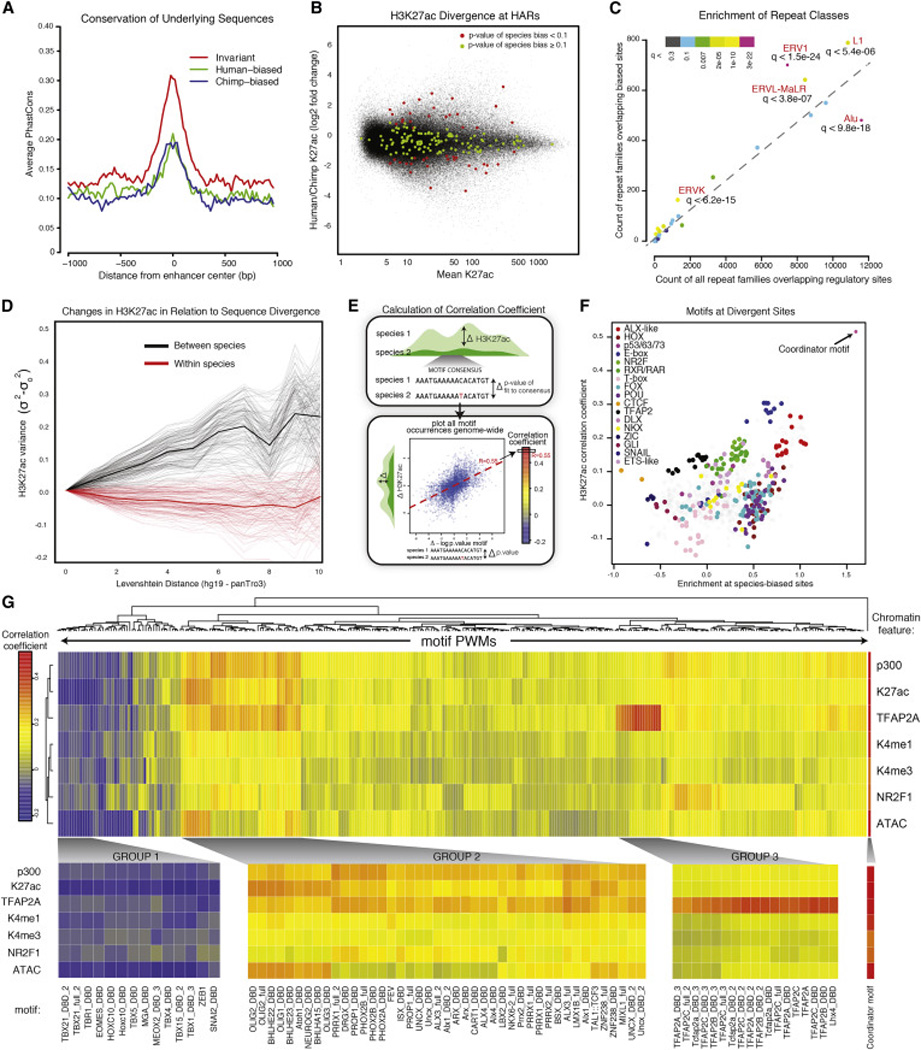
(A) Average PhastCons scores are shown for strong invariant enhancers (q-value > 0.98), strongly human-biased enhancers (q value < 0.0001) or strongly chimp-biased enhancers (q-value <0.0001) for 1kb surrounding each enhancer center.
(B) Degree of species bias (log2 fold change H3K27ac human/chimp, y axis) relative to enhancer strength (human-chimp averaged H3K27ac enrichment, x axis) for bulk CNCC elements (black) and elements overlapping HARs (color representing q-value of species-bias: q-value < 0.1 in red, q-value ≥ 0.1 in green).
(C) Counts of repeat families overlapping species-biased enhancers (y axis) relative to counts of repeat families overlapping all regulatory sites (x axis) are plotted. Q-score of enrichment for different repeat classes indicated by color.
(D) Pairwise H3K27ac variance σ2-σ2ld=0 at enhancers across samples, ranked by increasing sequence dissimilarity counted by Levenshtein distance (ld) between orthologous 200bp enhancers, relative to ld = 0. Comparison between samples of different species shown in black, same species shown in red (means represented by thick lines).
(E) Schematic showing method for deriving the correlation coefficient. For a given motif, each occurrence genome-wide containing a genetic change across species is plotted as Δ-log10 p-value (human/chimp) of the fit to consensus (x axis) vs. ΔH3K27ac for the overlying enhancer region (human/chimp) (y axis), then a line is fit. The slope of the line represents the correlation coefficient for that given motif and epigenomic modification genome-wide.
(F) Enrichments of classes of motifs at species-biased enhancers over all enhancers (log odds ratio, x axis) plotted relative to genome-wide correlation coefficient calculated for each motif (using H3K27ac), as described in panel E (y axis).
(G) Genome-wide correlation coefficients were calculated for whole databases of annotated motifs and multiple chromatin features, revealing motifs with large influence on epigenomic profiles. Correlation coefficients are bi-clustered per motif, and resulting changes in enrichment of chromatin features (p300, K27ac, TFAP2A, H3K4me1, H3K4me3, NR2F1, ATAC) at all enhancers containing mutated PWMs are represented by color. Individual subclusters are magnified below with corresponding motifs indicated.
