

Abstract
Complex organisms are faced with the challenge of generating and maintaining diverse cell types, ranging from simple epithelia, to neurons and motile immune cells1–3. To meet this challenge, a complex set of regulatory pathways controls nearly every aspect of cell growth and function, including genetic and epigenetic programming, cytoskeleton dynamics, and protein trafficking. The far reach of cell fate specification pathways makes it particularly catastrophic when they malfunction, both during development and for tissue homeostasis in adult organisms. Furthermore, the promise of stem cells as a therapeutic derives from their ability to deftly navigate the multitude of pathways that control cell fate4. How the molecular components that make up these pathways function to specify cell fate is beginning to become clear. Work from diverse systems suggests that the atypical Protein Kinase C (aPKC) is a key regulator of cell fate decisions in metazoans5–7. Here we examine some of the diverse physiological outcomes of aPKC’s function in differentiation, along with the molecular pathways that control aPKC, and those that are responsive to changes in its catalytic activity.
Graphical Abstract
PKC family kinases are ubiquitous components of cellular signaling pathways8,9. In animals, PKCs are commonly divided into three subfamilies (yeast contain a single PKC), including the conventional, novel, and atypical (Figure 1)8. This last group contains the iota (lambda in mice) and zeta isoforms in mammals, and a single isoform in flies and worms. All family members contain a catalytic domain at the COOH-terminus connected to NH2-terminal regulatory domains (Figure 1A). The downstream pathways that are regulated by each isoform is primarily determined by their kinase domain’s specificity, which determines the repertoire of substrates that they can phosphorylate. Upstream regulation of PKCs is determined by phosphorylation of the kinase domain and allosteric mechanisms that depend on interactions with specific elements contained within the NH2-terminal regulatory domain8.
Figure 1.

PKC family kinases and regulation and function of atypical Protein Kinase C. A. Schematic of the protein kinase C family showing domain architectures, demonstrating both common and unique aspects of each PKC family member (PS = pseudosubstrate; C1 and C2 are cysteine rich domains; PB1 Phox/Bem1 domain). B. Schematic of Par-mediated polarity mechanism. aPKC generates cellular polarity through phosphorylation and exclusion of cortically localized substrates (pink).
aPKC regulates differentiation in diverse physiological contexts
The physiological contexts in which aPKC participates in cellular differentiation are remarkably varied, suggesting that it is a central component of a fate specification machinery. In this section, we discuss several examples of systems where aPKC plays a known role in regulating cell fate with the goal of emphasizing the diverse physiology in which it can function.
In the mammalian pre-implantation embryo, aPKC activity is essential for development of extra-embryonic tissues such as the Primitive Endoderm (PrE)10,11. These tissues provide the connection with the mother and serve as signaling centers for subsequent embryonic patterning12. The PrE forms a highly organized epithelium at the exterior of the epiblast, which ultimately forms the fetus. However, PrE cells are originally specified in an apparently stochastic manner, intermingled with epiblast cells. Intriguingly, aPKC is essential for not only specification of the PrE fate, but segregation of the mixed PrE and epiblast cells. Following segregation, aPKC causes PrE cells to become highly polarized and to promote pro-survival signals in correctly sorted cells11.
In the fly central nervous system, aPKC regulates the balance between self-renewal of neuronal progenitors and their differentiation into neurons5,6. The Drosophila neuroblast (NB) is a cell that participates in the development of both the embryonic and larval nervous systems. NBs undergo repeated asymmetric cell divisions that produce as daughter cells a self-renewed NB and a Ganglion Mother Cell (GMC). The GMC subsequently divides once more which typically generates two cells that become neurons. NBs with incorrect levels of aPKC activity fail to asymmetrically divide and can exhibit characteristics of tumor cells, or alternatively can prematurely differentiate and concomitant loss of the progenitor pool. Excess aPKC activity leads to indefinite replication capacity13, whereas NB quiescence or premature differentiation are associated with inadequate aPKC activity14,15. aPKC is also polarized in mammalian neurons and is required for axonal-dendritic differentiation during development16,17. Perturbing aPKC or regulator of aPKC localization and activity leads to improper number of axons and dendrites. In addition to its role in development, aPKC-mediated asymmetric cell division is also essential for homeostasis in the adult gut18.
The central role of aPKC in cell fate determination is also supported by the severe consequences if it is improperly regulated. Overexpression of aPKC is observed in multiple cancers19, including hepatocellular carcinoma, pancreatic adenocarcinoma, and breast cancer. It has recently been shown that excess aPKC activity can overcome contact inhibited growth in epithelial cells and is sufficient for transformation20. It is interesting to note that the aPKC iota/lambda and zeta isoforms may have distinct functions in regulating proliferation based on their requirement in different cell types. For example, the iota/lambda isoform promotes the growth and metastasis of triple-negative breast cancers, a subtype defined by the absence of estrogen receptor, progesterone receptor and epidermal growth factor receptor 221. However, the zeta isoform is required for the mitogen induced growth of squamous cell carcinomas of the head and neck22. Whether these isoforms are indeed differentially regulated and/or act on distinct downstream pathways is an important outstanding question.
