

Abstract
AIM
The frequency and impact of symptoms experienced by patients with congenital, childhood, and juvenile-onset myotonic dystrophy (CDM/ChDM/JDM) is not documented. This report identifies symptomatic areas with the greatest disease burden in an international population of patients with early-onset myotonic dystrophy type-1 (DM1).
METHOD
We distributed surveys to parents of patients with CDM/ChDM/JDM. Patients with CDM/ChDM/JDM were members of the US National Registry of DM1 Patients and Family Members, the Canadian Neuromuscular Disease Registry, or the Swedish Health System. Surveys inquired about 325 symptoms and 20 themes associated with CDM/ChDM/JDM. Parents identified the importance of each symptom and theme to their affected child. The prevalence of each symptom and theme were compared across subgroups of patients. The statistical analysis was performed using Fisher’s exact and Kruskal–Wallis tests.
RESULTS
One hundred and fifty parents returned surveys. The most frequently reported symptomatic themes in children were issues involving communication (81.7%) and problems with hands or fingers (79.6%). Problems with communication and fatigue were the issues that were reported to have the greatest impact on childrens’ lives, while 24.1% of children reported cardiac disorders and 15.8% had problems with anesthesia.
INTERPRETATION
A range of symptoms contribute to the burden of disease faced by children with DM1. Many of these symptoms are under-recognized.
Myotonic dystrophy type-1 (DM1) is a multisystemic, autosomal dominant disorder caused by an unstable CTG repeat in the DMPK gene on chromosome 19q13.3.1–3 The CTG repeat length may expand from generation to generation and be associated with more severe disease manifestations and an earlier onset of symptoms.4 In adults, DM1 classically presents with distal weakness, early onset cataracts, and delayed muscle relaxation (myotonia).4 When manifestations appear at birth, patients are classified as having congenital myotonic dystrophy (CDM).5,6 Patients who manifest symptoms after 1 year of age and before 10 years old, are said to have childhood myotonic dystrophy (ChDM).5 Patients with symptom onset after age 10 years are considered to have juvenile-onset myotonic dystrophy (JDM). Despite these classifications, myotonic dystrophy likely represents a continuum of disease severity rather than a series of discrete pathogenic steps.
Patients with CDM may demonstrate their first symptoms during gestation, with reduced fetal movements and polyhydramnios.7 At birth, infants with CDM may present with hypotonia, respiratory distress, generalized weakness, failure to thrive, feeding difficulties, or a club-foot deformity.4,6,8 For infants requiring prolonged ventilation, current care indicates a mortality rate as high as 25% during the first year.9 Children with CDM frequently have intellectual disability and profound dysarthria.10–12 There is also a higher reported rate of autism spectrum disorder (ASD) and attention-deficit–hyperactivity disorder (ADHD) in these children.13
Individuals with ChDM with symptom onset shortly after 1 year of age experience many of the same symptoms as children with CDM. Studies have shown that individuals with ChDM frequently experience intellectual disability and behavioral disturbances such as ADHD and ASD.12–14 Among the current authors who care for patients with ChDM (NEJ, CC, A-BE, CRH, and RTM), our anecdotal clinical experience suggests that children with ChDM with a later symptom onset often have fewer cognitive symptoms despite having significant hand myotonia (delayed muscle relaxation after contraction) and fatigue.
Some patients with DM1 may develop their first symptom after the age of 10 years, but before adulthood (JDM). The symptom severity, manifestations, and comorbidities have not been well described in this cohort.
While, ideally, an individual patient is best suited to comment on the presence and severity of their symptoms, young age, developmental delays, or significant cognitive limitations can all limit the ability of a child with DM1 to identify and quantify the severity of their symptoms. In such cases, a surrogate, such as a parent, can provide valuable insight into their child’s condition.15
Previous studies have focused on the discrete symptomatic domains of children with DM1.8,12–14 The current study seeks to better understand DM1 disease burden and the relationship between disease manifestations and a patient’s quality of life as reported by DM1 parents.