Species-biased enhancers are enriched for specific classes of retroelements
Given that nearly half of the human genome is composed of transposable elements the majority of which invaded the primate lineage prior to the separation of humans and chimpanzees (Cordaux and Batzer, 2009), we hypothesized that a subset of species-biased orthologous enhancers may be transposon-derived. Interestingly, we found that while CNCC enhancers overlapped with many different classes of repeats, specific subclasses of endogenous retroviruses (ERV1, ERVL-MaLR and ERVK) as well as L1 elements were preferentially enriched at species-biased enhancers (Figure 4C), suggesting that these specific subclasses may harbor progenitor sequences that are prone to acquire CNCC enhancer activity over relatively short evolutionary distances.
Sequence substitutions within TF binding motifs at species-biased enhancers contribute to epigenomic divergence
Consistent with the expectation that species-specific biases are largely sequence-driven, we observed that the variance in H3K27ac between species at each enhancer scales proportionally with the degree of sequence dissimilarity (i.e., Levenshtein distance) at those orthologous sites, while the intra-species variance at the same regions remains unchanged (Figure 4D). Nonetheless, even at enhancers with detectable species bias, sequence substitutions were still infrequent - only ~3 to 6 substitutions per 500 bp enhancer - suggesting that a small number of mutations can confer substantial effects on overall enhancer activity, likely by affecting binding of key sequence-dependent TFs. We therefore interrogated how frequently sequence substitutions fall within particular classes of TF motifs and to what degree these mutations correlate, either positively or negatively, with changes in enhancer activity or other chromatin modifications (Figure 4E). This in essence leverages preexisting genetic variation like a large-scale mutagenesis screen.
Through this approach we identified a large set of both known and novel motifs for which deviation from the consensus was correlated with species-bias of H3K27ac and other epigenomic marks, implying functional consequences for these mutations. As expected, the correlations vary in frequency and in effect, with some motifs being frequent and having small effects (e.g., Forkhead factors), while others being infrequent but conferring large effects (e.g., TFAP2A), with one outlier motif being both very frequent and conferring large effects when mutated (see description of the ‘Coordinator’ motif below) (Figure 4F). Among our top hits we identified many motifs for TFs with known effects in NC regulation, including a set of TFAP2 motif variants that serve as a positive control for our approach, as we see a high correlation between TFAP2 motif mutations and inter-species divergence in TFAP2A ChIP signals at these sites (Figures 4G, Group 3). We previously showed that TFAP2A participates in establishment of active chromatin states at NC enhancers (Rada-Iglesias et al., 2012), and consistently we observed that divergence from the TFAP2A consensus also correlates with the loss of H3K27ac, co-activator binding and chromatin accessibility. Notably, TFAP2 motifs are depleted from species-biased sites, likely due to strong selective pressure to conserve TFAP2A function in the NC and possibly in other pleiotropic contexts (Figure 4F). Another interesting set of motifs, which are both frequent at species-biased sites and positively correlated with permissive chromatin states, are those recognized by ALX homeobox factors that are highly expressed in the face and mutated in severe frontonasal dysplasias in humans (Twigg et al., 2009).(Figure 4F, 4G - Group 2).
Intriguingly, we also identified a group of motifs whose mutations away from the consensus were correlated with a gain in chromatin accessibility and H3K27ac, suggesting that these motifs may recruit repressive factors with negative effects on overall enhancer activity. Examples of such motifs included the SNAI2 motif, which is bound by a known transcriptional repressor, the TBX-family motif bound by T-box factors, and other candidate negative regulators representing distinct TF classes, e.g. HIC1/2, MESP1, TCF3/4, and GLIS1 (Figures 4G, Group 1). These results suggest an unappreciated prevalence of repressive inputs in quantitative modulation of enhancer activity.
‘Coordinator’: a novel motif that is highly predictive of active chromatin states and species-bias
Surprisingly, one motif stood out as an outlier in this analysis as it was exceptionally enriched at divergent sites and was the most correlated with changes in all examined active chromatin features (Figures 4F, top right, 4G, far right). This sequence, which we termed the ‘Coordinator’ motif, is a 17bp-long motif, which we identified through de novo motif discovery from our CNCC-specific enhancers and was not previously annotated to a known regulatory complex. We note that portions of the Coordinator resemble an E-box and HOX-like motifs, however these represent large protein families and the particular factors that bind at this element remain to be identified.
Sequence analysis using INSIGHT, a tool to infer signatures of recent natural selection using human polymorphism data (Gronau et al., 2013), found evidence of positive selection at the Coordinator motif occurrences within species-biased enhancers, but not within invariant enhancers, suggesting that the motif and its cognate binder(s) have played a privileged role in recent enhancer divergence in primate CNCCs (Figure 5A). When we further dissected the motif by individual bases, we found that the correlations of each nucleotide with ChIP enrichments (both for histone modifications and TF ChIPs) recapitulated the information content of the motif itself as would be expected if Coordinator motif mutations were causal for the observed chromatin changes (Figure 5B). Fittingly, we found human mutations that strengthen the Coordinator motif within both human-biased enhancers tested in mouse transgenesis (Figure S6E). Globally, the Coordinator motif was preferentially enriched at distal regulatory elements rather than at promoters (Figure S6F), and further enriched at enhancers that were CNCC-specific as opposed to those that shared measurable H3K27ac in other tissue types (Figure 5C). Interestingly, we observe that LTR9 elements, a retroelement class enriched at species-biased enhancers, are 5x more likely to harbor a Coordinator motif variant than MER52A elements, a similar repeat class depleted from species-biased sites. Even at sites without activity in CNCCs, LTR9 sequences are 3.7x more likely to harbor a Coordinator-like motif than MER52A, consistent with the idea that a preexisting Coordinator-like progenitor sequence contributed to the recent adaptation of some retroelements for CNCC enhancer function. Lastly, we found that the Coordinator motif alone was able to drive activity in luciferase reporter assays in CNCCs (Figure 5D).
