

Abstract
Short-acting β2-agonist bronchodilators are the most common medications used in treating chronic obstructive pulmonary disease (COPD). Genetic variants determining bronchodilator responsiveness (BDR) in COPD have not been identified.
We performed a genome-wide association study (GWAS) of BDR in 5789 current or former smokers with COPD in one African American and four white populations. BDR was defined as the quantitative spirometric response to inhaled β2-agonists. We combined results in a meta-analysis.
In the meta-analysis, SNPs in the genes KCNK1 (P=2.02×10−7) and KCNJ2 (P=1.79×10−7) were the top associations with BDR. Among African Americans, SNPs in CDH13 were significantly associated with BDR (P=5.1×10−9). A nominal association with CDH13 was identified in a gene-based analysis in all subjects.
We identified suggestive association with BDR among COPD subjects for variants near two potassium channel genes (KCNK1 and KCNJ2). SNPs in CDH13 were significantly associated with BDR in African Americans.
Introduction
Chronic obstructive pulmonary disease (COPD) is a disorder characterized by progressive loss of lung function. It is currently the third leading cause of death world-wide, and the global burden of disease is expected to continue to rise(1). Although cigarette smoke is the greatest risk factor for COPD, recent studies have identified several genetic risk factors for this disease(2).
Inhaled bronchodilators, including β2-agonists, play a key role in COPD management guidelines. These medications act on smooth muscle receptors in bronchial airways to produce muscle relaxation and airway dilation, resulting in improved airflow through the lungs (1), and have been shown to alleviate COPD symptoms(3). The response to inhaled bronchodilators is measured by a change in the forced expiratory volume in one second (FEV1) using standardized spirometry before and after the administration of β2-agonists. Although COPD is characterized by relatively fixed airflow limitation, up to two-thirds of COPD patients will exhibit a positive response to an inhaled bronchodilator at any one time(4).
The quantitative response to inhaled β2-agonists is a heritable trait(5), and candidate gene studies have identified several genes suggestive of association with quantitative measures of bronchodilator responsiveness (BDR)(6, 7). In addition, candidate gene(8) and genome-wide association studies (GWAS) have identified variants associated with BDR in subjects with asthma (9–11). We hypothesized that genome-wide association studies would identify associations with BDR in COPD.
Subjects and Methods
Study subjects
Details of the COPDGene, ECLIPSE, GenKOLS, and NETT studies, including study procedures, genotyping, and quality control, have been reported(12–16). COPDGene subjects were current and former smoking non-Hispanic white (NHW) or African American (AA) from the U.S. ECLIPSE subjects were Caucasian current or former smokers from Europe, North American and New Zealand. GenKOLS subjects were current and former smokers from Norway. NETT subjects were white former smokers from the U.S. All subjects had moderate to severe COPD (GOLD stage 2 or greater(17)). Subjects were excluded if they had a recent COPD exacerbation.
Spirometry
All subjects completed a respiratory questionnaire and performed standardized spirometry according to American Thoracic Society or European Respiratory Society guidelines. COPDGene, NETT, and GenKOLS subjects were tested before and approximately 20 minutes after administration of 2 puffs (180 μg) of inhaled β2-agonist (albuterol/salbutamol). ECLIPSE subjects were tested before and 15 minutes after inhalation of 400 μg β2-agonist (albuterol/salbutamol).
BDR was measured using three quantitative metrics that have been previously reported(5). BDRABS, the absolute difference in pre- versus post- bronchodilator FEV1; BDRPRED, the absolute difference in pre- versus post-bronchodilator FEV1 as a percentage of FEV1 percent predicted; and BDRBASE, the absolute difference in pre versus post bronchodilator FEV1 as a percentage of baseline FEV1.