MATERIALS AND METHODS
Participants
We identified patients with myotonic dystrophy with an onset of symptoms before the age of 18 years. In the United States, individuals and parents were recruited from the National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy Patients and Family Members (www.urmc.rochester.edu/neurology/national-registry).16 In Canada, participants and parents were recruited from the Canadian Neuromuscular Disease Registry and from the site investigator’s (CC) neuromuscular clinic. In Sweden, participants were identified via rehabilitation center directors. We sent a survey to the parents of all identified patients with congenital, childhood, and juvenile-onset myotonic dystrophy (CDM/ChDM/JDM). The mother, father, or primary caregiver was given a 3-month period to complete and return the survey. The choice of which parent should complete the survey was left to the individual families. Paper surveys were distributed, though respondents could complete the survey by telephone in the United States and Canada, if desired.
The survey
The survey inquired about 325 potential symptoms of importance and the 20 symptomatic themes that these symptoms represent. These symptoms and themes were previously identified through qualitative interviews with patients with CDM/ChDM/JDM and their parents.17 The survey also inquired about symptoms and issues important to the population with adult-onset DM1.18 Surveys were constructed and worded for parents or caregivers to comment on the symptoms of their affected child. In cases where a parent had more than one child with CDM/ChDM/JDM, they were asked to complete a survey for each of their children with DM1. Demographics for both the parent and each child with DM1 were collected. In addition, parents were asked about their child’s medical history and developmental milestones. Results regarding these medical diagnoses (e.g. ADHD, intellectual disability, difficulty with anesthesia, cardiac problems) were based on parent report.
To create the Swedish version of the parent proxy surveys, a native English speaker who was fluent in Swedish translated the survey into Swedish. Two bilingual Swedish physicians reviewed the translation for clarity and grammar. A native Swedish speaker then back-translated the survey from Swedish to English.19 The two English versions were reviewed for compatibility and final corrections were made. For each symptom, parents identified if their child experienced the symptom and subsequently rated the relative impact the symptom had on the child’s life using a Likert scale.
All surveys and methods were approved by local ethics boards (Institutional Review Board).
Statistical analysis
The prevalence of each symptom and theme were calculated. For children who experienced an individual symptom, an average life impact score (mean impact) was calculated (0=‘My child experiences this but it does not affect his or her life’; 1=‘It affects my child’s life a little’; 2=‘It affects my child’s life moderately’; 3=‘It affects my child’s life very much’; and 4=‘It affects my child’s life severely’) and the population impact score (0–4) was calculated as the average life impact score of those endorsing the symptom, multiplied by the prevalence of the symptom.18
Analyses used parent responses and responses were categorized in several ways. These analyses sought to identify changes associated with age at onset, length of CTG repeat length, and age of the child at the time the parent completed the survey. First, responses were analyzed based on the disease onset of the patient with DM1: CDM (onset <1y); ChDM (onset between 1–10y); and juvenile DM1 (onset between 11–17y). Second, responses were categorized by the CTG repeat length of the child. Finally, we performed a cross-sectional analysis based on the age of the child at the time the survey was completed. These age groupings were chosen in consultation with a pediatric neuropsychologist based on developmental age. Age ranges included: 0 to 4 years, 5 to 7 years, 8 to 11 years, 12 to 17 years, and 18 years and older. To evaluate for the impact of disease onset (CDM, ChDM, or JDM), we calculated the life impact score for each symptomatic theme in our patient groups. Fisher’s exact tests were employed to compare the prevalence of each theme across all different subgroups. Kruskal–Wallis tests were used to compare the life impact scores across different subgroups. A two-tailed p value less than or equal to 0.05 was considered significant.
RESULTS
Demographics
In total, 150 parents responded from the 340 surveys distributed, giving an overall response rate of 44.1% (51% US registry, 44% Canadian, and 35% Swedish). Mothers completed the survey 68.7% of the time and fathers completed the survey 15.0% of the time; both parents assisted in completing the survey 5.4% of the time and another caregiver completed the survey 10.9% of the time. Of the mothers completing the survey, 67.3% had myotonic dystrophy. Of the fathers completing the survey, 13.6% had myotonic dystrophy. The demographic data of the children with DM1 is provided in Table I. Characteristics and medical comorbidities of the children with DM1, classified by CDM, ChDM, and JDM, are provided in Table II.