Cellular mechanisms of fate determination by aPKC
In this section, we examine the cellular mechanisms by which aPKC controls cell fate. As a regulator of cell polarity, aPKC is a member of the Par (partitioning defective) complex, which includes Par-3 (Bazooka in flies), and Par-623,24. Polarity is essential for many aspects of cell function, including aPKC’s role in cell fate specification in the Drosophila NB. Early in mitosis, asymmetrically dividing NBs begins to polarize such that by metaphase, aPKC and the rest of the Par complex localize to one half of the cell cortex, while neuronal fate specification factors localize to the other half5,6. Because the mitotic spindle is aligned with cortical polarity, the cytokinetic furrow bisects the two cortical domains: one daughter cell is formed from the cortex containing the Par complex, and the other forms from the cortex with differentiation factors bound. aPKC is a key output of Par complex activity, as it phosphorylates downstream targets to displace them into the cytoplasm25. These substrates can localize to cortical regions that lack the Par complex but are removed from the cortex once they enter the Par domain (Figure 1B). For at least several aPKC substrates, this mechanism appears to involve phosphorylation of short motifs enriched for basic and hydrophobic residues that directly interact with phospholipids26,27. Phosphorylation of the motif alters its electrostatic character thereby reducing the affinity for the membrane and causing displacement of the substrate into the cytoplasm.
Activating aPKC at the NB apical cortex is critical for restricting neuronal fate determinants to the basal cortex. These proteins include the coiled-coiled protein Miranda with its cargo protein, the transcription factor, Prospero (Pros; Prox1 in mammals), and the translational regulator Brain Tumor (Brat; TRIM3 in mammals), as well as the Notch signaling regulator Numb5,6. Following mitosis, these determinants induce conversion into a ganglion mother cell (GMC) by preventing self-renewal and promoting differentiation. Pros is a homedomain transcription factor that translocates to the GMC nucleus and activates genes that specify differentiation while repressing genes that are necessary for self-renewal5. High Pros expression in NBs is sufficient to drive their differentiation28 while intermediate levels induce quiescence29. Differentiation is aided by the translational repressor Brat, which regulates important proliferation signals including Cyclin E, β-Catenin, dMyc, and Mad30–32, and the repressor of Notch signaling, Numb14,33.
Besides excluding neuronal fate determinants from the self-renewed NB, aPKC also plays a direct role in maintaining NB fate. The transcription factor Zif represses NB formation and in NBs lacking Zif aPKC is unpolarized34. The aPKC gene contains Zif binding sites and Zif appears to repress aPKC expression. Furthermore, Zif is an aPKC substrate and phosphorylation prevents its entrance into the nucleus, forming a feedback loop that regulates aPKC expression and localization.
Regulation of aPKC during asymmetric cell division is controlled by a large network of regulatory factors. The Rho GTPase Cdc42 is a key regulator of the Par complex by binding 35–37. The neoplastic tumor suppressor Lgl is a negative regulator of aPKC localization and helps ensure that aPKC is restricted to the proper cortical region38,39. Dynamin associated protein-160 (Dap160) regulates both aPKC localization and kinase activity40. It co-localizes with the Par complex at the apical cortex of dividing NBs and interacts with both aPKC and Par6. Dap160, through an unknown mechanism, also helps ensure that aPKC is properly polarized and does not enter the basal cortical domain. Other factors that control aPKC activity and localization include Clueless41 and Canoe/afadin42,43, although the mechanisms by which they do so are poorly understood.
Another important cellular role for aPKC is in orienting the division axis. Several recent reviews cover aPKC function in oriented cell divisions in detail44,45.
Regulation of the cell cycle by aPKC
Cell fate specification can be tightly coupled to the cell division cycle. For example, in certain contexts a prolonged G1 cell cycle phase leads to differentiation, while a shortened G1 promotes proliferation (i.e. self-renewal)46. Recent evidence from the Xenopus neuroectoderm suggests that G1 is controlled in part by the inhibition of G1 specific cyclin/cdks47. Although many aPKC functions involve its activity at the cell cortex, aPKC is found in the nucleus of progenitor cells in this tissue48 consistent with a role in transcriptional regulation. This seems to be the case for at least one cell-cycle regulatory protein in Xenopus progenitor cells, p27xic. p27xic is a CIP/KIP protein family of cyclin-dependent kinase inhibitors (CDKIs) that prevents the G1 to S transition by inhibiting cyclin-dependent kinase 2 (Cdk2) through binding and sequestering it from the nucleus (Figure 2A). In this manner, the level of p27xic expression in the progenitor cells can indirectly affect the decision to proliferate or differentiate by controlling G1 length. But what controls the level of p27xic? Recent work has demonstrated that p27xic is an aPKC substrate and phosphorylation regulates its ability to inhibit the G1 to S transition. Phosphorylation prevents p27xic’s binding to Cdk2 providing a simple, but elegant method for coupling aPKC activity to cell cycle control, and ultimately the decision to proliferate or differentiate49 (Figure 2B).
Figure 2.

aPKC regulation of the cell cycle. A. When aPKC levels are low, p27Xic1 is able to elongate the G1 to S transition by binding to Cdk2, which can lead to differentiation in Xenopus neuroectoderm progenitor cells. B. When aPKC levels are high, p27Xic1 phosphorylation by aPKC blocks p27Xic1 binding of Cdk2, shortening the G1 to S transition to promote proliferation.