Genotyping
All subjects were genotyped using Illumina platforms (Human Hap550 for ECLIPSE and GenKOLS, Quad610 for NETT, and Human OmniExpress for COPDGene) as previously described(13, 15). We included all variants and subjects that passed quality control, based on cluster plots (genotyped) and imputation quality (R2 ≥ 0.80) for imputed SNPs, Hardy-Weinberg equilibrium (P-value), and missingness (% threshold). Imputation was performed using MaCH and minimac with 1000 Genomes phase I v 3 European reference panels for white subjects. Cosmopolitan reference panels were used for COPDGene AA subjects. Variants with a minor allele frequency (MAF) < 1% and R2 ≤ 0.80 were excluded from analysis. Ancestry-based principal components were generated for each study using EIGENSOFT2.0(18). We performed Taqman genotyping (Applied Biosystems, Foster City, CA) for the SNPs rs114132812 and rs115067260 among 23 and 38 African American COPDGene subjects respectively, who were imputed to be carriers of the minor allele.
Statistical analysis
Baseline subject demographics and outcome variables were analyzed in R (v2.15.1). We excluded 20 subjects with BDR variables more than six standard deviations from the mean. We performed linear regression analysis for the three BDR variables in PLINK(19) including genotyped and imputed SNPs, adjusting for age, gender, pack-years smoking history, and ancestry-based principal components. We combined the results from all five samples in a fixed-effects meta-analysis using METAL(20). We additionally performed a gene-based test of significance among the top 20% of all SNPs using VEGAS (a Versatile Gene-based test for Genome-wide Association Studies). This method performs gene-based association testing by assigning SNPs within 50kb of a gene in accordance with the hg18 assembly and then uses simulation to account for linkage disequilibrium. All genes are tested for association with the trait of interest(21). Using the top 20% of significantly associated SNPs, we used VEGAS software to test 13,675 genes. Based on this number, we established a Bonferroni significance threshold of 3.6×10−6. We analyzed the top genes from our GWAS as well as the top genes identified through the VEGAS analysis using the functional annotation tool, DAVID(22, 23).
We tested the association of seven asthma and three COPD SNPs previously associated with BDR in asthma GWAS (9, 11) and COPD candidate gene studies (ADRB2, EPHX1, and SERPINE2) (6) with BDR in our meta-analysis results. We additionally tested the association of two SNPs from the β2-agonist receptor gene, ADRB2 (Arg16Gly, rs1042713 and Gln27Glu, rs1042714). We tested our top SNPs for their association with lung function (FEV1/FVC and FEV1) in the four COPD populations with the broadest range of lung function values: COPDGene NHW and AA, GenKOLs, ECLIPSE. We examined our top BDR variants for their association with BDR in two asthma GWAS(9, 11).
Results
The demographic data for each study population are presented in Table 1. These outcomes appear to follow a normal distribution (Supplementary Figure 1). All three outcomes are significantly correlated in COPDGene NHW, but BDRABS and BDRPRED appear more correlated (R2=0.97), compared to BDRABS and BDRBASE (R2=0.85) or BDRBASE and BDRPRED (R2=0.88).
Table 1.
Study Subjects
| COPD Gene NHW | COPD Gene AA | ECLIPSE | GenKOLS | NETT | |
|---|---|---|---|---|---|
| N | 2792 | 811 | 1757 | 853 | 364 |
| Population origin | US | US | Europe/US/New Zealand | Norway | US |
| Age | 64.7 (8.2) | 59.0 (8.2) | 63.7 (7.1) | 65.6 (10.0) | 67.5 (5.8) |
| Males (%) | 55.6 | 54.7 | 66.9 | 60.0 | 64.8 |
| Pack-years | 56.2 (27.8) | 42.4 (23.1) | 50.3 (27.4) | 32.1 (18.5) | 66.1 (30.9) |
| Current smoker (%) | 34.7 | 60.8 | 35.4 | 46.9 | 0 |
| BMI (kg/m2) | 28.1 (6.1) | 28.0 (6.8) | 26.7 (5.6) | 25.3 (4.9) | 25.0 (3.5) |
| FEV1 pp (%) | 49.7 (18.0) | 52.3 (17.7) | 47.6 (15.6) | 50.7 (17.3) | 28.0 (7.4) |
| FEV1/FVC | 0.49 (0.13) | 0.53 (0.12) | 0.45 (0.12) | 0.51 (0.13) | 0.32 (0.06) |
| BDRABS (L) | 0.10 (0.15) | 0.07 (0.18) | 0.12 (0.14) | 0.10 (0.14) | 0.09 (0.08) |
| BDRBASE (%) | 9.2 (12.4) | 7.6 (14.2) | 10.7 (13.4) | 8.1 (11.7) | 13.4 (12.0) |
| BDRPRED (%) | 3.6 (4.9) | 2.89 (6.5) | 3.9 (4.8) | 3.4 (4.7) | 3.3 (3.0) |
Data are presented as mean (sd) or N (%). NHW: non-Hispanic white; AA: African American; FEV1pp: FEV1 percent predicted; BMI: Body mass index. BDRABS: (Post-BD FEV1 – Pre-BD FEV1); BDRBASE: (BDRABS/(Pre-BD FEV1*100)); BDRPRED: (BDRABS/(FEV1pp*100)).