Table I.
Demographic data
| Number of children with DM1 studied | 150 |
| Children with DM1, sex (%) | |
| Male | 61.3 |
| Female | 38.7 |
| Sex of parent with myotonic dystrophy (%) | |
| Male | 24.4 |
| Female | 70.9 |
| Unknown | 4.7 |
| Mean age of participant with DM1, y (SD) | 17.9 (10.2) |
| Mean age of disease onset, y (SD) | 3.4 (5.0) |
| Mean CTG repeat length | 959.0 |
| Race (%) | |
| Asian | 5.8 |
| Caucasian | 89.3 |
| Other | 4.9 |
| Hispanic | 6.7 |
| Mean gestational age of child with DM1 | 38 weeks |
| Complications during pregnancy | % Yes |
| Pre-eclampsia or eclampsia | 2.0 |
| Low birthweight | 18.7 |
| Polyhydramnios | 5.3 |
| Preterm labour | 11.3 |
| Percent unemployed >18y | 86.7 |
DM1, Myotonic dystrophy type-1.
Table II.
Comparison of developmental and intellectual characteristics of congenital, childhood, and juvenile-onset myotonic dystrophy
| CDM | ChDM | Juvenile DM |
|
|---|---|---|---|
| Numbera | 74 | 54 | 15 |
| Mean age, y (SD) | 13.36 (8.4) | 20.4 (9.2) | 31.4 (8.8) |
| Mean CTG (SD) | 1225.8 (526.4) | 660.09 (396.9) | 565.7 (129.8) |
| Mean age at onset (SD) |
0 | 5.1 (2.8) | 12.9 (1.3) |
| Reported walking age, mo (SD) |
24 (13.2) | 14 (4.1) | 12 (2.4) |
| Reported toilet training age, mo (SD) |
57 (42.1) | 35 (12.5) | 35 (17.4) |
| Reported speaking age, mo (SD) |
31 (25.6) | 21 (10.9) | 19 (10.2) |
| Intellectual impairment, % |
45.8 | 28.3 | 0 |
| ADHD, % | 13.7 | 28.3 | 6.7 |
| Autism spectrum disorder, % |
21.6 | 26.9 | 0 |
Seven parents did not provide an age at onset.
CDM, congenital myotonic dystrophy; ChDM, childhood myotonic dystrophy; DM, myotonic dystrophy; ADHD, attention-deficit–hyperactivity disorder.
Prevalence of themes and symptoms
The prevalence, average life impact score, and population impact score of all of the themes and symptoms for all participants with CDM/ChDM/JDM is provided in Table SI (online supporting information). The most frequent themes in all children were: communication issues (81.8%); problems with hands or fingers (79.6%); and fatigue (78.6%). The frequency of problems with hands and fingers, and fatigue was lower than that reported by patients with adult-onset DM1 who reported a frequency of 93.5% and 90.8% respectively.18 Among all the individual symptoms, the most frequently reported issues were hand weakness (85.1%), difficulty opening jars or bottles (84.3%), and learning difficulties (83.3%) (Table SI).
Table SII (online supporting information) provides the frequency of each theme categorized by the affected child’s current age, CTG repeat length, and sex. Theme prevalence in DM1 children by country is provided in Table SIII (online supporting information). Age was associated with an increased prevalence of many of the themes (Fig. 1a). Problems with the hands or fingers (p=0.002), emotional issues (p=0.001), fatigue (p=0.009), pain (p=0.001), inability to do activities (p=0.026), myotonia (p=0.004), gastrointestinal issues (p=0.014), and social issues (p<0.001) all increased and were dependent on age. Themes less prevalent in the 5- to 7-year-old age group compared to either the 0- to 4-year-old or 8- to 11-year-old age group were problems with shoulders or arms (p=0.005), changed body image (p<0.001), difficulty thinking (p=0.030), problems choking or swallowing (p=0.008), and sleep issues (p=0.001).
Figure 1.
