

Abstract
The blood-brain barrier (BBB) is a critical component of the central nervous system (CNS) that regulates the flux of material between the blood and the brain. Because of its barrier properties, the BBB creates a bottleneck to CNS drug delivery. Human in vitro BBB models offer a potential tool to screen pharmaceutical libraries for CNS penetration as well as for BBB modulators in development and disease, yet primary and immortalized models respectively lack scalability and robust phenotypes. Recently, in vitro BBB models derived from human pluripotent stem cells (hPSCs) have helped overcome these challenges by providing a scalable and renewable source of brain microvascular endothelial cells (BMECs). We have demonstrated that hPSC-derived BMECs exhibit robust structural and functional characteristics reminiscent of the in vivo BBB. Here, we provide a detailed description of the methods required to differentiate and functionally characterize hPSC-derived BMECs to facilitate their widespread use in downstream applications.
1.0 INTRODUCTION
1.1 Scope
The brain vasculature is key to central nervous system (CNS) health through its regulation of molecular and cellular traffic across the blood-brain interface. In doing so, brain blood vessels also create a barrier to ions, drugs, pathogens, and CNS-damaging toxins. This so-called blood-brain barrier (BBB) restricts the delivery of CNS therapeutics, thereby hindering CNS drug development. An increasing number of studies have also indicated BBB dysfunction in many CNS-related pathologies, including Alzheimer’s disease [1], stroke [2], and traumatic brain injury [3].
At the cellular level, the BBB is comprised of brain-specific endothelial cells that line the brain vasculature. These brain microvascular endothelial cells (BMECs) are distinct from other tissue-specific endothelium through expression of a unique subset of tight junction proteins, nutrient transporters, and efflux transporters [4,5] that play important roles in BBB physiology. For instance, tight junction proteins limit paracellular diffusion, nutrient transporters regulate the flow of energy and amino acids across the CNS, and efflux transporters remove a wide array of therapeutics from the CNS [4,6]. In vitro BBB tissue models, including primary cells isolated from brain capillaries [7–10] and immortalized primary BMEC lines [11–13], recapitulate many properties of the in vivo BBB. However, species-specific differences in BMEC efflux transporter expression reduce the utility of animal-derived in vitro BBB modeling for human health applications [14,15]. In addition, scarce availability of primary human BMECs [16] and reduced barrier properties of immortalized human BMEC lines limit application of these models [13].
Recent advancements in stem cell technology have overcome several of these material challenges leading to protocols whereby BMEC-like cells are derived from expandable and renewable human stem cell sources. These sources include cord blood-derived hematopoietic stem cells [17], circulating cord blood-derived endothelial progenitors [18], and human pluripotent stem cells (hPSCs) [19]. Human PSC-derived BMECs exhibit physically tight barriers similar to the in vivo BBB and functionally active transporters, including the most well characterized transporter, P-glycoprotein (P-gp). In addition, hPSC-derived BMECs exhibit permeabilities to a range of small molecules that correlate well with those observed in rodents [19]. Human PSC-derived BMECs also offer the unique ability to examine cellular signaling events involved in human BBB development as undifferentiated hPSCs undergo cell fate decisions towards BMECs. Disease phenotypes with putative BMEC involvement may also be examined using induced pluripotent stem cell (iPSC) lines derived from patients.
The hPSC-derived BMECs are produced through simultaneous differentiation of endothelial and neural progenitors [19]. This mixed, co-differentiation environment recapitulates some aspects of early BBB development [19], including components of WNT signaling activation [20–22] and responsiveness to retinoic acid (RA) signaling [23], a pathway implicated in BBB regulation [24]. After 8 days of differentiation, the mixed neural progenitor/endothelial population is subcultured onto collagen/fibronectin. Endothelial cells selectively attach to the collagen/fibronectin matrix while neural progenitor cells do not and are thus removed following medium exchange, yielding virtually pure BMEC monolayers. Supplementing differentiating BMEC cultures with RA further enhances hPSC-derived BMEC tightness to levels comparable to the in vivo BBB [23]. BMECs also exhibit responsiveness to other cells from the neurovascular unit, such as pericytes and astrocytes and neurons, offering the ability to examine cellular crosstalk at the BBB [19,23]. In this article, we detail optimized differentiation strategies as well as the characterization required to validate hPSC-derived BMEC monocultures.
1.2 Overview of BMEC Differentiation and Characterization Procedures
The hPSC-derived BMEC differentiation occurs in a four-step process to generate pure BMEC populations. 1) Human PSCs are seeded as singularized cells onto Matrigel-coated plates. 2) Culture density is quantified to ensure it falls within the optimal range, and hPSCs are then differentiated to a mixed endothelial cell/neural progenitor cell culture by switching cells to unconditioned medium (UM) for 6 days (D6) (Figure 1a). 3) Endothelial cells are next selectively expanded by switching to endothelial cell (EC) medium supplemented with RA (EC + RA medium) from D6-8 (Figure 1a). By D8 of the differentiation, endothelial cells express BBB-specific tight junction proteins—including Claudin-5, Occludin, and ZO-1, the nutrient transporter GLUT-1, and the efflux transporter P-gp [19]. 4) To purify BMECs, mixed cultures are subcultured at D8 onto collagen/fibronectin-coated filters or plates; BMECs selectively adhere to the collagen/fibronectin substrate while the neural cells do not, resulting in a virtually pure BMEC monolayer by D9. BMECs subcultured on filters can be used to measure transendothelial electrical resistance (TEER) starting at D9 of the differentiation, while other functional assays such efflux transporter activity are usually performed on D10 when TEER reaches a maximum (Figure 1a). Cells cultured on plates are typically used for verifying BMEC quality by immunocytochemistry at D10, including expression of endothelial cell markers—VE-cadherin and PECAM-1—tight junction proteins, the nutrient transporter Glut-1, and efflux transporters—P-gp, BCRP, and MRP1 (Figure 2).
Figure 1.

Differentiation of IMR90-C4 iPSCs to BMECs. a) Timeline for BMEC differentiation and subculture onto plates and filters. b) Bright field images illustrate morphology and density at i) D-2 ii) D0, iii) D2, and iv) D8 of the differentiation. Scale bar equals 500 μm. c) Immunocytochemistry analysis of neural tracts that develop in between endothelial cell clusters at D8 of differentiation. Confluent cell monolayers at D8 of differentiation, shown i) by DAPI nuclear stain, express ii) the endothelial cell marker VE-cadherin (red, Santa Cruz, Clone BV9, Ms IgG2a) in regions surrounding nestin positive neural tracts (green, Millipore, Clone 10C2, Ms IgG1). Scale bar equals 200 μm.