Figure 5. Properties of the novel “Coordinator” motif.
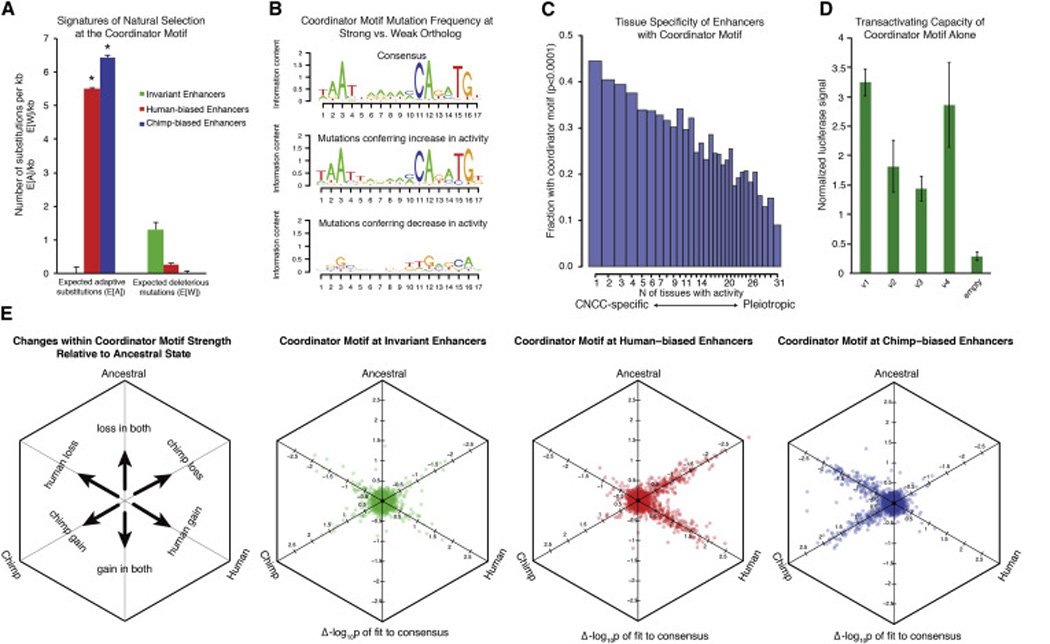
(A) Expected number of adaptive substitutions (E[A]) per kilobase and expected number of deleterious mutations E[W] per kilobase were calculated for all sites of the Coordinator motif at invariant enhancers (green), at human-biased enhancers (red) and at chimp-biased enhancers (blue) using default INSIGHT parameters (Gronau et al., 2013). Significance indicated by * (p<0.01). Overall fractions of nucleotides under selection (ρ) not shown (ρinvariant =0.66, p<0.01; ρhuman-biased = 0.015, p<0.01; ρchimp-biased = 0.019, p<0.01).
(B) Position weight matrix of the Coordinator consensus sequence from top 3000 CNCC specific enhancers is shown (top) relative to logo of mutations preferred at more acetylated (H3K27ac) alleles (middle) versus mutations at less acetylated alleles (bottom).
(C) Enhancers were scored for H3K27ac ChIP-seq enrichments from 30 public data set cell types and binned by number of tissues with activity (1 to 31). The fraction of enhancers per bin with recognizable Coordinator motif (p-value < 0.0001) is indicated on y axis.
(D) Four different versions (V1-4) of the Coordinator motif were cloned in tandem into luciferase reporter vectors and tested for transactivation activity in human CNCCs. Luciferase was normalized relative to renilla transfection control.
(E) Comparison of sequence changes within the Coordinator motif with a reconstructed human-chimp ancestral outgroup. Changes in fit to the Coordinator consensus compared to the ancestral ortholog (−log10p-value) were plotted as orthographic projections along space diagonals for all occurrences of the motif for both human and chimpanzee lineages at different classes of sites. Overlapping data points were offset for better visualization. Schematic shown on the far left.
Sequence analysis reveals the recent evolutionary history of Coordinator motif changes
Our results suggest that nucleotide changes within Coordinator motif sites represent an important class of ‘causative’ mutations predictably associated with gain or loss of CNCC enhancer activity. Thus, by comparing the fit to the consensus for Coordinator-like motifs with a reconstructed ancestral outgroup, we can infer the polarity of enhancer activity change in each lineage relative to the common human-chimp ancestor. Using this strategy, we observed that human-biased enhancers contain Coordinator-like sequences that were equally prone to: (i) a gain in the fit in the human lineage (n=300) or (ii) a loss in fit in the chimp lineage (n=255) relative to the ancestral state (Figure 5E). However, human-biased enhancers contain almost no examples where there was a gain of Coordinator fit in the chimp lineage or loss in the human lineage, an important validation of our analysis. Conversely, we see that chimp-biased enhancers are similarly prone to gains of the Coordinator motif in the chimp lineage (n=218) versus losses in the human lineage (n=255) and again, with almost no gains in human or losses in chimp. Thus, there appears to be no preferred direction of enhancer divergence in either lineage since the split from our common ancestor for this class of sites. We also applied our analysis to hominin outgroups such as Denisovans and Neanderthals and found that, as expected given the much more recent split from the common ancestor, these lineages primarily share the human-like variants of the Coordinator motif at species-biased sites (Figure S6G). Therefore, even for individuals substantially more diverged than any modern human, most changes are present in the hominin lineage relative to the human-chimp ancestor. However, there is a small set of changes that are unique to humans compared to other hominins, and those clearly merit further exploration.