The BDR outcomes were statistically different when comparing the four Caucasian populations (P<0.05 for ANOVA for all three BDR outcomes). However, these small differences are unlikely to be clinically significant. We additionally compared the NHW and AA subjects from COPDGene. The AA subjects demonstrated significantly lower bronchodilator responsiveness for all three outcomes, and these results remained significant, albeit small, after adjusting for age, gender, and pack-years (BDRABS: 0.10 L vs 0.07 L, P = 0.004; BDRPRED: 3.59% v 2.80%, P=0.02; BDRBASE: 9.24% vs 7.60%, P=0.007).
Table 2 presents the most significant SNPs from the GWAS in the non-Hispanic white subjects from COPDGene. The top SNP annotated to a gene is presented in this table. The full list of SNPs with P < 1×10−6 are presented in Supplementary Tables 1–3. SNP rs17575208, located upstream from the gene EPHA7 on chromosome 6, was significantly associated with BDRABS (β=0.11, P=8.29×10−9). This variant was also associated with BDRPRED (β=3.22, P=1.03×10−7) and BDRBASE (β=7.06, p=5.64×10−6), though the P-values were not genome-wide significant.
Table 2.
Most significant variants and genes from the GWAS of COPDGene non-Hispanic white subjects
| SNP | Chr | Nearest Gene | Beta (se) | Allele | FRQ | P value |
|---|---|---|---|---|---|---|
| BDRABS | ||||||
| rs17575208 | 6 | EPHA7 | 0.11 (0.02) | A | 0.99 | 8.92×10−9 |
| rs7778219* | 7 | LOC285889 | −0.03 (0.01) | G | 0.11 | 1.62×10−7 |
| rs2367245 | 17 | KCNJ2 | 0.02 (0.01) | G | 0.57 | 1.33×10−6 |
| rs115310518 | 5 | CDH18 | 0.064 (0.014) | T | 0.98 | 2.20×10−6 |
| rs78008396 | 6 | PARK2 | −0.075 (0.016) | A | 0.98 | 2.92×10−6 |
| rs7932838* | 11 | SOX6 | 0.05 (0.01) | G | 0.03 | 3.34×10−6 |
| rs11775549 | 8 | NKX2-6 | −0.044 (0.0095) | G | 0.94 | 3.39×10−6 |
| rs34342951 | 8 | NKX6-3 | −0.021 (0.005) | G | 0.72 | 3.42×10−6 |
| rs11260945 | 1 | IGSF21 | −0.046 (0.01) | G | 0.95 | 3.74×10−6 |
| rs73671623 | 8 | STC1 | −0.044 (0.010) | G | 0.94 | 4.01×10−6 |
| rs56323342 | 11 | ME3 | −0.040 (0.009) | G | 0.95 | 4.21×10−6 |
| rs56010187 | 16 | HS3ST4 | −0.023 (0.005) | G | 0.78 | 5.65×10−6 |
| rs1335517 | 14 | C14ORF37 | 0.027 (0.006) | T | 0.85 | 5.85×10−6 |
| rs181350634 | 3 | IFT80 | 0.09 (0.02) | A | 0.99 | 6.29×10−6 |
| rs7552783* | 1 | KCNK1 | −0.0197 (0.004) | G | 0.28 | 7.10×10−6 |
| BDRBASE | ||||||
| rs1032243 | 3 | MIR548A3 | −1.96 (0.40) | A | 0.77 | 9.59×10−7 |
| rs326981 | 5 | MTRR | −2.48 (0.51) | G | 0.89 | 1.32×10−6 |
| rs4772755 | 13 | LINC00460 | −1.75 (0.36) | G | 0.70 | 1.36×10−6 |
| rs6943859* | 7 | KLHDC10 | 3.0 (0.62) | C | 0.07 | 1.84×10−6 |
| rs10242432 | 7 | ZC3HC1 | −2.98 (0.62) | G | 0.93 | 1.85×10−6 |