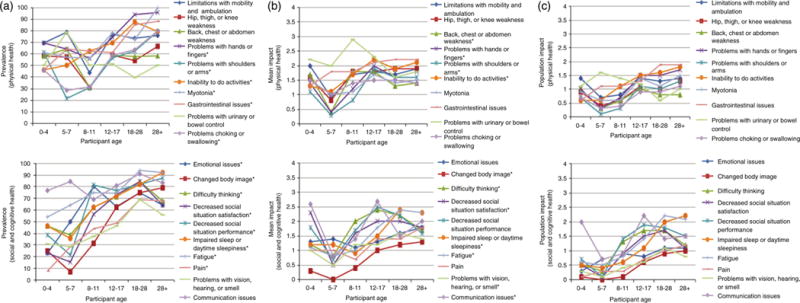
The impact of congenital, childhood, and juvenile-onset myotonic dystrophy by age. (a) Prevalence of themes by age. (b) Mean Likert response by age. (c) Impact score by age. *Indicates p<0.05.
The frequency of many themes was associated with CTG repeat length (Fig. 2a). Among these themes, proximal leg weakness (p=0.02), truncal weakness (p=0.04), and problems with urinary or bowel control (p=0.003) were all more frequent in children with high CTG repeat while myotonia (p=0.001) was less frequent in patients with longer CTG repeat expansions. A higher frequency of some themes was associated with a paternal inheritance of the disease. These themes included: emotional issues (p=0.03), changes in body image (p=0.02), social issues (p=0.01), and impaired sleep (p=0.008) (Table SII).
Figure 2.
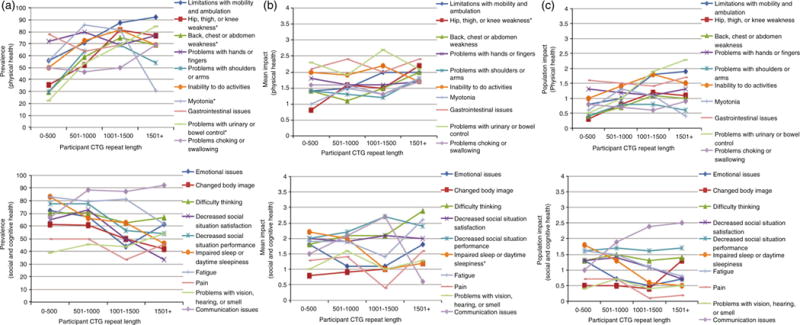
The association of congenital, childhood, and juvenile-onset myotonic dystrophy with CTG repeat length. (a) Prevalence of themes by CTG repeat length. (b) Mean Likert response by CTG repeat length. (c) Impact score by CTG repeat length. *Indicates p<0.05.
Impact of themes and symptoms
Average life impact scores
In children with DM1, the highest life impact scores were, gastrointestinal issues (2.1), problems with urinary or bowel control (2.1), and decreased performance in social situations (2.0) had the highest average life impact scores. These themes are divergent from those themes that have the greatest effect on adult-onset DM1 patients. In our previous study, patients with adult-onset DM1 identified fatigue (2.5), mobility limitations (2.4), and an inability to do specific activities (2.4) as the themes with the highest average life impact scores.18 Similar to the frequency of themes, the impact of some themes was associated with age (Fig. 1b). These themes included: problems with hands or fingers (p=0.001), communication issues (p=0.002), impaired sleep (p<0.001), fatigue (p<0.001), and changed body image (p=0.046).
The average life impact of one theme, proximal leg weakness, was associated with CTG repeat length (p=0.033).
The average life impact of only one theme, pain, was associated with which parent had myotonic dystrophy. In instances where the father had the disease, pain had a higher life impact score for the child (p=0.04) (Table SII).
Population impact
The themes with the greatest population impact scores were: communication issues (1.6), fatigue (1.5), and gastrointestinal issues (1.5). When evaluating individual symptoms, learning difficulties (2.0), reliance on family members (1.9), and difficulty with math (1.7) had the highest population impact scores (Table SI).
During subgroup analysis, the population impact score of themes in children with DM1 reaching adulthood (18-to 28-year-old age group) varied depending on when they first developed symptoms (i.e. if the patient had CDM, ChDM or JDM) (Fig. S1, online supporting information). Participants with adult CDM and ChDM had higher population impact scores than participants with for all themes with the exception of emotional issues and problems with urinary and bowel control (Fig. S1).