Species-biased enhancers flank genes that show species-biased expression
Recent studies suggest that gene expression levels are more evolutionarily conserved than utilization of cis-regulatory elements, and can be buffered by redundant or compensatory elements regulating the same loci (Hong et al., 2008; Odom et al., 2007; Schmidt et al., 2010; Vierstra et al., 2014; Wong et al., 2014). Nonetheless, at least some of the species-biased enhancers should be associated with transcriptional changes at nearby genes if they are responsible for morphological variation. To test this, we performed RNA-seq analyses of our human and chimp CNCC populations and identified genes whose expression significantly diverged between, but not within species. We found that genes with significantly divergent expression between humans and chimpanzees are strongly enriched for nearby species-biased enhancers, with human-biased genes flanked by human-biased enhancers, and chimp-biased genes flanked by chimp-biased enhancers (Figure 6A). In addition, we observed that the fraction of species-biased genes (but not the degree of the expression bias) scales with the number of flanking enhancers biased towards the same species (Figure 6B).
Figure 6. Clusters of regulatory divergence overlap loci with crucial roles in trait variation and are predictive of expression bias.
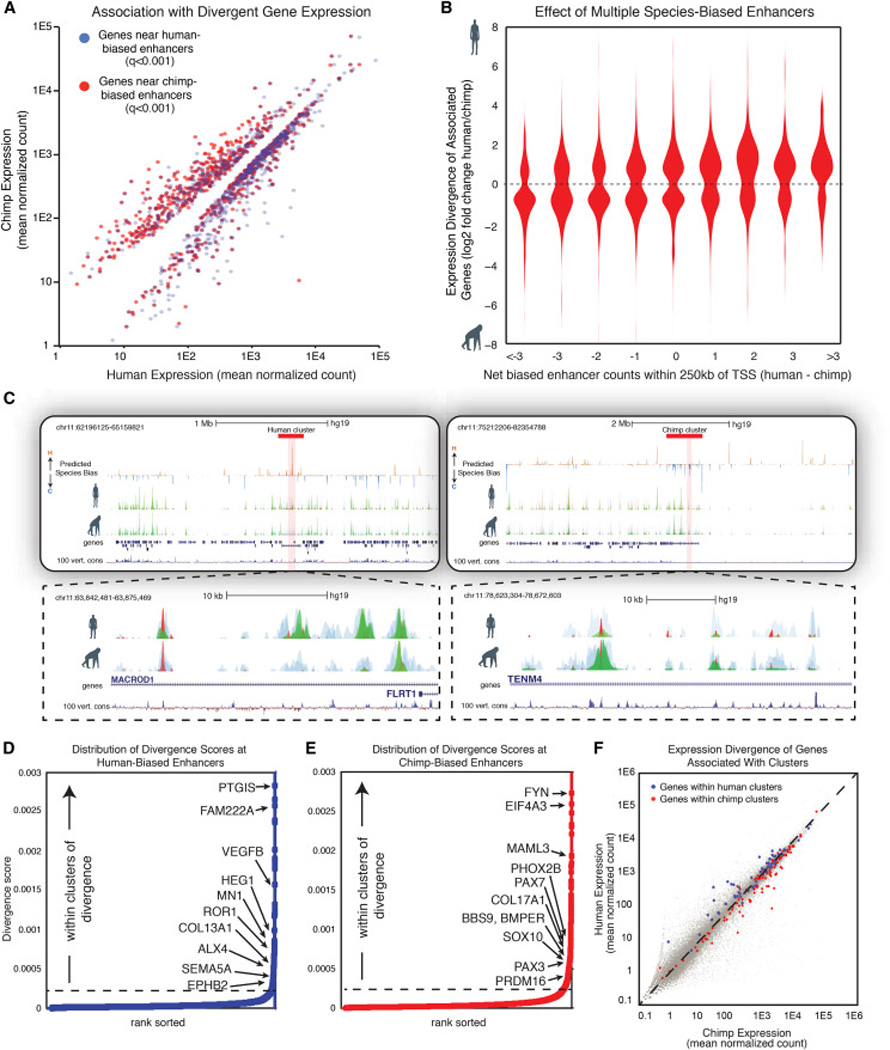
(A) Mean normalized human expression (x axis) versus mean normalized chimp expression (y axis) for genes associated with human-biased enhancers (q-value <0.001, blue) or with chimp-biased enhancers (q-value <0.001, red). Only genes with significant inter-species expression change (padj-value <0.1) shown.
(B) Violin plots showing log2 fold change human/chimp H3K27ac enrichment at orthologous enhancers binned by total count of biased enhancers (total number of human-biased enhancers minus total number of chimp-biased enhancers) within 250kb of promoter regions for genes with significant differences in expression across species (padj-value <0.1).
(C) Representative browser tracks showing clusters of species-biased enhancers. Top panel shows broad view with predicted species-bias track (human-biased in orange, chimp-biased in blue) and the corresponding H3K4me1 (blue), p300 (red), and H3K27ac (green) from 2 individuals of each species shown in overlay. Boundaries of the cluster are indicated by a red block. Close-up of an individual cluster of biased enhancers shown below. All chromatin features are mapped to hg19.
(D–E) Distribution of divergence scores at human-biased enhancers (D) and chimp-biased enhancers (E). Selected genes falling within identified clusters are highlighted next to the enhancer in the cluster with highest divergence score.
(F) Mean normalized human expression (x axis) versus mean normalized chimp expression (y axis) for genes within or flanking human-biased enhancer clusters (blue) or chimp-biased enhancer clusters (red).