Figure 2.

Immunocytochemistry of BMEC Markers in BMEC Monolayers at D10. a) Expression of endothelial cell markers PECAM-1 (Thermo Scientific ‘Labvision’, Cat# RB-10333, Rb polyclonal) and VE-cadherin (Santa Cruz, BV9, Ms IgG2a); tight junction and associated proteins Claudin-5 (Invitrogen, Clone 4C3C2, IgG1), Occludin (Invitrogen, Clone OC-3F10, IgG1), ZO-1; and the nutrient transporter Glut1 (Thermo Scientific ‘Labvision’, Clone SPM498, Mouse IgG2a). Scale bar equals 50 μm. b) Expression of efflux transporters P-gp (Thermo Scientific ‘Neomarkers’, anti-p170 Clone F4, Mouse IgG1), MRP1 (Millipore, Clone QCRL1, Mouse IgG1), and BCRP (Millipore, MAB4155 clone 5D3, Mouse IgG2b) at D10. Scale bar equals 50 μm.
2.0 MATERIALS
2.1 Human PSC Lines Validated for BBB Differentiation
IMR90-C4 (WiCell WB008)
WA09 (WiCell RB-001)
iPSC DF19-9-11T.H. (WiCell WB0219)
2.2 Materials
Costar™ cell culture plates (96, 48, 24, 12, or 6 well plates (Corning 3596, 3548, 3524, 3513, 3516)
15 mL conical tubes (Fisher 05-539-5)
Microfuge tubes (Fisher 02-681-272)
500 mL filter-top bottles (Thermo 566-0020)
Corning Transwell polyester filters 12 or 24 well (Sigma CLS3460, CLS3450)
5 mL and 10 mL glass pipettes
2.3 Equipment
Hemacytometer
Pipette aid
Fluorescent plate reader
EVOM with STX electrodes
Biosafety cabinet
Incubator (37°C and 5% carbon dioxide), containing a rotating platform
2.4 Reagents
mTeSR™1 (Stem Cell Technologies 05850)
Matrigel™, growth factor reduced (BD Biosciences 354230)
Versene (Invitrogen 15040-066)
ROCK inhibitor, Y27632 dihydrochloride (Tocris 1254)
Accutase (Invitrogen A1110501)
Trypan blue (Invitrogen 15250-061)
DMEM/F12 (Invitrogen 11330-057)
Nonessential amino acids (NEAA) (Invitrogen 11140-050)
Glutamax (Invitrogen 35050-061)
Knockout serum replacement (KOSR) (Invitrogen 10828-028)
β-Mercaptoethanol (Sigma M3148)
Human endothelial serum-free medium (hESFM) (Invitrogen 11111)
Platelet poor plasma-derived serum, bovine (PDS) (Fisher 50-443-029)
Human basic fibroblast growth factor (bFGF) (R&D Systems 233-FB)
All-trans retinoic acid (Sigma R2625)
Fibronectin (Sigma F1141)
Collagen IV (Sigma C5533)
Glacial acetic acid (Sigma 537020)
DMSO (Sigma D2650)
Methanol (Fisher A412-4)
32% paraformaldehyde in water (VWR 100496-496)
PBS (Sigma D8537)
Goat serum (Sigma G9023)
Triton-X 100 (Sigma T8787)
DAPI (Life Technologies D1306) diluted to 5 mg/mL in deionized water
Sodium fluorescein (Sigma F6337)
Rhodamine 123 (Sigma 83702)
Cyclosporin A (CsA) (Sigma C1832)
RIPA buffer (Sigma R0278)
HBSS buffer (Life Technologies 14025092)
Ethanol (Fisher BP2818-4)
Tissue-grade sterile water (Sigma 59900C)
2.5 Material Preparation
All the following preparations are made within a biosafety cabinet under sterile conditions unless otherwise indicated.
2.5.1 Aliquoting Matrigel
Matrigel rapidly forms a gel above 4°C and cannot be aliquoted once it has gelled. Therefore, when aliquoting Matrigel, work quickly and on ice. Follow manufacturer instructions for aliquoting Matrigel into 2.5 mg aliquots for coating five 6-well plates. Store aliquots at −20 to −80°C.
2.5.2 Matrigel-Coated Plates
Matrigel-coated plates can be made up to seven days in advance of starting a differentiation or passaging stem cells for maintaining pluripotent stem cell cultures. Coated plates are typically stored at 37°C until ready to use, up to one week. To coat a 6-well plate, resuspend 0.5 mg of Matrigel in 6 mL DMEM/F12 and aliquot 1 mL solution per well of a 6 well plate. If plates are not used within 24 hours, add an additional 1 mL of DMEM/F12 per each well of a 6-well plate to prevent Matrigel-coated plates from drying out. Dispose any plates with dried coating solution.
2.5.3 UM
In a 500 mL filter unit, combine 392.5 mL DMEM/F12 with 100 mL of KOSR, 5 mL of non-essential amino acids, 2.5 mL of Glutamax, and 3.5 μL of β-mercaptoethanol. Sterile-filter components and store at 4°C for up to two weeks.
2.5. EC + RA Medium
Resuspend lyophilized bFGF in 0.1% BSA in PBS to a final concentration of 100 μg/mL. Store 50 μL aliquots at −20°C. Filter sterilize PDS before use and store 1 mL aliquots at −20°C. Resuspend RA in DMSO to 10 μM and store in 20 μL aliquots at −20°C. To make EC medium, add 1 mL of filter-sterilized PDS to 100 mL of hESFM and bring to 20 ng/mL bFGF (1:5000 dilution of stock bFGF solution). Filter sterilize before use. EC medium stores for one week. Add 10μM RA to EC medium just prior to feeding cells.
NOTE: RA is light sensitive. Use EC + RA medium immediately (do not store).