Social factors
In total, 87.7% of parents reported that their child needed some form of special assistance in school. This assistance included speech therapy (55.3% of children), occupational therapy (40.7%), physical therapy (35.3%), smaller classroom size (42.7%), or test modification (42%). Augmentive speech methods were used by 19.2% of the children (sign language, device, etc.). Children with DM1 currently over the age of 18 years had significant limitations in their ability to attain employment (Table I).
CDM subgroup analysis
Of the participants with CDM, 77% had hypotonia at birth, 25.7% had clubfoot, and 42.5% were reported to have difficulty with breathing at birth. For those requiring ventilatory assistance (n=43), the mean duration of ventilation was 3 weeks (SD 1.9).
Medical complications
When assessing for anesthesia complications, 15.8% of patients with early-onset DM1 had difficulty with general anesthesia, 56.8% were reported to have no difficulty with anesthesia, and 27.4% had not yet had anesthesia. Diabetes was diagnosed in 2.7% of the children. Cardiac arrhythmias were reported in 24.1% of the population with CDM. The majority of children did not require use of assistive devices (ankle braces, canes or walkers, wheelchairs, or motorized wheelchairs) (59.3%). Patients with CMD who did require assistive devices used: ankle or leg braces (22.7%); canes or walkers (10.7%); wheelchairs (14.0%); and motorized wheelchairs (6.0%). A history of scoliosis was reported in 15.9% of children, with 1.4% of the total population with CDM requiring surgery because of this complication.
DISCUSSION
This study provides parental insight into the disease burden and progression of disease experienced by patients with early-onset myotonic dystrophy (CDM, ChDM, and JDM). DM1 parents highlight the wide spectrum of symptomatic disease in early-onset DM1, and identify common comorbid conditions potentially amendable to screening.
Children with early-onset DM1, particularly those with CDM, had a high rate of intellectual disability, ADHD, and ASD compared to the general population. The general population has an intellectual disability rate of 0.71%.20 Here, parents reported a 45.8% frequency of intellectual disability in children with CMD and a 28.3% rate in children with ChDM. In addition, the frequency of ASD in children with CDM and ChDM was 21.6% and 26.9% respectively, compared to a published frequency of 9% in the general population.21 For ADHD, the prevalence in the general population is 5%, compared to 13.1% and 28.1% in the population with CDM and ChDM respectively.22
It is noteworthy that the reported frequency of cardiac conduction defects was 24% in the children with DM1 in our study. This finding, and the association between cardiac conduction defects and sudden death in DM123 may warrant early screening for conduction defects in younger patients with DM1. Similarly, the high rate of reported anesthesia complications reinforces previous recommendations regarding careful pre-, peri-, and post-anesthesia care in all patients with DM1.24
This cross-sectional study provides a preliminary evaluation of symptom progression during pediatric-onset DM1. There are clusters of symptoms that have the greatest impact on patients before the age of 5 years. Some of these symptoms reportedly improved during the next age cohort (5–7y) before again progressing. We suspect that this reported course of disease progression is caused by many factors. These factors likely include standard milestone acquisition by children, adaptive strategies adopted by DM1 children and parents, possible parent report bias, and perhaps pathological disease progression associated with DM1. Additional longitudinal studies are required for clarification.
Many of the symptoms and themes affecting the burden of disease in childhood-onset myotonic dystrophy are different than those reported in adult-onset myotonic dystrophy.18 This is likely related to parent symptom perception and the relatively more severe phenotype associated with early-onset myotonic dystrophy. The differences between the clinical manifestations and disease burden experienced by adult-onset DM1 and childhood-onset DM1 underscores the need to approach patients with childhood-onset myotonic dystrophy differently and with a focus on the issues that are of greatest importance to the given population.
Specifically, our survey results highlight that communication problems are important in children with DM1. Treating clinicians who recognize this issue in patients with DM1 may consider an early referral for speech therapy. Moreover, the reports of cardiac arrhythmias in our young patients with DM1 suggest that early cardiac monitoring should be considered. Finally, the significant cognitive, behavioral, and social issues identified reinforce the role of a multidisciplinary approach in caring for these children.