Clusters of regulatory divergence flank loci involved in intra-human facial variation
Interestingly, we found that strongly divergent enhancers were not distributed at random throughout the genome, but instead were likely to fall in close genomic proximity to other species-biased enhancers matching in polarity (Figure S7A), suggesting that divergent enhancers fall into regulatory clusters. To systematically locate these clusters, we calculated a genome-wide divergence score using a moving window over the nearest ~10 enhancers for each species, integrating both the degree and genomic span of divergent enhancers in series (Figure S7B). This strategy revealed a low baseline encompassing the bulk of interspersed species-biased enhancers (examples of Chr11 in Figure S7C–D, top panels), but exposed a subset of regions throughout the genome (~1–4 per chromosome) with a marked increase in their divergence score resulting from presence of dense clusters of strongly biased enhancers (Figure 6C). Importantly, we find that these clusters of divergence do not emerge simply by chance due to increased frequency of enhancers near highly active CNCC genes (Figures S7C and S7D).
When ranking all human- and chimp-biased enhancers according to their divergence score, we observed an inflection in the distribution (Figures 6D for human, 6E for chimp). Using this inflection point as a cutoff, we identified 32 human and 65 chimp clusters of divergence, spanning genomic windows of on average ~500kb and encompassing ~11.9% of all species-biased enhancers. Of note, while some clusters overlapped super-enhancers in CNCCs, most super-enhancers were not identified as a species-biased cluster and many species-biased clusters did not encompass super-enhancers, indicating that these two entities are distinct (Whyte et al., 2013).
We speculate that these species-biased enhancer clusters represent broad cis-regulatory regions under strong evolutionary pressure to diverge, and hypothesize that they may contain genes with central roles in the regulation of NC-associated phenotypes. Indeed these regions fall immediately over or next to genes that are critical in facial morphogenesis, including PRDM16, MN1, COL17A1, EDNRA, PAX3, PAX7, SOX10 and ALX4. Intriguingly, of five chromosomal regions linked to normal-range human facial variation in GWAS, three (PRDM16, COL13A1 and PAX3) fall directly within these regions of high divergence. Importantly, the clusters were highly predictive of changes in nearby gene expression for the bulk of the associated genes in the region (Figure 6E), suggesting that either 1) multiple genes in the vicinity are under coordinated selection for these super-divergent regions to emerge or, more likely, that 2) strong selection on one or a few target genes could drive changes in the local enhancer landscape that have secondary effects on other genes in the vicinity. Altogether, we provide evidence that highly divergent clusters of tissue-specific enhancers may promote inter-species and intra-species phenotypic variation.
Resource for studies of human morphological evolution
In addition to informing the basic mechanisms underlying the cis-regulatory divergence of human and chimpanzee NC, our study also provides a rich resource for future investigations of morphological evolution of human craniofacial traits. Ontology annotations of all significantly species-biased enhancers reveal strong associations with processes important for various craniofacial structures that are diverging in human and chimps (Figure 7A). As examples, we highlight some of the most interesting divergent candidate genes in Figure 7B. These featured loci show species-biased expression in our RNA-seq and also map to regions with species-biased enhancer divergence, and are emphasized due to their known associations with CNCC development and/or facial morphology. Nonetheless, it is important to bear in mind that the biases in gene expression and enhancer states highlighted in Figure 7 refer to the relative change between human and chimpanzee CNCCs, without ascribing the polarity of the change with respect to the ancestral status.
Figure 7. Species-biased enhancers are associated with genes affecting craniofacial structures.
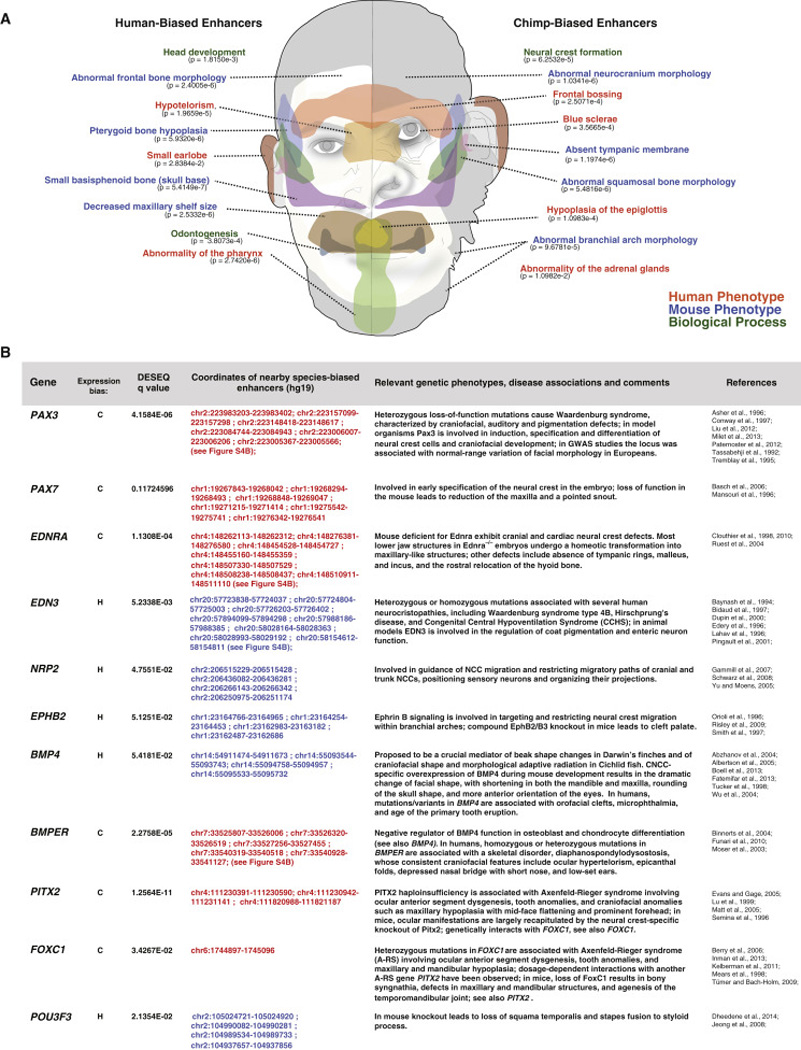
(A) GREAT term enrichments and associated facial regions indicated for human-biased enhancers (q<0.01, baseMean>300) and chimp-biased enhancers (q <0.01, baseMean>300), binomial raw p-values shown below. Ontology categories are color-coded (human phenotypes = red, mouse phenotypes = blue, biological processes = green).