2.5.5 4:1:5 collagen/fibronectin/water Extracellular Matrix (ECM) Coating Solution
Dissolve 5 mg of collagen IV in 5 mL of sterile-filtered 0.5 mg/mL acetic acid. Mix 5 parts sterile tissue-grade water with 4 parts collagen IV (1 mg/mL) and 1 part fibronectin (1 mg/mL). This solution is used to coat Transwell filters for measuring TEER or molecular permeability (e.g. sodium fluorescein). The 4:1:5 collagen/fibronectin /water ECM coating solution can be diluted 5x in sterile tissue-grade water to coat cell culture plates for immunocytochemistry or efflux transporter assays.
2.5.6 Immunolabeling blocking buffer
Dilute goat serum in PBS to the appropriate concentration for immunolabeling with the desired antibody (see Table 3). To immunolabel an intracellular epitope with paraformaldehyde-fixed cells, add 0.1% Triton-X 100 to the blocking buffer to permeabilize cells.
Table 3.
Antibodies and Staining Conditions for Immunocytochemistry
| Antibody | Vendor, clone or product#, species | Fixative | Dilution | Block | Secondary Antibody |
|---|---|---|---|---|---|
| PECAM-1 | Thermo Sci (Labvision), RB-10333, Rb polyclonal | MeOH only | 1:25 | 10% PBSG | Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11034) |
| Glut-1 | Thermo Sci (Labvision), SPM498, Mouse IgG2a | MeOH | 1:100–1:500 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| VE-cadherin | Santa Cruz, BV9, Ms IgG2a | MeOH | 1:25 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) Goat anti-Mouse IgG2a Secondary Antibody, Alexa Fluor® 594 conjugate (Life A-21135) |
| Santa Cruz, F-8, IgG1 | MeOH | 1:100 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) | |
| Occludin | Invitrogen, OC-3F10, IgG1 | MeOH | 1:200 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| ZO-1 | Invitrogen, 40–2200, Rb polyclonal | MeOH | 1:100 | 10-% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| Claudin-5 | Invitrogen, 4C3C2, IgG1 | MeOH | 1:50 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| P-gp | Thermo Scientific (Neomarkers), p170 (F4), Mouse IgG1 | MeOH | 1:25 | 10% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| BCRP | Millipore, MAB4155 clone 5D3, Mouse IgG2b-kappa | 4% PFA only | 1:50 | 10% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| MRP1 | Millipore, MAB4100 clone QCRL1, Mouse IgG1 | MeOH | 1:25 | 40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| Nestin | Millipore, 10C2, Ms IgG1 | MeOH | 1:500 | 40% PBSG | Goat anti-Mouse IgG1 Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-21121) |
| Antibody | Vendor, clone or product #, species | Fixative | Dilution | Block | Secondary Antibody |
|---|---|---|---|---|---|
| PECAM-1 | Thermo Sci (Labvision), RB-10333, Rb polyclonal | MeOH only | 1:25 | 10% PBSG | Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11034) |
| Glut-1 | Thermo Sci (Labvision), SPM498, Mouse IgG2a | MeOH | 1:100–1:500 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| VE-cadherin | Santa Cruz, BV9, Ms IgG2a | MeOH | 1:25 | 10–40% PBSG | Goat anti-Mouse IgG2a Secondary Antibody, Alexa Fluor® 594 conjugate (Life A-21135) |
| Santa Cruz, F-8, IgG1 | MeOH | 1:100 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) | |
| Occludin | Invitrogen, OC-3F10, IgG1 | MeOH | 1:200 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| ZO-1 | Invitrogen, 40–2200, Rb polyclonal | MeOH | 1:100 | 10-% PBSG | Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11034) |
| Claudin-5 | Invitrogen, 4C3C2, IgG1 | MeOH | 1:50 | 10–40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| P-gp | Thermo Scientific (Neomarkers), p170 (F4), Mouse IgG1 | MeOH | 1:25 | 10% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| BCRP | Millipore, MAB4155 clone 5D3, Mouse IgG2b | 4% PFA only | 1:50 | 10% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| MRP1 | Millipore, MAB4100 clone QCRL1, Mouse IgG1 | MeOH | 1:25 | 40% PBSG | Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-11001) |
| Nestin | Millipore, 10C2, Ms IgG1 | MeOH | 1:500 | 40% PBSG | Goat anti-Mouse IgG1 Secondary Antibody, Alexa Fluor® 488 conjugate (Life A-21121) |
2.5.7 Sodium fluorescein
Resuspend sodium fluorescein to a stock concentration of 10 mM in PBS. Store at 4°C in the dark. To make a working concentration, dilute 1:1000 in EC medium (final concentration: 10 μM).
NOTE: Sodium fluorescein is light sensitive. Avoid prolonged exposure to light.
2.5.8 Rhodamine 123
Resuspend Rhodamine 123 to a stock concentration of 10 mM in ethanol. Store at 4°C in the dark. To make a working concentration, dilute 1:1000 in HBSS buffer (final concentration: 10 μM).
NOTE: Rhodamine 123 is light sensitive. Avoid prolonged exposure to light.
2.5.9 CsA
Resuspend CsA to a stock concentration of 10 mM in DMSO. Store at 4°C. To make a working concentration, dilute 1:1000 in HBSS buffer (final concentration: 10 μM).
3.0 PROCEDURE FOR MAINTAINING hPSCS
Human pluripotent stem cells are cultured using the mTeSR1-Matrigel system according to WiCell’s feeder-independent stem cell protocols. When undifferentiated hPSC colonies reach 80% confluence (typically 3–5 days after passage), cells are dissociated using Versene and passaged at 1:6–1:20 split ratios onto Matrigel-coated 6-well plates. Human pluripotent stem cells can typically undergo BMEC differentiation up to passage 50. Consider thawing earlier passage hPSCs when the current line reaches passage 45.
Aspirate medium from cells.
Wash well with Versene by adding 1 mL of cold Versene to each well of a 6-well plate for hPSCs undergoing passaging, and subsequently aspirate Versene from well.
Add 1 mL of Versene to well. Incubate well at 37°C for 7 minutes.
While cells are dissociating, prepare plates that cells will be passaged onto by labeling pre-coated Matrigel plates with the passage date, cell line, passage number, and split ratio. Aspirate Matrigel off the plates and add 1 mL of mTeSR1 to each well of the labeled 6-well plates.
-
Using a 5 mL glass pipette, gently dissociate hPSCs from well by gently spraying 2 mL of mTeSR1 over the cells, collecting the dissociated cell off the plate and spraying the plate again for a total of 2–3x.