When analyzing the disease burden of patients with early-onset DM1 who reach adulthood, participants who had CDM had higher population impact scores for most themes compared to those who had ChDM and JDM. This observation supports the possible association between a later disease onset and a reduced disease severity later in life.
There are limitations to this study. All items were parent reported (e.g. the presence or absence of cardiac conduction defects) and not confirmed by a review of medical records. This is specifically of concern in relation to the report of medical comorbidities, including the use of a tracheostomy, pacemaker, and the occurrence of anesthesia complications. Additional research should further investigate the prevalence of these issues as well as the use of a gastrostomy tube and non-invasive ventilation in the population with DM1. Second, though the response rate was adequate,25 it is likely that the respondents do not perfectly represent the world’s population of early-onset DM1 parents. While three countries were represented (and provided similar parent responses), it is unknown how participants from other countries would have responded to similar survey questions. Third, our data directly represents the opinions of myotonic dystrophy patient caregivers. While it is probable that caregivers can provide a reasonable overview of the disease burden faced by their children, it is possible that certain components of disease burden are known only to individual patients and would be misrepresented by our methods. Additionally, this data is partially provided by parents with DM1, and their disease manifestations may bias their report. Additional studies will need to evaluate the correlation between children with myotonic dystrophy and caregiver reports of symptoms.
This large cross-sectional, multi-national study helps to better understand the disease progression and disease burden experienced by patients with early-onset myotonic dystrophy. Future studies will need to evaluate which factors and treatments can best reduce the disease burden faced by this population.
Supplementary Material
Figure SI: The impact of congenital, childhood, and juvenile-onset myotonic dystrophy in adulthood. (a) The impact score for physical health themes in participants aged 18 to 28 years, as separated by symptom onset. (b) The impact score for physical health themes in participants aged 18 to 28 years, as separated by symptom onset.
Table SI: Parent-reported responses to individual and theme items, including the prevalence, mean impact, and population impact score.
Table SII: Prevalence and impact of themes by age, (CTG) repeat length, and inheritance pattern.
Table SIII: Theme responses separated by country of origin.
What this study adds.
Communication issues, fatigue, and problems with hands and fingers are important in childhood-onset myotonic dystrophy.
The age at onset, CTG repeat length, and age of the child affect the severity of symptoms.
Up to 24% of children with childhood-onset myotonic dystrophy have cardiac problems at a young age.
Acknowledgments
This research is supported by the Myotonic Dystrophy Foundation. The funding source did not participate in study design, data analysis, or manuscript decisions.
ABBREVIATIONS
- ASD
Autism spectrum disorder
- CDM/ChDM/JDM
Congenital, childhood, and juvenile-onset myotonic dystrophy
- DM1
Myotonic dystrophy type-1
Footnotes
The authors have stated that they had no interests that might be perceived as posing a conflict or bias.
SUPPORTING INFORMATION
The following additional material may be found online.
References
- 1.Fu YH, Pizzuti A, Fenwick RG, Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–8. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–5. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 3.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 4.Harper P. Myotonic Dystrophy. 3rd. London: W.B. Saunders; 2001. [Google Scholar]
- 5.Koch MC, Grimm T, Harley HG, Harper PS. Genetic risks for children of women with myotonic dystrophy. Am J Hum Genet. 1991;48:1084–91. [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell C, Levin S, Siu VM, Venance S, Jacob P. Congenital myotonic dystrophy: Canadian population-based surveillance study. J Pediatr. 2013;163:120–5. e1–3. doi: 10.1016/j.jpeds.2012.12.070. [DOI] [PubMed] [Google Scholar]