Transcriptional programming by aPKC
Hedgehog (HH) signaling is important for cell fate decisions that specify the animal body plan50. In the absence of HH ligand, Patched (Ptch) represses HH signaling through inhibition of the receptor Smoothened (Smo)51 (Figure 3). When HH binds Ptch at the membrane, transcriptional activators such as GLI (Cubitus interruptus in Drosophila) become active51. This pathway can regulate stem cell proliferation versus differentiation decisions52 and is often reactivated during the initiation and progression of cancers such as basal cell carcinomas (BCCs) and lung squamous cell carcinomas (LSCCs)53,54. Binding to Ptch requires numerous HH post-translational modifications including specific proteolysis followed by palmitoylation by HH acyl transferase (HHAT)51. Once HH ligand binds to Ptch, Ptch no longer inhibits Smo, resulting in translocation of GLI to the nucleus and subsequent activation of proliferative genes50. Recently, aPKC has been found to regulate multiple points within the HH pathway. Activity of aPKC leads to upregulation of the HH ligand, phosphorylation of the receptor Smoothened, and activation of the bifunctional transcriptional regulator of HH signaling, GLI55–57 (Figure 3).
Figure 3.

aPKC regulation of Hedgehog signaling. In basal cell carcinomas (BCCs) and lung squamous cell carcinomas (LSCCs) aPKC is able to phosphorylate GLI (BCCs) and SOX2 (LSCCs) transcription factors. These phosphorylations can lead to positive feedback, upregulating HH signaling genes including HHAT and aPKC itself. This activation can occur independently of HH ligand receptor binding. In the Drosophlia developing wing, aPKC phosphorylates the Smoothened receptor to regulate its activity and its subsequent proper development.
There are multiple mechanisms by which aPKC regulates HH signaling. First, expression of the HHAT enzyme is dependent on aPKC activity. This control occurs by aPKC’s phosphorylation of SOX2, an important transcriptional regulator of stem cell maintenance. SOX2 modification by aPKC allows it to bind the HHAT promoter region57 (Figure 3). This leads to an increase in functional HH ligands. Upregulation of HHAT by aPKC can be important for tumorigenic growth by maintaining stemness, as has been demonstrated for LSCC oncospheres57.
HH signaling can also be regulated by aPKC downstream of the Ptch receptor. The GLI1 transcription factor is an aPKC substrate55, and, as with SOX2, phosphorylation activates transcription of GLI1 target genes including aPKC itself (Figure 3). This positive feedback loop can lead to the development and progression of basal cell carcinomas (BCCs) independent of Smo activation of GLI155 (Figure 3). Currently, Smo inhibitors are used to treat BCCs but the tumors can develop resistance50,58. Inhibition of aPKC signaling inhibits BCC tumor-growth indicating that inhibitors could have therapeutic potential for treating BCCs58. In Drosophila, aPKC phosphorylates Smo and GLI (Cubitus interruptus in Drosophila) to polarize them basolaterally, thereby promoting HH signaling during early wing development56. However, the molecular mechanism by which aPKC activity is controlled during HH signaling remains unclear.
aPKC regulation of Wnt signaling
Many tissues, such as the epidermal and intestinal epithelia, undergo rapid turnover requiring constant differentiation from precursor cells for tissue maintenance. In mammalian epidermal models, aPKC regulates cell fate by ensuring proper division orientation59. Adult intestinal stem cells are continually replenishing the cells of the epithelium, which is turned over ever 3–5 days60,61. In these adult stem cell models, precise regulation of β-Catenin (Wnt signaling) and Yap (Hippo pathway) is required for maintenance of tissue homeostasis and prevention of tumor initiation and progression1,62.
In the absence of Wnt ligands, β-Catenin is degraded by the “destruction” complex composed of the tumor suppressor adenomatous polyposis coli (APC), scaffolding protein Axin, glycogen synthase-3 (GSK-3β) and casein kinase 1 (CK1). While the complex is intact, β-Catenin is phosphorylated by GSK-3β and degraded by the proteasome63. In the absence of nuclear β-Catenin, downstream Wnt-dependent target genes are not transcribed, inhibiting proliferative and growth signals (Figure 4). When Wnt is bound to the receptor Frizzled (FZD) and a co-receptor, Axin is thought to be degraded and the destruction complex dissociates, concomitantly stabilizing β-Catenin levels, allowing for nuclear translocation and binding to co-activator TCF/LEF proteins. Ultimately, this leads to the transcription of Wnt-dependent target genes63 (Figure 4). Wnt signaling has been implicated in polarity through interactions with the Par complex in migratory cells64. Recent work has shed light on how aPKC might be playing a direct roll in Wnt signaling.
Figure 4.

aPKC regulation of Wnt signaling. aPKC is part of the destruction complex, where it can phosphorylate Catenin to prime it for (2) GSK-3 phosphorylation and subsequent proteasomal degradation. aPKC is also able to phosphorylate YAP, leading to proteasomal degradation. Loss of aPKC or Wnt binding leads to disassembly of the destruction complex and activation of Wnt signaling favoring a proliferative state. The fate of aPKC once the destruction complex is inactivated is unknown.