NOTE: cells will easily dissociate without need to touching the tip to the well surface or scraping cells off the plate. If 70% of cells do not come off after gently spraying mTeSR1 over the wells 3x, considering incubating cells in Versene for an additional 1–2 minutes at 37°C.
Collect cells in a 15 mL conical tube.
Calculate volume of dissociated cells for appropriate seeding volume. One 6-well Matrigel plate prepared in step 4 will require an additional 6 mL of cells (1 mL of cells for each well). For example, if splitting at a 1:12 ratio, 1 mL of cells collected in step 6 will passage into 6 wells of a 6-well plate. Therefore, add 1 mL of dissociated cells to 5 mL of mTeSR1 to obtain the appropriate 1:12 split ratio for one 6-well plate.
Add 1 mL of cells diluted to the appropriate volume in step 7 to each well the prepared Matrigel plates from step 4. The total volume will be 2 mL of cells per well of a six well plate.
Place plates in the incubator and even distribute cells across plates by shaking back-to-forth, the left-to-right, and then pausing for 3 seconds. Repeat the shaking for a total of three cycles of back-to-forth, left-to-right shaking.
4.0 PROCEDURE FOR DIFFERENTIATING BMECs
4.1: Singularized hPSC seeding and expansion for BMEC differentiation, day minus 3 (D-3)
Begin singularized cell seeding procedure when undifferentiated hPSCs have reached an appropriate split density (i.e. when hPSC colonies reach 80% confluence).
Aspirate medium from cells.
Add 1 mL cold Accutase to each well of hPSCs.
-
Incubate at 37°C until all the cells have completely dissociated (approximately 7 minutes).
NOTE: Prolonged Accutase time (>7 min) can decrease hPSC viability
-
Using a p1000, collect cells, and spray gently over the surface 2–3x to dislodge any remaining cells.
NOTE: Spraying wells too vigorously can decrease cell viability for seeding hPSCs.
Dilute cells 5x in mTeSR1 (i.e. add 1 mL cell suspension in 4 mL mTeSR1).
Spin down cells for 5 minutes at 1000 rpm.
Aspirate medium and resuspend cells in 1 mL mTeSR1 per each well of cells. Thoroughly triturate 5–6 times using p1000 to yield singularized cell suspension.
Take 20 μL of cells to count, drawing from the middle of the sample to prevent bias from settling cells.
In microfuge tube, combine 20 μL cells with 20 μL Trypan Blue (1:1 dilution). Mix well.
-
Count cells on hemacytometer. Exclude blue cells (dead cells) from cell count.
NOTE: One well of a 6-well plate of hPSCs usually yields between 1 – 2.5 million cells depending on the density at split.
-
Calculate appropriate volume of cells to add to each well of a 6-well plate. The ideal number of cells to seed is that which yields approximately 300,000 cells per well of a 6-well plate (30,000 cells/cm2) at the start of differentiation.
NOTE: Seeding number can vary from passage to passage depending on the growth rate of cells. Typical seeding number for IMR90-C4 iPSCs is between 75,000 – 125,000 cells per well of a 6-well plate (7,500 – 12,500 cells/cm2).
-
Add cells to each well of a 6-well plate containing 2 mL mTeSR1 + 10 μM ROCK inhibitor Y27632 per well, always drawing cells from the middle of the sample to prevent bias due to cell settling. Re-triturate as needed.
One well of the newly seeded plate will be used as a counting well at start of differentiation. Make sure to seed enough wells for both counting and experiments.
-
Place plate in 37°C incubator, and shake plate back and forth, pause, shake left-to-right, pause, and repeat shaking cycle 3x total to distribute cells evenly.
NOTE: Do not allow medium in plates to swirl. This will lead to low cell attachment in the center of the well and high cell densities at the edges of the wells.
-
Approximately 24 hours later (i.e., D-2), remove mTeSR1 + ROCK inhibitor and replace with 2 mL mTeSR1 in each well.
NOTE: Stem cells treated with ROCK inhibitor should have a spread, mesenchymal-like morphology (Figure 1b, panel i).
Approximately 24 hours later (i.e., D-1), aspirate the medium and add 2 mL mTeSR1.
4.2 UM Phase: Inducing BMEC/Neural Co-Differentiation (D0–D6)
On D0, repeat section 4.1 steps 1–6 for one well of a 6-well plate (the “counting well”)
Aspirate medium and resuspend cells in mTeSR1 to yield an appropriate cell density for counting on the hemacytometer (generally 300 μL is sufficient).
Take 20 μL of cells to count, drawing from the middle of the sample to prevent bias from settling cells.
In microfuge tube, combine 20 μL of cells with 20 μL Trypan blue.
-
Count cells on hemacytometer. Exclude blue cells (dead cells) from cell count.
NOTE: Starting density at the initiation of differentiation (D0) is extremely important for high quality BMEC differentiations [25]. Optimal number of cells is approximately 300,000 cells per well of a 6-well plate (30,000 cells/cm2) for IMR90-C4 cells. Between 250,000 – 400,000 cells per well of a 6-well plate (25,000 – 40,0000 cells/cm2) is acceptable. If the starting density is below 25,000 cells/cm2 consider waiting one more day before starting the differentiation. Ideal starting density is depicted visually in Figure 1b, panel ii.
Switch remaining wells of cells from mTeSR1 to UM (2 mL per well of a 6-well plate) to initiate differentiation.
Change medium every day using 2 mL of UM per well from D1–5.
4.3 Expanding BMEC Population (D6–8)
-
On D6, aspirate medium and add 2 mL of EC + RA medium per well. Medium is not changed on D7.
NOTE: RA is typically added at D6 while cells are maintained in EC medium and during the subculturing step at D8. If an experiment may be negatively impacted by RA addition, BMECs can be successfully differentiated without RA supplementation D6 and D8 during subculture [19]. However, BMECs will exhibit lower VE-cadherin and occludin expression at D8 of the differentiation and lower TEER [23].