| rs8032265* | 15 | C15orf60 | 3.63 (0.78) | A | 0.05 | 3.18×10−6 |
| rs2367245 | 17 | KCNJ2 | 1.57 (0.34) | G | 0.57 | 3.34×10−6 |
| rs8108918* | 19 | VAV1 | 1.74 (0.37) | T | 0.26 | 3.98×10−6 |
| rs1004790 | 2 | LOC645949 | −5.77(1.26) | A | 0.98 | 4.66×10−6 |
| rs17574208 | 6 | EPHA7 | 7.06 (1.53) | A | 0.99 | 5.64×10−6 |
| BDRPRED | ||||||
| rs17575208 | 6 | EPHA7 | 3.22 (0.60) | A | 0.99 | 1.03×10−7 |
| rs7778219* | 7 | LOC285889 | −1.03 (0.20) | G | 0.11 | 3.90×10−7 |
| rs11775549 | 8 | NKX2-6 | −1.53 (0.31) | G | 0.94 | 6.99×10−6 |
| rs73671623 | 8 | STC1 | −1.53 (0.31) | G | 0.94 | 1.27×10−6 |
| rs2367245 | 17 | KCNJ2 | 0.629 (0.13) | G | 0.57 | 2.53×10−6 |
| rs78008396 | 6 | PARK2 | −2.50 (0.53) | A | 0.98 | 3.07×10−6 |
| rs115310518 | 5 | CDH18 | 2.07 (0.45) | T | 0.98 | 4.63×10−6 |
| rs1032243 | 3 | MIR548A3 | −0.73 (0.16) | A | 0.77 | 4.30×10−6 |
| rs1335517 | 14 | C14orf37 | 0.89 (0.20) | T | 0.85 | 5.65×10−6 |
Presenting the top SNP from each top gene with P < 5×10−6; Adjusted for age, sex, pack-years and principal components; Results filtered for MAF > 1%, R2> 0.80.
Genotyped SNP.
Chr: Chromosome; FRQ: reference allele frequency; Allele: reference allele. The full list of SNPs with P < 5×10−6 is presented in the supplementary material.
Table 3 presents the top SNPs annotated to genes having P < 5×10−6 for the COPDGene AA subjects. The full list of SNPs with P < 5×10−6 are presented in Supplementary Tables 4–6. Variants in the gene CDH13 were significantly associated with BDRABS (rs115067260; β=0.17±0.03, P=5.05×10−9) and BDRPRED (rs114132812, β=7.63±1.32, P=1.19×10−8), and showed suggestive association with BDRBASE (rs77347308 β=−17.14±3.39, P=5.35×10−7). In addition, a genotyped SNP in the gene SGCD was significantly associated with BDRBASE (rs10056066, β=7.12±1.29, P=4.86×10−8), and several rare imputed SNPs in the gene GOLGA8B were associated with the outcome BDRPRED (rs76677753, β=9.49±1.67, P=1.9×10−8). A recent GWAS using COPD subjects from the Lung Health Study population identified the variants in the gene SGCD as associated with airway responsiveness measured by methacholine challenge test in a physiologically distinct asthma cohort(24). While the response to inhaled methacholine is distinct from the response to inhaled bronchodilators, similar mechanisms of smooth muscle activation could be involved. We tested the two top SGCD SNPs from that study in our AA population. The SNP rs456290 was associated with BDRBLINE (P=0.02), and the SNP rs2642660 approached replication (P=0.08). These SNPs were not associated with BDRBLINE in the NHW population, or in the meta-analysis.