(B) Table of highlighted divergently-expressed genes showing direction of bias (human-biased versus chimp-biased indicated by H or C, respectively), DESeq adjusted p-value of expression divergence, coordinates of nearby species-biased enhancers with corresponding bias (hg19), description of genetic phenotypes, disease associations, comments, and relevant references.
Our divergently-expressed genes are known to be involved in multiple, distinct developmental processes that cooperate to influence differential allocation of CNCCs in facial primordia and, in turn, contribute to species-specific morphology (Fish et al., 2014). These processes (and associated species-biased genes) include: (i) CNCC specification (e.g. PAX3, PAX7), (ii) migration and guidance of CNCC migratory paths (e.g. EPHB2, NRP2, EDNRA, EDN3), (iii) modulation of CNCC proliferation at facial primordia (e.g. BMP4), and (iv) regulation of CNCC differentiation (e.g. PITX2). Moreover, heterozygous mutations in many of these genes (e.g. PAX3, PITX2, FOXC1, EDN3, BMPER) are associated with human syndromes that include craniofacial manifestations, suggesting that altered gene dosage can drive both morphological variation between species and, below a certain threshold, disease-associated malformations (Figure 7B). Furthermore, many phenotypes of the highlighted genes affect aspects of head morphology that have diverged between humans and chimps (e.g., size of the mandible and maxilla, skull shape, and pigmentation) (Figure 7B and Discussion). Altogether, our study provides a wealth of candidate loci for further deep exploration in studies of human evolution and variation.
Discussion
Our study utilizes primate cellular models to provide a comprehensive map of human and chimp regulatory divergence in a tissue with central relevance to the development of the head and face. We show that a common mechanism of regulatory divergence in higher primates is quantitative modulation of orthologous elements, driven largely through small numbers of sequence changes that perturb tissue-specific TF binding motifs. This is consistent with previous studies from closely related Drosophila or mouse species demonstrating that large effects can be conferred by a small number of mutations affecting direct and cooperative binding of key TFs (Bradley et al., 2010; He et al., 2011; Stefflova et al., 2013). Interestingly, we find that not all TF binding sites contribute equally to regulatory divergence – in fact, we identify a broad spectrum of regulatory motifs that vary in frequency and effect, suggesting a mechanism through which evolution can fine-tune cis-regulation across an enhancer landscape. One outlier in our analysis is the Coordinator motif, a de novo consensus sequence that is strongly predictive of the surrounding chromatin features and is highly enriched at species-biased enhancers. We speculate that the factor(s) that recognize the Coordinator motif play a privileged role in the establishment of enhancer competence in this cell context, reminiscent of the Drosophila TAGteam motif bound by a pioneer factor Zelda (Liang et al., 2008; Satija and Bradley, 2012). Furthermore, we find evidence of repressive inputs into quantitative modulation of enhancer activity, with a sizable number of motifs whose gain in strength negatively correlates with acquisition of permissive chromatin states.
Our work provides a rich framework for future gene-centric studies on the developmental mechanisms of human morphological evolution. Indeed, our approach identified loci that are known to profoundly affect NC development and craniofacial morphology, often in a dosage-sensitive manner. For example, we observed that two genes involved in CNCC specification, PAX3 and PAX7, are expressed at higher levels in chimps and are associated with clusters of chimp-biased enhancers. In mice, mutations of these TFs lead to reduction of pigmentation and snout length (Pax3) (Tremblay et al., 1995), and reduction of maxilla and pointed snout (Pax7) (Mansouri et al., 1996), features that are consistent with smaller jaw size and hypopigmentation of humans as compared to chimps. Furthermore, humans are sensitive to alterations of PAX3 dosage, as haploinsufficiency of this gene is associated with craniofacial, auditory and pigmentation defects (Waardenburg Syndrome, OMIM #193510) and genetic variants at this locus have been identified in GWAS studies as regulators of normal-range facial shape (Liu et al., 2012; Paternoster et al., 2012). Thus, variation in PAX3 and PAX7 levels represents an attractive possible mechanism for mediating facial shape divergence between humans and chimpanzees.