4.4 Subculturing BMECs onto collagen/fibronectin coated filters and plates at D8
-
Prepare collagen/fibronectin-coated plates and filters for subculturing BMECs for each specific assay. Coat Transwells with 4:1:5 collagen/fibronectin/water ECM solution at least 4 hours at 37 °C, or up to 24 hours in advance of subculturing. ECM coating solution volume depends on plate/filter size (Table 1). Coat plates with 4:1:5 collagen/fibronectin/water ECM coating solution for at least 1 hour or up to 24 hours at 37 °C. To save reagents, 4:1:5 collagen/fibronectin/water ECM coating solution can be diluted 5x in sterile water for coating cell culture plates, but should not be diluted for filters.
NOTE: 12-well or 24-well Transwells can be used for measuring TEER. 12-well Transwells are used for measuring sodium fluorescein permeability, 24-well plates are used for measuring efflux transporter activity via accumulation assays, and 96-well plates are used for immunocytochemistry.
Aspirate plates/filters and allow to dry in a sterile hood. Plates only need to dry for 5 minutes, while Transwell filters require 20 minutes.
Rehydrate collagen/fibronectin-coated filters by adding half the working volume/well with hESFM (Table 2). For example, rehydrate one six-well with 1 mL of hESFM.
Retrieve cells from incubator and aspirate spent medium.
Add 1 mL cold Accutase per well of cells at D8.
-
Incubate at 37°C until 90% of cells begin to visibly detach from the plate (either released from surface or rounded up from the surface).
NOTE: Proper Accutase incubation time is critical for maximum TEER. Typical dissociation time is 30 minutes, but can vary between 20 minutes to 1 hour depending on the passage. It is recommended to monitor cells following 10 minutes of Accutase treatment, viewing cells every five minutes until approximately 90% of the cells detach. If Accutase time is too short, cells will not be singularized and will peel off as sheets or clusters of cells. Ultimately, these clusters will result in poor BMEC monolayers (visualized under a bright field microscope as clusters of cells growing overtop the BMEC monolayer) and reduced TEER. On the other hand, extended Accutase incubation may decrease cell viability and result in subconfluent monolayers on Transwell filters.
NOTE: If using a “no-RA” differentiation scheme, Accutase incubation time is typically shorter (10–20 minutes).
Following Accutase incubation, use a p1000 to collect cells and spray gently over surface 2–3x to dislodge any remaining cells. Triturate briefly to break up cell clumps. Take a 20 μL aliquot of cells to count on hemacytometer.
Count cells on hemacytometer by diluting 20 μL of cells with 20 μL of Trypan blue, excluding dead cells (blue cells).
Dilute cells 1:5 in hESFM and centrifuge for 5 minutes at 1000 RPM.
-
Aspirate medium and resuspend cells in the appropriate volume of EC + RA medium (for working volumes, see Table 2). Thoroughly triturate 3–4 times to yield a singularized cell suspension.
-
For immunocytochemistry, seed 300,000 cells per well of a 96-well plate. For accumulation assay, seed 250,000 cells per well of a 24-well plate. For 12- and 24-well filters, seed 1.1 million cells per 12-well Transwell filter and 330,000 cells/24-well Transwell filter (i.e., 1 million cells/cm2).
For example, to seed a 12-well filter using 3 million cells from a single well of a 6-well plate, resuspend cells in 1 mL EC + RA medium, add 167 μL of EC + RA medium to each filter, followed by 333 μL cells, making a final volume of 0.5 mL in top chamber of a 12-well filter.
NOTE: If using a “no-RA” differentiation scheme, do not add RA to the medium at D8.
-
-
Place plate or filters in 37°C incubator, shaking plate back and forth, waiting 2 seconds, shaking side-to-side, waiting two seconds, and repeating this cycle 3x to distribute cells evenly.
NOTE: This shaking method prevents the medium from swirling, which will lead to uneven cell attachment in the filter center.
-
24 hours later (i.e., D9), aspirate spent medium and add appropriate volume of hESFM + 1% PDS (without bFGF or RA).
NOTE: If bFGF and RA are not withdrawn from the media, the maximum TEER will be significantly reduced.
Table 1.
Volumes for coating 4:5:1 collagen/fibronectin/water ECM coating solution onto cell culture plates
| Plate/filter type for subculture phase | Volume of 4:1:5 solution |
|---|---|
| 24-well plate | 200 μL |
| 96-well plate | 50 μL |
| 12-well Transwell filter | 200 μL |
| 24-well Transwell filter | 100 μL |
Table 2.
Seeding densities and working volumes for subculturing BMECs
| Plate type for subculture phase | Seeding Densities | Working volume/ well |
|---|---|---|
| 24-well | 2.5 × 105 cells/well | 0.5 mL |
| 96-well | 3 × 105 cells/well | 0.2 mL |
| 12-well transwell filter | 1.0 × 106 cells/filter | Apical chamber – 0.5 mL Basolateral chamber – 1.5 mL |
| 24-well transwell filter | 3.3 × 105 cells/filter | Apical chamber – 200 μL Basolateral chamber – 800 μL |
4.5 Expected Morphology During Differentiation
Stage-specific densities and morphologies are provided to illustrate expected cell densities at the optimal starting density at D0 UM, which is approximately 30,000 hPSCs/cm2 for IMR90-C4 iPSCs (Figure 1a,b). Cells will approach confluence by D2 in UM and result in the formation of nestin+ neural mounds or “tracts” that can be seen by D8 (Figure 1). Between the neural tracts, VE-cadherin+ endothelial cells can be seen at D8 (Figure 1c) when cells are treated with EC + RA. VE-cadherin expression remains low at D8 if RA is not supplemented during the EC medium phase (D6–D8) [23]. Subconfluent cultures by D8 of the differentiation are typically a result of low starting densities (<25,000 hPSCs/cm2) at D0 and exhibit reduced endothelial cell yields at D8 of the differentiation [25]. This can also result in low endothelial cell yield and purity at D10. If cells are subconfluent at D8, consider seeding hPSCs at higher densities at D-3.
5.0 VALIDATING CELL QUALITY VIA IMMUNOCYTOCHEMISTRY
5.1 Immunolabeling BMECs
Validation is typically performed at D10 of differentiation.
Aspirate spent medium and wash once with 100 μL of PBS per well of a 96-well plate.
Fix with 50 μL of either 4% paraformaldehyde (diluted in PBS) or 100% ice-cold methanol per well of a 96-well plate for 15 minutes at room temperature. Fixation will depend on antibody (Table 3).
-
Wash 3x with 100 μL of PBS per well of a 96-well plate.