- 7.Zaki M, Boyd PA, Impey L, Roberts A, Chamberlain P. Congenital myotonic dystrophy: prenatal ultrasound findings and pregnancy outcome. Ultrasound Obstet Gynecol. 2007;29:284–8. doi: 10.1002/uog.3859. [DOI] [PubMed] [Google Scholar]
- 8.Canavese F, Sussman MD. Orthopaedic manifestations of congenital myotonic dystrophy during childhood and adolescence. J Pediatr Orthop. 2009;29:208–13. doi: 10.1097/BPO.0b013e3181982bf6. [DOI] [PubMed] [Google Scholar]
- 9.Campbell C, Sherlock R, Jacob P, Blayney M. Congenital myotonic dystrophy: assisted ventilation duration and outcome. Pediatrics. 2004;113:811–16. doi: 10.1542/peds.113.4.811. [DOI] [PubMed] [Google Scholar]
- 10.Ekstrom AB, Hakenas-Plate L, Tulinius M, Wentz E. Cognition and adaptive skills in myotonic dystrophy type 1: a study of 55 individuals with congenital and childhood forms. Dev Med Child Neurol. 2009;51:982–90. doi: 10.1111/j.1469-8749.2009.03300.x. [DOI] [PubMed] [Google Scholar]
- 11.Sjogreen L, Engvall M, Ekstrom AB, Lohmander A, Kiliaridis S, Tulinius M. Orofacial dysfunction in children and adolescents with myotonic dystrophy. Dev Med Child Neurol. 2007;49:18–22. doi: 10.1017/s0012162207000060.x. [DOI] [PubMed] [Google Scholar]
- 12.Wozniak JR, Mueller BA, Bell CJ, Muetzel RL, Lim KO, Day JW. Diffusion tensor imaging reveals widespread white matter abnormalities in children and adolescents with myotonic dystrophy type 1. J Neurol. 2013;260:1122–31. doi: 10.1007/s00415-012-6771-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekstrom AB, Hakenas-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:918–26. doi: 10.1002/ajmg.b.30698. [DOI] [PubMed] [Google Scholar]
- 14.Angeard N, Jacquette A, Gargiulo M, et al. A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1) Neuromuscul Disord. 2011;21:468–76. doi: 10.1016/j.nmd.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Eiser C, Morse R. Can parents rate their child’s health-related quality of life? Results of a systematic review. Qual Life Res. 2001;10:347–57. doi: 10.1023/a:1012253723272. [DOI] [PubMed] [Google Scholar]
- 16.Hilbert JE, Kissel JT, Luebbe EA, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD) Contemp Clin Trials. 2012;33:302–11. doi: 10.1016/j.cct.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson NE, Luebbe E, Eastwood E, Chin N, Moxley RT, 3rd, Heatwole CR. The impact of congenital and childhood myotonic dystrophy on quality of life: a qualitative study of associated symptoms. J Child Neurol. 2014;29:983–6. doi: 10.1177/0883073813484804. [DOI] [PubMed] [Google Scholar]
- 18.Heatwole C, Bode R, Johnson N, et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1) Neurology. 2012;79:348–57. doi: 10.1212/WNL.0b013e318260cbe6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terwee CB, Roorda LD, de Vet HC, et al. Dutch-Flemish translation of 17 item banks from the patient-reported outcomes measurement information system (PROMIS) Qual Life Res. 2014;23:1733–41. doi: 10.1007/s11136-013-0611-6. [DOI] [PubMed] [Google Scholar]
- 20.Boyle CA, Boulet S, Schieve LA, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127:1034–42. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- 21.Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C, Prevention. Prevalence of autism spectrum disorder among children aged 8 years – autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63:1–21. [PubMed] [Google Scholar]
- 22.Association AP. Diagnostic and Statistical Manual of Mental Disorders (5th edition) DSM-5. Washington: American Psychiatric Association; 2013. [Google Scholar]
- 23.Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688–97. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- 24.Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth. 2013;23:794–803. doi: 10.1111/pan.12120. [DOI] [PubMed] [Google Scholar]
- 25.Baruch Y. Response rate in academic studies – a comparative analysis. Hum Rel. 1999;52:421–38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure SI: The impact of congenital, childhood, and juvenile-onset myotonic dystrophy in adulthood. (a) The impact score for physical health themes in participants aged 18 to 28 years, as separated by symptom onset. (b) The impact score for physical health themes in participants aged 18 to 28 years, as separated by symptom onset.
Table SI: Parent-reported responses to individual and theme items, including the prevalence, mean impact, and population impact score.
Table SII: Prevalence and impact of themes by age, (CTG) repeat length, and inheritance pattern.
Table SIII: Theme responses separated by country of origin.