We also find evidence that genes already known to affect facial morphology in other species, such as BMP4, are diverging in higher primates as well. BMP4 is the most well understood example of a factor that influenced craniofacial morphological change during evolution, as it has been implicated in mediating changes in beak morphology in Darwin’s finches (Abzhanov et al., 2004), and in jaw shape in Cichlid fish (Albertson et al., 2005). We were therefore intrigued to note that BMP4 is associated with strongly human-biased enhancers and is expressed at higher levels in humans than in chimps. Conversely, expression of the BMP4 inhibitor BMPER was significantly chimp-biased and showed dramatic strengthening of the local chimp enhancer landscape. What would be the potential effects of elevated BMP4 expression on primate facial development? Interestingly, in the mouse, CNCC-specific overexpression of BMP4 results in a dramatic change of facial shape, with shortening of both the mandible and maxilla, rounding of the skull, and more anterior orientation of the eyes (Bonilla-Claudio et al., 2012) – morphological changes that resemble those observed between human and chimps. Thus, the same molecular mechanism that has been postulated to influence beak morphology in Darwin’s finches may also contribute to our uniquely human facial features.
Even more intriguing, of five chromosomal regions that have been associated with normal-range human facial variation in GWAS, three (PRDM16, COL13A1 and PAX3) coincide with clusters of species-biased enhancers uncovered in our study (Liu et al., 2012; Paternoster et al., 2012), suggesting a significant overlap between loci regulating intra- and inter-species variation of facial shape in higher primates. We therefore hypothesize that other divergent clusters identified in our study represent novel candidates for loci involved in the regulation of facial shape in humans. More broadly, we suggest that comparisons of human regulatory landscapes with those of a closely related primate in any tissue of interest may provide an effective strategy to identify candidate loci involved in normal-range and disease-associated variation.
Experimental procedures
CNCC derivation
Pluripotent lines were differentiated into CNCC as previously described (Rada-Iglesias et al., 2012), details provided in Supplemental Methods.
Chromatin Immunoprecipitation (ChIP) and preparation of ChIP-seq libraries
ChIPs were performed using approximately 0.5–1 × 107 CNCCs per experiment, as previously described (Bajpai et al., 2010; Rada-Iglesias et al., 2011, 2012). Antibodies used for ChIPs are listed in the Supplemental methods. Sequencing libraries were prepared starting from 30ng of ChIP DNA using the NEBNext Multiplex Oligos for Illumina kit (Cat# E7335S). Libraries were multiplexed 4–6 samples per lane for 1×50bp Next-Gen sequencing on Illumina HiSeq platform. Raw and processed data will be deposited in NCBI Gene Expression Omnibus (GEO) (accession number pending).
Quantitative analysis of H3K27ac ChIP-seq and identification of divergence
All sequencing reads were aligned to both reference genomes (hg19 and panTro3) using default settings with bowtie2.2.4, regardless of species of origin. Modal peak positions for candidate regulatory elements were determined using a mean shift procedure, described in the Supplemental Methods. To obtain count statistics for each H3K27ac ChIP alignment we counted read coverage in 1.6kb window surrounding modal peak positions. ENCODE blacklisted regions and outlier regions with high counts in control input sequences relative to ChIP were removed as artifacts. Scores for visualization and classification of remaining ChIPs were obtained using a kernel density estimate, as previously described (Buecker et al., 2014).
Calculations of species bias were inferred with DESeq2, based on the read counts from all replicates of H3K27ac at candidate enhancers from three human lines (one hESC, two iPSC) and two chimp lines (two iPSC). DESeq2 analysis was performed separately for panTro3 and hg19 counts, then conservatively, the higher p-adj value and lower abs(log2FoldChange) of the analysis from either hg19 or panTro3 were assigned to each regions, while rare regions with discordant calls were excluded from list of biased sites (less than 0.1%).
Supplementary Material
Acknowledgments
We thank D. Kingsley, R. Greenberg, J. Buenrostro and Wysocka lab members for comments on the manuscript. We also thank R. Aho and J.D. Benazet for help with imagining of mouse transgenic embryos and interpretation of staining patterns, as well as P. Nano and E. Grow for help generating reporter reagents. This work was supported by W.M. Keck Foundation, Innovation Fund, and NIH R01 GM095555 (J.W.), U01 DE024430 (J.W. and L.S.) and CIRM training grant (TG2-01159) (S.L.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ. Bmp4 and morphological variation of beaks in Darwin’s finches. Science. 2004;305:1462–1465. doi: 10.1126/science.1098095. [DOI] [PubMed] [Google Scholar]
- Albertson RC, Streelman JT, Kocher TD, Yelick PC. Integration and evolution of the cichlid mandible: the molecular basis of alternate feeding strategies. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16287–16292. doi: 10.1073/pnas.0506649102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attanasio C, Nord AS, Zhu Y, Blow MJ, Li Z, Liberton DK, Morrison H, Plajzer-Frick I, Holt A, Hosseini R, et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science. 2013;342:1241006. doi: 10.1126/science.1241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent NC formation. Nature. 2010;463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsborough A, Wood BA. Cranial morphometry of early hominids: facial region. Am. J. Phys. Anthropol. 1988;76:61–86. doi: 10.1002/ajpa.1330760107. [DOI] [PubMed] [Google Scholar]