aPKC has now been identified as a component of destruction complex that interacts with Yap and β-Catenin65. While best known for their role in Hippo pathway signaling66, Yap and Yaz also interact with the destruction complex62,67. aPKC phosphorylates both β-Catenin and Yap, preventing their nuclear accumulation, thereby inhibiting Wnt and Hippo downstream targets required for proliferation and cell growth65 (Figure 4). β-Catenin must be phosphorylated at its aPKC phosphorylation site (either by aPKC or another kinase) before GSK-3β can act on it68,69. Yap activity is increased by aPKC, in a manner that is at least partially independent from canonical Hippo signaling. In Drosophila, GSK-3β regulates polarity by phosphorylating aPKC, which targets it for proteasomal degradation70 suggesting crosstalk between these pathways.
aPKC’s regulation of JAK/Stat
Janus Kinase (JAK) and Signal transducer and activator of transcription (Stat) are important growth regulators that play a prominent role in development and tumor progression71–73. Numerous signaling pathways activate JAK/Stat by inducing JAK recruitment to Stat and subsequent Stat phosphorylation. During IL6 cytokine activation, phosphorylation of the Stat3 isoform by JAK leads to Stat3 nuclear translocation where it activates proliferation and survival genes and represses differentiation genes74. Stat3 has also been implicated in the maintenance of cancer stem cells (CSCs)75,76.
In a recent study, aPKC activity was found to activate Stat3 in a mammalian model of breast cancer77. Activation occurs via aPKC’s interaction with the NF-κb signaling pathway, which is up regulated in many human cancers78. In this system, aPKC becomes active in the cytoplasm after loss of polarity where it activates IKK ultimately causing increased IL6 production79,80. This leads to a positive feedback loop associated with proliferation and tumor progression (Figure 5). Up-regulation of IL6 by active aPKC in unpolarized cells also occurs in Drosophila models that combine polarity loss with oncogenic transformations81. In fact, constitutively active aPKC is sufficient to induce IL6 (Upd in Drosophila) expression, although the effect is dependent on the Drosophila ortholog of YAP (Yki)81. Whether or not aPKC induces IL6 through YAP via the canonical Hippo pathway signaling or as part of the destruction complex (i.e. Wnt signaling), remains to be resolved.
Figure 5.

aPKC regulation of JAK/Stat signaling. Loss of polarity leads to cytoplasmic aPKC which causes activation of IKKβ, degradation of IB, and translocation of p65 to the nucleus to upregulate IL6 production. The increase in IL6 leads to a positive feedback loop with JAK/Stat3 signaling, which, when unregulated, leads to proliferation and tumor progression in a breast cancer model.
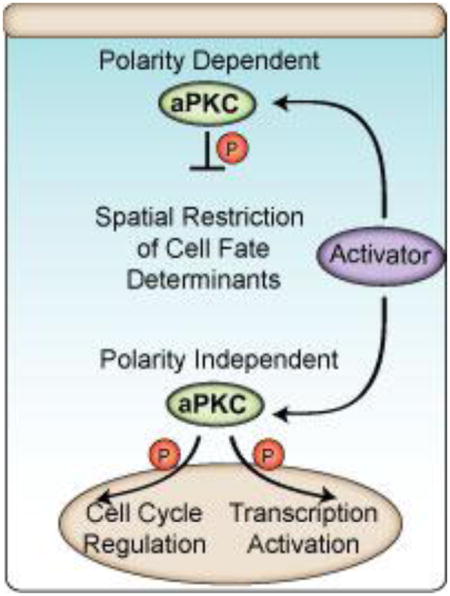
The above examples suggest that the output of aPKC activity is dependent on the cellular context. For example, while aPKC promotes self-renewal and cell growth in neuroblasts, it seems to inhibit self-renewal in the intestinal epithelium. This conundrum highlights the necessity of discovering and understanding the mechanisms that regulate aPKC activity in spatial and temporal manner in these diverse cell and organismal contexts.
Regulation of aPKC: localized activity
The cellular mechanisms by which aPKC regulates differentiation suggest a high degree of both catalytic and spatial control9. For example, the cortical exclusion of fate determinants in polarized neuroblasts during asymmetric cell division requires that aPKC activity is not only tightly coupled to the cell cycle, but that it is localized to a specific cortical domain. The central role of aPKC in many differentiation pathways means that incorrect activity levels could lead to improper fate specification or proliferation, as described in the previous sections. In general, PKC family enzymes are controlled primarily by kinase domain phosphorylation and allosteric mechanisms (Figure 1A). In aPKCs, the kinase domain is phosphorylated at the activation loop and turn motif8. A third site present in other PKC isoforms, known as the hydrophobic motif, is a non-phosphorylatable residue in aPKCs. The activation loop is modified by Phosphoinositide-Dependent Kinase 1 (PDK1)8. For some time, PKC turn motifs were thought to be modified as the result of autophosphorylation, but elegant work by Parker and co-workers demonstrated that the turn motif is phosphorylated by an exogenous kinase82. Interestingly, at least in some contexts, this kinase can be the mammalian target of rapamycin 2 complex (mTORC2)83,84. However, the physiological role of these phosphorylations in regulating aPKC remains unclear. They may be constitutive, “priming” modifications85, and recent structural evidence even suggests that they may not be required for activity86.