Table 3.
Most significant variants and genes from the GWAS of COPDGene African American subjects
| SNP | Chr | Nearest Gene | Beta (se) | Allele | FRQ | P value |
|---|---|---|---|---|---|---|
| BDRABS | ||||||
| rs115067260 | 16 | CDH13 | 0.17 (0.03) | A | 0.97 | 5.05×10−9 |
| rs140948272 | 17 | PITPNA | 0.33 (0.06) | C | 0.99 | 5.24×10−9 |
| rs76677753 | 15 | GOLGA8B | 0.25 (0.05) | A | 0.99 | 7.96×10−8 |
| rs78060357 | 7 | PLXNA4 | 0.08 (0.02) | A | 0.90 | 8.06×10−8 |
| rs145442019 | 16 | HSBP1 | 0.203 (0.038) | A | 0.99 | 8.58×10−8 |
| rs13345720* | 19 | RNASEH2A | −0.05 (0.010) | C | 0.34 | 1.28×10−7 |
| rs114871691 | 12 | BTBD11 | 0.114 (0.02) | A | 0.95 | 1.32×10−7 |
| rs60085550 | 1 | OR6F1 | 0.157 (0.030) | G | 0.97 | 1.76×10−7 |
| rs2295599* | 6 | SYCP2L | 0.07 (0.014) | A | 0.10 | 5.31×10−7 |
| rs149447163 | 2 | UBR3 | 0.191 (0.038) | G | 0.98 | 5.62×10−7 |
| rs145986148 | 16 | KCNG4 | 0.169 (0.034) | A | 0.98 | 6.59×10−7 |
| rs12529809 | 6 | ELOVL2 | −0.07 (0.01) | G | 0.88 | 8.09×10−7 |
| rs387092 | 16 | MLYCD | 0.20 (0.04) | A | 0.99 | 9.90×10−7 |
| rs4742936 | 9 | ABCA1 | 0.088 (0.018) | C | 0.94 | 9.95×10−7 |
| BDRBASE | ||||||
| rs10056066* | 5 | SGCD* | 7.12 (1.29) | A | 0.07 | 4.86×10−8 |
| rs143376495 | 6 | TFAP2B | −19.47 (3.63) | G | 0.99 | 1.06×10−7 |
| rs2295599* | 6 | SYCP2L* | 6.0 (1.13) | A | 0.10 | 1.38×10−7 |
| rs11651753* | 17 | MIR4315-2 | 4.22 (0815) | T | 0.22 | 2.81×10−7 |
| rs75762663 | 13 | PRR20E | −19.22 (3.80) | A | 0.99 | 5.34×10−7 |
| rs77347308 | 16 | CDH13 | −17.14 (3.39) | A | 0.99 | 5.35×10−7 |
| rs77094751 | 18 | SALL3 | −10.23 (2.03) | G | 0.96 | 5.65×10−7 |
| rs62385074 | 5 | LCP2 | −10.17 (2.03) | G | 0.96 | 6.32×10−7 |
| rs141697850 | 4 | TRAM1L1 | −15.05 (3.01) | C | 0.99 | 7.02×10−7 |
| rs12529809 | 6 | ELOVL2 | −5.674 (1.14) | G | 0.88 | 7.85×10−7 |
| rs2575515 | 4 | GRXCR1 | 3.525 (0.71) | T | 0.47 | 8.33×10−7 |
| rs7848679 | 9 | ABCA1 | 6.96 (1.41) | G | 0.93 | 9.31×10−7 |
| rs76999017 | 2 | INPP5D | −8.263 (1.67) | G | 0.95 | 9.70×10−7 |
| BDRPRED | ||||||
| rs114132812 | 16 | CDH13 | 7.63 (1.32) | A | 0.98 | 1.19×10−8 |
| rs76677753 | 15 | GOLGA8B | 9.49 (1.67) | A | 0.99 | 1.90×10−8 |
| rs13345720* | 19 | RNASEH2A | −1.83 (0.35) | C | 0.34 | 1.60×10−7 |
| rs114871691 | 12 | BTBD11 | 4.116 (0.79) | A | 0.95 | 2.06×10−7 |
| rs72653721 | 6 | SYCP2L | −2.746 (0.53) | C | 0.88 | 2.91×10−7 |
| rs147388556 | 1 | LPHN2 | 4.17 (0.81) | T | 0.96 | 3.32×10−7 |
| rs11245387 | 10 | FAM175B | −1.76 (0.34) | T | 0.40 | 3.49×10−7 |
| rs145442019 | 16 | HSBP1 | 7.10 (1.39) | A | 0.99 | 3.55×10−7 |
| rs143859190 | 2 | GALNT13 | 8.10 (1.58) | T | 0.99 | 3.67×10−7 |
| rs188637550 | 8 | MTUS | 6.89 (1.36) | T | 0.98 | 4.60×10−7 |
| rs78060357 | 7 | PLXNA4 | 2.88 (0.57) | A | 0.90 | 5.08×10−7 |
| rs12529809 | 6 | ELOVL2 | −2.64 (0.52) | G | 0.88 | 5.37×10−7 |