NOTE: Certain epitopes are particularly susceptible to weak immunolabeling when cells are fixed and stored for longer than 2 days prior to staining. It is advised to label cells the same day cells are fixed.
-
Block cells for 60 minutes at room temperature in blocking solution, 50 μL per well of a 96-well plate (Table 3).
NOTE: Methanol fixation permeabilizes the cells, but paraformaldehyde fixation does not. If cells were fixed in paraformaldehyde and are being probed for an intracellular antigen, add 0.1% Triton X-100 to the blocking solution (during blocking step only) to permeabilize the cells.
Dilute primary antibodies in blocking solution and incubate overnight at 4°C on rocker, 50 μL per well of a 96-well plate.
Wash 3x in PBS, 100 μL per well of a 96-well plate.
Dilute secondary antibodies 1:200 in blocking solution and incubate for 1 hour at room temperature on rocker, 50 μL per well of a 96-well plate. Protect from light.
Aspirate antibody solution and add DAPI nuclear counter stain, diluted 1:5000 in PBS for 15 minutes on rocker.
Wash 3x in PBS, 100 μL per well of a 96-well plate, and visualize marker expression and localization on a fluorescence microscope (Figure 2).
5.2 Expected immunocytochemistry results
Typical results for BMEC monolayers at D10 of the differentiation are shown in Figure 2. BMECs should exhibit confluent monolayers on filters and plates. All cells should express endothelial adherens proteins VE-cadherin and PECAM-1, tight junction proteins Occludin and Claudin-5, glucose transporter Glut-1, and efflux transporters P-gp, BCRP, and MRP-1 by D10 (Figure 2). Low or weak labeling of multiple markers may indicate a low-quality differentiation. To assess differentiation quality, consider coupling immunocytochemistry results with functional assays, including TEER and/or the sodium fluorescein permeability assay.
6.0 PROCEDURE FOR ASSESSING BARRIER TIGHTNESS BY TEER
6.1 Measuring TEER Follow EVOM manufacturer’s instructions to measure TEER
NOTE: Drop in temperatures can artificially raise TEER. It is recommended to measure TEER one plate at a time to avoid temperature decreases caused prolonged time outside the incubator. In addition, EVOM electrodes can disrupt cell monolayer integrity and artificially reduce TEER if they scratch the cell surface. Therefore, avoid directly contacting the EVOM electrodes with the filter surface.
6.2 Expected TEER Results
IMR90-C4 iPSCs typically exhibit a maximum TEER of >2000 Ω×cm2 48 hours after subculture, at D10 of differentiation (Figure 3). Note that differentiating BMECs without RA supplementation in EC medium will result in a maximum TEER in the range of 100–500 Ω×cm2 [19,23]. Maximum TEER below 2000 Ω×cm2 at D10 in RA-treated BMECs often correlates with poor BMEC monolayers. This often results from suboptimal Accutase time or improper hPSC starting density at D0 UM. While maximum TEER of IMR90-C4 iPSCs is generally unaffected by hPSC starting density, WA09 human embryonic stem cells (hESCs) and DF19-9-11T iPSCs demonstrate a sharp decrease in TEER outside the optimum hPSC starting density [25]. Consider evaluating a range of hPSC starting densities to find a starting density that maximizes TEER for each cell line.
Figure 3.

Functional measurement of BMEC passive barrier as shown by TEER following subculture for IMR90-C4 iPSCs. Error bars represent the standard deviation of triplicate Transwell filters.
7.0 ASSESSING BARRIER TIGHTNESS BY PERMEABILITY TO SODIUM FLUORESCEIN
7.1 Measuring Sodium Fluorescein Permeability Sodium fluorescein permeability is typically assessed at D10 in cells cultured in 12-well Transwell filters
At D8, subculture cells onto collagen/fibronectin-coated Transwells at a density of 1 million cells/cm2. To determine clearance slopes and Pe values, a blank, collagen/fibronectin-coated Transwell must be included in the experiment.
Measure TEER 48-hours post-subculture (D10).
-
Remove medium and replace with pre-warmed hESFM medium containing 1% PDS (500 μL into the top chamber and 1500 μL at the bottom chamber of a 12-well Transwell filter). Let incubate for 60 minutes at 37°C.
NOTE: The incubation step is critical, as low medium temperature can affect the cell monolayer baseline barrier.
Measure TEER after pre-incubation to identify starting barrier tightness. Dilute sodium fluorescein to 10 μM in EC medium. Remove medium from the top chamber and replace with 500 μL of compound in all samples and blank transwells.
At 15, 30, 45, and 60 minutes, collect 150 μL from the bottom chamber and place into a 96-well plate. Pipette gently up and down 2–3 times with micropipette before extracting 150 μL. Replace the missing volume with 150 μL pre-warmed cell culture medium into the bottom chamber.
-
At the 60 minute time point, collect an additional 150 μL from the top chamber and transfer to a 96-well plate. Place an additional 150 μL of EC medium without sodium fluorescein for background subtraction.
NOTE: The top solution chamber may need to be diluted to prevent oversaturated signal.
Read the fluorescence in the fluorescent plate reader (485 nm excitation/530 nm emission).
-
Correct signal by removing background and background and accounting for signal loss from sampling bottom chamber.
For example, after subtracting background, the 15 minute sample exhibits a signal of 1000 relative fluorescence units (RFU) and the 30 minute sample exhibits a signal of 1500 RFU. The corrected signal at 30 minutes is (1500 RFU + (1000 RFU X 150 μL/1500 μL)) = 1500 + 100 = 1600 RFU. When calculating the 45 minute corrected value, use 1600 RFU as the 30 minute sample value.
-
Calculate the clearance volume at each time point.
Clearance volume = (VB *( SB,t)) / (ST,60 min), where VB is the volume of bottom chamber (1500 μL); SB,t is the corrected signal of bottom chamber at time, t; ST,60 min is the signal of top chamber at 60 minutes.
Calculate the linear slope of clearance volume vs. time using Excel using linear regression for both the culture (mc) and the blank filter (mf).
Calculate sodium fluorescein permeability using the following formula: 1/Pe = 1/mf –1/(mc) [26]. Pe (cm/min) = [(1/(1/Pe)) / 1000]/Area.
7.2 Anticipated sodium fluorescein permeability values
Typical sodium fluorescein permeability is below 1×10−6 cm/sec when TEER is above 2,000 Ωxcm2.