In addition to modification of the kinase domain by phosphorylation, PKCs can also be regulated through allosteric mechanisms by binding of upstream pathway components to the NH2-terminal regulatory domain9. Although aPKCs have a different complement of upstream regulators compared to their conventional and novel counterparts, they share several important regulatory elements (Figure 1A). Perhaps most important is the “pseudosubstrate”, which has many of the sequence characteristics of a normal substrate so that it can bind in the kinase domain active site, but an alanine at the position that would be phosphorylated prevents progression through the catalytic cycle8,87. Determining how the pseudosubstrate is removed from the kinase domain’s active site is a key part of understanding PKC activation mechanisms, but other domains, such as the C1, may also directly repress kinase activity87,88. The C1 cysteine rich domain is directly COOH-terminal to the pseudosubstrate in all PKCs, and in the single structure of a full-length PKC, the C1 binds a lobe of the kinase domain where it could potentially inhibit activity89. aPKC’s regulatory domain is distinguished from the other family members by the presence of a PB1 domain that heterodimerizes with certain PB1s from other proteins, and a COOH terminal PDZ ligand sequence8,90–92.
The Rho GTPase Cdc42 indirectly regulates aPKC by binding to the Par complex member Par-6. GTP-bound Cdc42 interacts with the semi-crib and PDZ domain of Par-6 causing a conformational change, which is essential for aPKC polarization35–37. Par-6 contains a PB1 domain that binds aPKC’s PB1 and this interaction, via an unknown mechanism, displaces the pseudosubstrate from the kinase domain active site87,92,93 (Figure 6). Interestingly, Par-6 is overexpressed in breast cancer cells and induces their proliferation94. Par-6 is also required to recruit aPKC to the cortex, where lipid binding can play a direct role in the activation of aPKC downstream of phosphatidylionositol 3-kinase (PI3K) by binding phosphatidylinositol 3,4,5-phosphate (PIP3)95–97. The lipid ceramide also activates aPKC by directly interacting with the kinase domain, an interaction that is important for junction formation in epithelia and signaling during cellular stress conditions98,99. Coupling of aPKC protein-protein and protein-lipid interactions to activation provides an elegant mechanism for ensuring that aPKC is active at the right place and time. Cdc42 may also play a direct role in controlling aPKC’s kinase activity as the Par-6 semi-CRIB and PDZ are important for full activation of aPKC by Par-6, further coupling aPKC localization and activity to the NB apical cortex35,37,100. In the Drosophila neuroblast, loss of either Par-3 or Par-6 leads to improper aPKC localization, defective asymmetric cell division, and improper development15,35,101.
Figure 6.
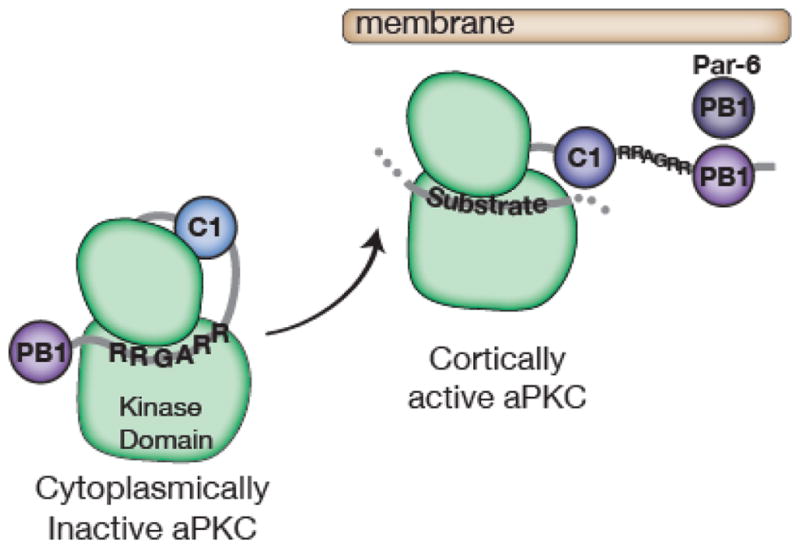
Regulation of aPKC localization and activity. Par-6’s interaction with aPKC’s PB1 domain disrupts the pseudosubstrate’s (sequence = RRGARR) inhibition of the kinase domain. The C1 domain may also play a role in regulating aPKC kinase activity.
While Cdc42 and Par-6 are critical for increasing the amount of cortically localized, active aPKC, the neoplastic tumor suppressor Lgl is an important repressor of localization and activity that helps ensure the basal cortical domain remains free of aPKC (and therefore bound to neuronal fate determinants)38,39. The mechanism by which Lgl inhibits aPKC has remained enigmatic. In NBs lacking Lgl activity, aPKC activity is no longer restricted to the apical cortex leading to an increase in proliferation and a loss of apico-basal polarity39. aPKC counteracts Lgl’s repression by phosphorylating it and displacing it into the cytoplasm38. How Lgl inhibits aPKC’s localization to the basal cortex remains unknown.
Concluding remarks
How cellular diversity is generated during development is one of the most fundamental questions in Biology. Once development is complete, homeostasis requires the constant activity of progenitor cells to replenish rapidly turned over differentiated products. Each of these processes is highly intertwined with proliferation pathways, such that defects are commonly associated with tumorigenesis. Our understanding of the molecular mechanisms that control cell fate decisions is still in its infancy, but it is now clear that the atypical members of the PKC kinase family are involved in many aspects of fate specification. Some of these functions relate to aPKC’s activity in regulating cell polarity, but there are newly identified polarity-independent aPKC functions (both in normal and pathological Biology) that are essential for conferring proper cell identity. We expect that many more aPKC substrates and downstream pathways remain to be found, and that fitting them into the puzzle of cell fate determination will help provide a more complete picture of this fundamental process. Furthermore, the mechanisms that govern the localized activity of aPKC are just now being uncovered and will no doubt be important for understanding the diversity of physiological contexts in which aPKC functions.