- Bonilla-Claudio M, Wang J, Bai Y, Klysik E, Selever J, Martin JF. Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal. Development. 2012;139:709–719. doi: 10.1242/dev.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, Li X-Y, Trapnell C, Davidson S, Pachter L, Chu HC, Tonkin LA, Biggin MD, Eisen MB. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol. 2010;8:e1000343. doi: 10.1371/journal.pbio.1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, LeDouarin NM. Development and evolution of the NC: an overview. Dev. Biol. 2012;366:2–9. doi: 10.1016/j.ydbio.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:838–853. doi: 10.1016/j.stem.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CE, Blekhman R, Marioni JC, Gilad Y. Gene expression differences among primates are associated with changes in a histone epigenetic modification. Genetics. 2011;187:1225–1234. doi: 10.1534/genetics.110.126177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. Cranial NC cells on the move: their roles in craniofacial development. Am. J. Med. Genet. A. 2011;155A:270–279. doi: 10.1002/ajmg.a.33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Leng J, Yin J, Reilly SK, DeMare LE, Emera D, Ayoub AE, Rakic P, Noonan JP. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell. 2013;154:185–196. doi: 10.1016/j.cell.2013.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JL, Sklar RS, Woronowicz KC, Schneider RA. Multiple developmental mechanisms regulate species-specific jaw size. Dev. Camb. Engl. 2014;141:674–684. doi: 10.1242/dev.100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompel N, Prud’homme B, Wittkopp PJ, Kassner VA, Carroll SB. Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature. 2005;433:481–487. doi: 10.1038/nature03235. [DOI] [PubMed] [Google Scholar]
- Gronau I, Arbiza L, Mohammed J, Siepel A. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol. Biol. Evol. 2013b;30:1159–1171. doi: 10.1093/molbev/mst019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, Zeitlinger J. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat. Genet. 2011;43:414–420. doi: 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hong J-W, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS. Exploring the genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev. 2014;29:15–21. doi: 10.1016/j.gde.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Jheon AH, Schneider RA. The cells that fill the bill: NC and the evolution of craniofacial development. J. Dent. Res. 2009;88:12–21. doi: 10.1177/0022034508327757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Wiebe V, Pääbo S. Extensive Nuclear DNA Sequence Diversity Among Chimpanzees. Science. 1999;286:1159–1162. doi: 10.1126/science.286.5442.1159. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Liang H-L, Nien C-Y, Liu H-Y, Metzstein MM, Kirov N, Rushlow C. The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature. 2008;456:400–403. doi: 10.1038/nature07388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DE. Sphenoid shortening and the evolution of modern human cranial shape. Nature. 1998;393:158–162. doi: 10.1038/30227. [DOI] [PubMed] [Google Scholar]
- Liu F, van der Lijn F, Schurmann C, Zhu G, Chakravarty MM, Hysi PG, Wollstein A, Lao O, de Bruijne M, Ikram MA, et al. A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet. 2012;8:e1002932. doi: 10.1371/journal.pgen.1002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Stoykova A, Torres M, Gruss P. Dysgenesis of cephalic NC derivatives in Pax7−/− mutant mice. Dev. Camb. Engl. 1996;122:831–838. doi: 10.1242/dev.122.3.831. [DOI] [PubMed] [Google Scholar]
- Marchetto MCN, Narvaiza I, Denli AM, Benner C, Lazzarini TA, Nathanson JL, Paquola ACM, Desai KN, Herai RH, Weitzman MD, et al. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature. 2013;503:525–529. doi: 10.1038/nature12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, MacIsaac KD, Rolfe PA, Conboy CM, Gifford DK, Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster L, Zhurov AI, Toma AM, Kemp JP, St Pourcain B, Timpson NJ, McMahon G, McArdle W, Ring SM, Smith GD, et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am. J. Hum. Genet. 2012;90:478–485. doi: 10.1016/j.ajhg.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, Pennacchio LA. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, Fitzpatrick DR, Afzal V, et al. Human-specific gain of function in a developmental enhancer. Science. 2008;321:1346–1350. doi: 10.1126/science.1159974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human NC. Cell Stem Cell. 2012;11:633–648. doi: 10.1016/j.stem.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Bradley RK. The TAGteam motif facilitates binding of 21 sequence-specific transcription factors in the Drosophila embryo. Genome Res. 2012;22:656–665. doi: 10.1101/gr.130682.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D, Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, Wortham M, Tewari AK, London D, Song L, Lee B-K, Iyer VR, et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012;8:e1002789. doi: 10.1371/journal.pgen.1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoor F, Wood B, Zonneveld F. Implications of early hominid labyrinthine morphology for evolution of human bipedal locomotion. Nature. 1994;369:645–648. doi: 10.1038/369645a0. [DOI] [PubMed] [Google Scholar]
- Stefflova K, Thybert D, Wilson MD, Streeter I, Aleksic J, Karagianni P, Brazma A, Adams DJ, Talianidis I, Marioni JC, et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013;154:530–540. doi: 10.1016/j.cell.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay P, Kessel M, Gruss P. A transgenic neuroanatomical marker identifies cranial NC deficiencies associated with the Pax3 mutant Splotch. Dev. Biol. 1995;171:317–329. doi: 10.1006/dbio.1995.1284. [DOI] [PubMed] [Google Scholar]
- Twigg SRF, Versnel SL, Nürnberg G, Lees MM, Bhat M, Hammond P, Hennekam RCM, Hoogeboom AJM, Hurst JA, Johnson D, et al. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am. J. Hum. Genet. 2009;84:698–705. doi: 10.1016/j.ajhg.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Rynes E, Sandstrom R, Zhang M, Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R, et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014;346:1007–1012. doi: 10.1126/science.1246426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, et al. Enhancer Evolution across 20 Mammalian Species. Cell. 2015;160:554–566. doi: 10.1016/j.cell.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ES, Thybert D, Schmitt BM, Stefflova K, Odom DT, Flicek P. Decoupling of evolutionary changes in transcription factor binding and gene expression in mammals. Genome Res. 2014 doi: 10.1101/gr.177840.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Zhou X, Cain CE, Myrthil M, Lewellen N, Michelini K, Davenport ER, Stephens M, Pritchard JK, Gilad Y. Epigenetic modifications are associated with inter-species gene expression variation in primates. Genome Biol. 2014;15:547. doi: 10.1186/s13059-014-0547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.