8.0 ASSESSING P-GP ACTIVITY VIA RHODAMINE 123 ACCUMULATION ASSAYS
8.1 Accumulation assay procedure
The P-gp substrate Rhodamine 123 and P-gp-specific inhibitor CsA can be used to read out functional transporter activity. Accumulation assay is typically performed at D10 of differentiation.
-
At D8, purify cells onto collagen/fibronectin-coated 24-wells at a density of 250,000 cells per 24-well. Use 8 wells per experiment:
4x wells without CsA: 3x for fluorescence measurements, 1x for counting post-experiment (if normalizing samples to cell number).
4x wells with CsA: 3x for fluorescence measurements, 1x for counting post-experiment (if normalizing samples to cell number).
On day of experiment (typically D10), pre-warm HBSS buffer in 37°C water bath.
Wash wells 1x with 0.5 mL HBSS per 24-well.
-
Pre-incubate cells with 0.5 mL HBSS buffer with or without 10 μM CsA for 1 hour at 37°C on orbital shaker set to 35 rpm.
-
Half the cells are pre-treated with inhibitor, the other half are not.
NOTE: To diminish error, mix inhibitor and buffer in 15 mL conical rather than adding a small volume of inhibitor to each well.
-
-
Incubate cells with 0.5 mL buffer with 10 μM Rhodamine 123, with or without CsA, for 2 hours at 37°C on orbital shaker.
Half the cells are treated with inhibitor (same as in step 4), the other half are not.
Aspirate solution (can be done in non-sterile environment).
Wash 2x with 500 μL/well cold PBS.
-
Add 200 μL RIPA buffer per 24-well.
For normalizing to cell number, add 200 μL Accutase to counting well.
Place plate on rocker for 10 minutes at room temperature, protected from light.
-
Measure fluorescence on plate reader, excitation/emission = 485/530 nm.
If plate reader cannot accommodate 24-well plates, use a P200 to transfer lysate to 96-well plate.
NOTE: Work carefully to minimize bubbles, which can cause error in fluorescence measurement.
Normalize samples to either protein concentration (i.e., via BCA assay on cell lysates) or cell number (counted via hemacytometer).
9.1 Anticipated P-gp activity via Rhodamine 123 accumulation assay results
To measure whether or not the efflux transporters are active in differentiated BMECs, a substrate-based efflux transport measurement may be used. Upon inhibiting BMECs with the P-gp inhibitor CsA, the intracellular accumulation of the fluorescent P-gp substrate Rhodamine 123 typically increases by 40–100% (Figure 4). While 10 μM CsA has not been observed to be toxic to BMECs, this should be confirmed by normalizing fluorescence values to cell counts or BCA assay. Finally, care should be taken with each wash to ensure that cells are not sheared and washed off the plate.
Figure 4.
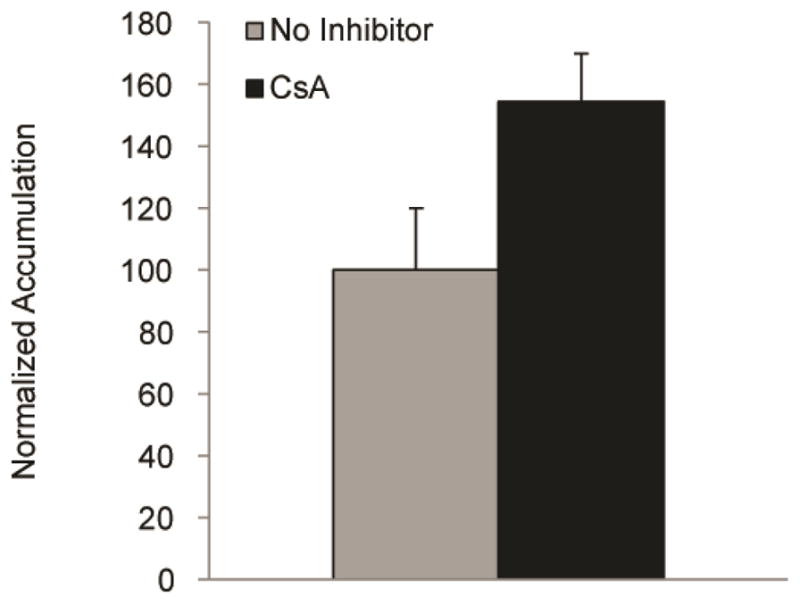
Assessment of efflux transporter activity via P-gp accumulation assays for IMR90-C4 derived BMECs at D10. Accumulation is normalized to the no inhibitor samples. Error bars represent the standard deviation of triplicate wells.
10.0 CONCLUDING REMARKS
The aforementioned assays can be used to differentiate and to validate hPSC-derived BMECs. However, specific experiments may require additional assays, many of which have been previously established for hPSC-derived BMECs. For instance, hPSC-derived BMECs express additional functional efflux transporters, including BCRP and MRP [19,23,25]. BMECs also exhibit responsiveness to other cell types of the neurovascular unit—including fetal-neural progenitor-derived neurons [23], astrocytes [19,23], and pericytes [23]— the ability to form vascular tubes, and internalize acetylated-LDL [19].
Using protocols described in this article, hPSC can be singularized and differentiated to virtually pure BMEC monolayers for screening drug compound libraries, investigating signaling mechanics implicated in BBB development and regulation, and modeling disease phenotypes. For instance, hPSC-derived BMEC responsiveness to peripheral cell types of the neurovascular unit demonstrate this system’s capability to interrogate hPSC-derived BMEC responsiveness to known and novel signaling pathways implicated in neurovascular unit cross-talk in BBB development and maintenance. In addition, while in vitro BBB models have been used previously to model vascular pathologies in CNS diseases, including Alzheimer’s disease [1,27], hypoxia [28], and microbial infiltration of the CNS [29], hPSC-derived BMECs may offer the potential to examine BBB dysfunction implicated in patient-specific iPSC-derived BMECs. This could offer special significance in examining genetic mutations that exacerbate vascular dysfunction in CNS pathology. Future directions of the model may also include investigating the mechanism of retinoic acid in enhancing BMEC phenotypes. RA is a potent hindbrain-patterning agent and has been demonstrated to promote neuronal and glial differentiations in vitro and regulate somatic motor development in chick embryo studies [30]. Therefore, RA-mediated effects on neural progenitor cells present during BMEC differentiation may affect ultimate BMEC barrier enhancement properties.