Highlights.
atypical Protein Kinase C (aPKC) regulates stem cell fate decision using multiple mechanisms.
aPKC polarizes fate determinant proteins during asymmetric cell division.
aPKC shortens the cell cycle to promote cell proliferation.
aPKC phosphorylates conserved transcription factors to regulate cell fate decisions.
aPKC controls cell fate decision by polarity dependent and independent mechanisms.
Acknowledgments
We would like to thank the members of the Prehoda lab for helpful discussions and critical reading of this manuscript. This work was supported by NIH grant R01 GM068032 (K.E.P).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clevers H, Loh KM, Nusse R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346:1248012. doi: 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- 2.Kohwi M, Doe CQ. Temporal Fate Specification and Neural Progenitor Competence During Development. Nat Rev Neurosci. 2013;14:823–838. doi: 10.1038/nrn3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seita J, Weissman IL. Hematopoietic Stem Cell: Self-renewal versus Differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–653. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- 6.Homem CCF, Knoblich JA. Drosophila neuroblasts: a model for stem cell biology. Dev Camb Engl. 2012;139:4297–4310. doi: 10.1242/dev.080515. [DOI] [PubMed] [Google Scholar]
- 7.Tepass U. The Apical Polarity Protein Network in Drosophila Epithelial Cells: Regulation of Polarity, Junctions, Morphogenesis, Cell Growth, and Survival. Annu Rev Cell Dev Biol. 2012;28:655–685. doi: 10.1146/annurev-cellbio-092910-154033. [DOI] [PubMed] [Google Scholar]
- 8.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosse C, et al. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 10.Dard N, Le T, Maro B, Louvet-Vallée S. Inactivation of aPKClambda reveals a context dependent allocation of cell lineages in preimplantation mouse embryos. PloS One. 2009;4:e7117. doi: 10.1371/journal.pone.0007117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saiz N, Grabarek JB, Sabherwal N, Papalopulu N, Plusa B. Atypical protein kinase C couples cell sorting with primitive endoderm maturation in the mouse blastocyst. Dev Camb Engl. 2013;140:4311–4322. doi: 10.1242/dev.093922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saiz N, Plusa B. Early cell fate decisions in the mouse embryo. Reprod Camb Engl. 2013;145:R65–80. doi: 10.1530/REP-12-0381. [DOI] [PubMed] [Google Scholar]
- 13.Caussinus E, Gonzalez C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat Genet. 2005;37:1125–1129. doi: 10.1038/ng1632. [DOI] [PubMed] [Google Scholar]
- 14.Lee CY, et al. Drosophila Aurora-A kinase inhibits neuroblast self-renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev. 2006;20:3464–3474. doi: 10.1101/gad.1489406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolls MM, Albertson R, Shih HP, Lee CY, Doe CQ. Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J Cell Biol. 2003;163:1089–1098. doi: 10.1083/jcb.200306079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Insolera R, Chen S, Shi SH. Par proteins and neuronal polarity. Dev Neurobiol. 2011;71:483–494. doi: 10.1002/dneu.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi SH, Jan LY, Jan YN. Hippocampal Neuronal Polarity Specified by Spatially Localized mPar3/mPar6 and PI 3-Kinase Activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- 18.Goulas S, Conder R, Knoblich JA. The Par complex and integrins direct asymmetric cell division in adult intestinal stem cells. Cell Stem Cell. 2012;11:529–540. doi: 10.1016/j.stem.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muthuswamy SK, Xue B. Cell polarity as a regulator of cancer cell behavior plasticity. Annu Rev Cell Dev Biol. 2012;28:599–625. doi: 10.1146/annurev-cellbio-092910-154244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archibald A, Al-Masri M, Liew-Spilger A, McCaffrey L. Atypical protein kinase C induces cell transformation by disrupting Hippo/Yap signaling. Mol Biol Cell. 2015;26:3578–3595. doi: 10.1091/mbc.E15-05-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paul A, et al. PKCλ/ι signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ. 2014;21:1469–1481. doi: 10.1038/cdd.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen EEW, et al. Protein kinase C zeta mediates epidermal growth factor-induced growth of head and neck tumor cells by regulating mitogen-activated protein kinase. Cancer Res. 2006;66:6296–6303. doi: 10.1158/0008-5472.CAN-05-3139. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein B, Macara IG. The PAR Proteins: Fundamental Players in Animal Cell Polarization. Dev Cell. 2007;13:609–622. doi: 10.1016/j.devcel.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- 25.Prehoda KE. Polarization of Drosophila Neuroblasts During Asymmetric Division. Cold Spring Harb Perspect Biol. 2009;1:a001388. doi: 10.1101/cshperspect.a001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong W, et al. A conserved polybasic domain mediates plasma membrane targeting of Lgl and its regulation by hypoxia. J Cell Biol. 2015;211:273–286. doi: 10.1083/jcb.201503067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey MJ, Prehoda KE. Establishment of Par-Polarized Cortical Domains via Phosphoregulated Membrane Motifs. Dev Cell. 2015;35:199–210. doi: 10.1016/j.devcel.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayraktar OA, Boone JQ, Drummond ML, Doe CQ. Drosophila type II neuroblast lineages keep Prospero levels low to generate large clones that contribute to the adult brain central complex. Neural Develop. 2010;5:26. doi: 10.1186/1749-8104-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai SL, Doe CQ. Transient nuclear Prospero induces neural progenitor quiescence. eLife. 