Highlights.
Codifferentiation of human pluripotent stem cells (hPSCs) to endothelial cells with neural progenitor cells yields endothelial cells that express blood-brain barrier (BBB) markers.
hPSC-derived brain microvascular endothelial cells subcultured onto collagen/fibronectin extracellular matrix form virtually pure monolayers with functional barrier tightness and efflux transporter activity reminiscent of brain vasculature in vivo.
hPSC-derived brain microvascular endothelial cells are a scalable and renewable cell source for modeling the human BBB in vitro.
Acknowledgments
The protocols developed for this article were supported by US National Institutes of Health (NIH) grants NS056249 (E.V.S.), AA020476 (E.V.S.), EB007534 (S.P.P.), NS083688 (EVS and SPP), NS085351 (SPP and EVS), the US National Science Foundation (NSF) grant EFRI-0735903 (S.P.P.), the Takeda Pharmaceuticals New Frontier Science Program (EVS and SPP). M.J.S. is a recipient of the NIH Biotechnology Training Program Fellowship and H.K.W. is a recipient of the NSF Graduate Research Fellowship. We would like to acknowledge Ethan S. Lippman, a recipient of the NIH Chemistry Biology Interface traineeship (T32 GM008505) and Samira M. Azarin, a recipient of the NSF Graduate Research Fellowship, for conceiving the strategy and initial characterization of hPSC-derived BMECs. We would like to thank Abraham Al-Ahmad for his work in characterizing the effects of RA in enhancing hPSC-derived BMECs.
We thank the WiCell Research Institute for providing research support.
ABBREVIATIONS
- BBB
blood-brain barrier
- CNS
central nervous system
- hPSCs
human pluripotent stem cells
- BMECs
brain microvascular endothelial cells
- P-gp
P-glycoprotein
- RA
retinoic acid
- UM
unconditioned medium
- EC
endothelial cell
- EC + RA medium
EC medium supplemented with RA
- TEER
transendothelial electrical resistance
- NEAA
nonessential amino acids
- KOSR
knockout serum replacement
- hESFM
human endothelial serum-free medium
- PDS
platelet poor plasma-derived serum
- bFGF
basic fibroblast growth factor
- ECM
extracellular matrix
- iPSCs
induced pluripotent stem cells
- hESCs
human embryonic stem cells
- RFU
relative fluorescence units
- CsA
Cyclosporin A
Footnotes
12.0 AUTHOR CONTRIBUTIONS
M.J.S., H.K.W., S.G.C., and T.Q. derived and analyzed the example data provided for this methods summary article. M.J.S., H.K.W., S.G.C., S.P.P, and E.V.S. contributed to writing the article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6.0 REFERENCES
- 1.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33:1500–13. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 5.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 6.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowman PD, Betz AL, DAR, Wolinsky JS, Penney JB, Shivers RR, et al. Primary culture of capillary endothelium from rat brain. In Vitro. 1981;17:353–362. doi: 10.1007/BF02618147. [DOI] [PubMed] [Google Scholar]
- 8.Gaillard PJ, Voorwinden LH, Nielsen JL, Ivanov A, Atsumi R, Engman H, et al. Establishment, functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci. 2001;12:215–222. doi: 10.1016/s0928-0987(00)00123-8. [DOI] [PubMed] [Google Scholar]
- 9.Mischeck U, Meyer J, Galla HJ. Characterization of gamma-glutamyl transpeptidase activity of cultured endothelial cells from porcine brain capillaries. Cell Tissue Res. 1989;256:221–226. doi: 10.1007/BF00224737. [DOI] [PubMed] [Google Scholar]
- 10.DeBault LE, Kahn LE, Frommes SP, Cancilla PA. Cerebral microvessels and derived cells in tissue culture: isolation and preliminary characterization. In Vitro. 1979;15:473–487. doi: 10.1007/BF02618149. [DOI] [PubMed] [Google Scholar]
- 11.Roux F, Durieu-Trautmann O, Chaverot N, Claire M, Mailly P, Bourre JM, et al. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J Cell Physiol. 1994;159:101–113. doi: 10.1002/jcp.1041590114. [DOI] [PubMed] [Google Scholar]
- 12.Song L, Pachter J. Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. Vitr Cell Dev Biol - Anim. 2003;39:313–320. doi: 10.1290/1543-706X(2003)039<0313:COMBME>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 13.Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 14.Syvänen S, Lindhe Ö, Palner M, Kornum BR, Rahman O, Långström B, et al. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos. 2009;37:635–643. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 15.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem. 2011;117:333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 16.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6:650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 17.Cecchelli R, Aday S, Sevin E, Almeida C, Culot M, Dehouck L, et al. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS One. 2014;9:e99733. doi: 10.1371/journal.pone.0099733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponio JB-D, El-Ayoubi F, Glacial F, Ganeshamoorthy K, Driancourt C, Godet M, et al. Instruction of Circulating Endothelial Progenitors In Vitro towards Specialized Blood-Brain Barrier and Arterial Phenotypes. PLoS One. 2014;9:e84179. doi: 10.1371/journal.pone.0084179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lippmann ES, Azarin SM, Kay JE, Nessler Ra, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–91. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–50. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 21.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. doi: 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizee MR, Wooldrik D, Lakeman KA, van Het Hof B, Drexhage JA, Geerts D, et al. Retinoic Acid Induces Blood-Brain Barrier Development. J Neurosci. 2013;33:1660–1671. doi: 10.1523/JNEUROSCI.1338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids Barriers CNS. 2015;12:13. doi: 10.1186/s12987-015-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrière N, Demeuse PH, Garcia E, Regina A, Debray M, Andreux JP, et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J Neurochem. 2005;93:279–289. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- 27.Sagare AP, Bell RD, Zlokovic BV. Neurovascular Dysfunction and Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harb Perspect Med. 2012;2:a011452. doi: 10.1101/cshperspect.a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol. 2014;171:1210–30. doi: 10.1111/bph.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KS. Mechanisms of microbial traversal of the blood-brain barrier. Nat Rev Microbiol. 2008;6:625–634. doi: 10.1038/nrmicro1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci. 2007;8:755–65. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]