2014;3:e03363. doi: 10.7554/eLife.03363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–1253. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 31.Harris RE, Pargett M, Sutcliffe C, Umulis D, Ashe HL. Brat Promotes Stem Cell Differentiation via Control of a Bistable Switch that Restricts BMP Signaling. Dev Cell. 2011;20:72–83. doi: 10.1016/j.devcel.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komori H, Xiao Q, McCartney BM, Lee CY. Brain tumor specifies intermediate progenitor cell identity by attenuating β-catenin/Armadillo activity. Dev Camb Engl. 2014;141:51–62. doi: 10.1242/dev.099382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knoblich JA, Jan LY, Jan YN. Asymmetric segregation of Numb and Prospero during cell division. Nature. 1995;377:624–627. doi: 10.1038/377624a0. [DOI] [PubMed] [Google Scholar]
- 34.Chang KC, et al. Interplay between the transcription factor Zif and aPKC regulates neuroblast polarity and self-renewal. Dev Cell. 2010;19:778–785. doi: 10.1016/j.devcel.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Atwood SX, Chabu C, Penkert RR, Doe CQ, Prehoda KE. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J Cell Sci. 2007;120:3200–3206. doi: 10.1242/jcs.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garrard SM, et al. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 2003;22:1125–1133. doi: 10.1093/emboj/cdg110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutterer A, Betschinger J, Petronczki M, Knoblich JA. Sequential Roles of Cdc42, Par-6, aPKC, and Lgl in the Establishment of Epithelial Polarity during Drosophila Embryogenesis. Dev Cell. 2004;6:845–854. doi: 10.1016/j.devcel.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- 39.Lee CY, Robinson KJ, Doe CQ. Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature. 2006;439:594–598. doi: 10.1038/nature04299. [DOI] [PubMed] [Google Scholar]
- 40.Chabu C, Doe CQ. Dap160/intersectin binds and activates aPKC to regulate cell polarity and cell cycle progression. Dev Camb Engl. 2008;135:2739–2746. doi: 10.1242/dev.024059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goh LH, et al. Clueless regulates aPKC activity and promotes self-renewal cell fate in Drosophila lgl mutant larval brains. Dev Biol. 2013;381:353–364. doi: 10.1016/j.ydbio.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 42.Choi W, Harris NJ, Sumigray KD, Peifer M. Rap1 and Canoe/afadin are essential for establishment of apical-basal polarity in the Drosophila embryo. Mol Biol Cell. 2013;24:945–963. doi: 10.1091/mbc.E12-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Speicher S, Fischer A, Knoblich J, Carmena A. The PDZ protein Canoe regulates the asymmetric division of Drosophila neuroblasts and muscle progenitors. Curr Biol CB. 2008;18:831–837. doi: 10.1016/j.cub.2008.04.072. [DOI] [PubMed] [Google Scholar]
- 44.Lu MS, Johnston CA. Molecular pathways regulating mitotic spindle orientation in animal cells. 2013;140:1843–1856. doi: 10.1242/dev.087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vorhagen S, Niessen CM. Mammalian aPKC/Par polarity complex mediated regulation of epithelial division orientation and cell fate. 2014;328:296–302. doi: 10.1016/j.yexcr.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Lange C, Calegari F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle. 2010;9:1893–1900. doi: 10.4161/cc.9.10.11598. [DOI] [PubMed] [Google Scholar]
- 47.Lange C, Huttner WB, Calegari F. Cdk4/CyclinD1 Overexpression in Neural Stem Cells Shortens G1, Delays Neurogenesis, and Promotes the Generation and Expansion of Basal Progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 48.Sabherwal N, et al. The apicobasal polarity kinase aPKC functions as a nuclear determinant and regulates cell proliferation and fate during Xenopus primary neurogenesis. Development. 2009;136:2767–2777. doi: 10.1242/dev.034454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabherwal N, Thuret R, Lea R, Stanley P, Papalopulu N. aPKC phosphorylates p27Xic1, providing a mechanistic link between apicobasal polarity and cell-cycle control. Dev Cell. 2014;31:559–571. doi: 10.1016/j.devcel.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 51.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 52.Ruiz i Altaba A, Sánchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002;2:361–372. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- 53.Ng JMY, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011;11:493–501. doi: 10.1038/nrc3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rubin AI, Chen EH, Ratner D. Basal-Cell Carcinoma. N Engl J Med. 2005;353:2262–2269. doi: 10.1056/NEJMra044151. [DOI] [PubMed] [Google Scholar]
- 55.Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang K, et al. Hedgehog-regulated atypical PKC promotes phosphorylation and activation of Smoothened and Cubitus interruptus in Drosophila. Proc Natl Acad Sci. 2014;111:E4842–E4850. doi: 10.1073/pnas.1417147111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Justilien V, et al. The PRKCI and SOX2 Oncogenes are Co-amplified and Cooperate to Activate Hedgehog Signaling in Lung Squamous Cell Carcinoma. Cancer Cell. 2014;25:139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atwood SX, et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell. 2015;27:342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niessen MT, et al. aPKCλ controls epidermal homeostasis and stem cell fate through regulation of division orientation. J Cell Biol. 2013;202:887–900. doi: 10.1083/jcb.201307001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 61.Jiang H, Edgar BA. Intestinal stem cells in the adult Drosophila midgut. Exp Cell Res. 2011;317:2780–2788. doi: 10.1016/j.yexcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Azzolin L, et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 63.Stamos JL, Weis WI. The β-Catenin Destruction Complex. Cold Spring Harb Perspect Biol. 2013;5:a007898. doi: 10.1101/cshperspect.a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 65.Llado V, et al. Repression of Intestinal Stem Cell Function and Tumorigenesis through Direct Phosphorylation of β-Catenin and Yap by PKCζ. Cell Rep. 2015;10:740–754. doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Azzolin L, et al. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151:1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 68.Amit S, et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu C, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 70.Colosimo PF, Liu X, Kaplan NA, Tolwinski NS. GSK3beta affects apical-basal polarity and cell-cell adhesion by regulating aPKC levels. Dev Dyn Off Publ Am Assoc Anat. 2010;239:115–125. doi: 10.1002/dvdy.21963. [DOI] [PubMed] [Google Scholar]
- 71.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 72.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–267. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 75.Magee JA, Piskounova E, Morrison SJ. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 77.Guyer RA, Macara IG. Loss of the Polarity Protein Par3 Activates Stat3 Signaling via an aPKC/NF-κB/IL-6 Axis in Mouse Mammary Cells. J Biol Chem. 2015 doi: 10.1074/jbc.M114.621011. jbc.M114.621011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 79.Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Win HY, Acevedo-Duncan M. Atypical protein kinase C phosphorylates IKKαβ in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 2008;270:302–311. doi: 10.1016/j.canlet.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 81.Bunker BD, Nellimoottil TT, Boileau RM, Classen AK, Bilder D. The transcriptional response to tumorigenic polarity loss in Drosophila. eLife. 2015;4 doi: 10.7554/eLife.03189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cameron AJM, Escribano C, Saurin AT, Kostelecky B, Parker PJ. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat Struct Mol Biol. 2009;16:624–630. doi: 10.1038/nsmb.1606. [DOI] [PubMed] [Google Scholar]
- 83.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Facchinetti V, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tobias IS, et al. Protein kinase Cζ exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. Biochem J. 2015 doi: 10.1042/BJ20151013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang C, Shang Y, Yu J, Zhang M. Substrate recognition mechanism of atypical protein kinase Cs revealed by the structure of PKCι in complex with a substrate peptide from Par-3. Struct Lond Engl 1993. 2012;20:791–801. doi: 10.1016/j.str.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 87.Graybill C, Wee B, Atwood SX, Prehoda KE. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J Biol Chem. 2012;287:21003–21011. doi: 10.1074/jbc.M112.360495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopez-Garcia LA, et al. Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem Biol. 2011;18:1463–1473. doi: 10.1016/j.chembiol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 89.Leonard TA, Różycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C βII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hirano Y, et al. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem. 2004;279:31883–31890. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- 91.Lamark T, et al. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 92.Noda Y, et al. Molecular recognition in dimerization between PB1 domains. J Biol Chem. 2003;278:43516–43524. doi: 10.1074/jbc.M306330200. [DOI] [PubMed] [Google Scholar]
- 93.Ivey RA, Sajan MP, Farese RV. Requirements for pseudosubstrate arginine residues during autoinhibition and phosphatidylinositol 3,4,5-(PO4)3-dependent activation of atypical PKC. J Biol Chem. 2014;289:25021–25030. doi: 10.1074/jbc.M114.565671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nolan ME, et al. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008;68:8201–8209. doi: 10.1158/0008-5472.CAN-07-6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bandyopadhyay G, et al. Dependence of insulin-stimulated glucose transporter 4 translocation on 3-phosphoinositide-dependent protein kinase-1 and its target threonine-410 in the activation loop of protein kinase C-zeta. Mol Endocrinol Baltim Md. 1999;13:1766–1772. doi: 10.1210/mend.13.10.0364. [DOI] [PubMed] [Google Scholar]
- 96.Standaert ML, et al. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem. 1997;272:30075–30082. doi: 10.1074/jbc.272.48.30075. [DOI] [PubMed] [Google Scholar]
- 97.Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3 activate PKC-zeta by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry (Mosc) 2001;40:249–255. doi: 10.1021/bi0018234. [DOI] [PubMed] [Google Scholar]
- 98.Bourbon NA, Yun J, Kester M. Ceramide Directly Activates Protein Kinase C ζ to Regulate a Stress-activated Protein Kinase Signaling Complex. J Biol Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- 99.Wang G, Krishnamurthy K, Umapathy NS, Verin AD, Bieberich E. The Carboxyl-terminal Domain of Atypical Protein Kinase Cζ Binds to Ceramide and Regulates Junction Formation in Epithelial Cells. J Biol Chem. 2009;284:14469–14475. doi: 10.1074/jbc.M808909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin D, et al. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- 101.Petronczki M, Knoblich JA. DmPAR-6 directs epithelial polarity and asymmetric cell division of neuroblasts in Drosophila. Nat Cell Biol. 2001;3:43–49. doi: 10.1038/35050550. [DOI] [PubMed] [Google Scholar]