| rs188524320 | 8 | PCM1 | 7.01 (1.40) | C | 0.99 | 6.39×10−7 |
| rs4742936 | 9 | ABCA1 | 3.28 (0.66) | C | 0.94 | 7.17×10−7 |
| rs17116498 | 8 | FGL1 | 7.07 (1.42) | C | 0.99 | 7.32×10−7 |
| rs139218005 | 8 | ATP6V1H | 4.935 (0.89) | A | 0.97 | 9.74×10−7 |
Presenting the top SNP from each top gene with P < 1×10−6; Adjusted for age, sex, pack-years and principal components; Results filtered for MAF > 1%, R2> 0.80.
Genotyped SNP.
Chr: Chromosome; FRQ: reference allele frequency; Allele: reference allele. The full list of SNPs with P < 5×10−6 is presented in the supplementary material.
The CDH13 SNPs were in LD (rs115067260 and rs114132812, R2=0.60, D’=1.0). These variants were monomorphic in the Caucasian populations. We tested additional variants within CDH13 in the COPDGene NHW subjects for association with BDR. Rs4783331 was nominally associated with BDRABS (β=0.11±0.02, P=9.39×10−5), and five additional SNPs in this gene had P<0.001.
We tested our model assumptions of normal distributions of the BDR traits, focusing on BDRABS in the African Americans. The BDR traits appeared to fit a normal distribution (Supplementary figure 1). We additionally examined the residuals from linear regression for the BDR outcomes in the African American subjects, adjusted for age, pack-years, and gender, which appeared normally distributed and were consistent with our model assumption (Supplementary figure 2). We performed an inverse normal transformation of the BDRABS phenotype, and tested this trait in a GWAS. The CDH13 SNPs, rs115067260 (P=4.46×10−6) and rs114132812 (P=5.10×10−6), remained the top associations. Taqman genotyping verified the imputation accuracy of the CDH13 SNPs. 22/23 imputed carriers of the SNP rs114132812 were heterozygous for the minor allele, and for rs115067260, 35/37 subjects were verified as heterozygous and one subject homozygous for the minor allele.
We performed a meta-analysis of the results of all five study populations (Table 4, Figure 1). A SNP in the potassium channel, subfamily K, Member 1 gene (KCNK1) demonstrated suggestive association with the outcome BDRABS (β =−0.0142, P=2.02×10−7). A SNP upstream from the gene KCNJ2 (rs9898686) showed suggestive association with all three traits: BDRABS, (β =0.014, P=2.05×10−7), BDRBASE (β=1.26, P=1.83×10−7) and BDRPRED (β =0.44, P=1.22×10−6). Variants in the gene MC5R (melanocortin 5 receptor) were suggestively associated with BDRPRED (rs12956045, β= −0.45, P=4.69×10−7). Several other variants in the KCNJ2 region also demonstrated nominal association, and these variants were in linkage disequilibrium (R2>0.80). SNPs upstream from KCNJ2 were recently identified as associated with lung function (measured by FEV1) in a joint meta-analysis of SNP and SNP-by-smoking effects in a population-based sample(25). We examined the top SNP from that analysis, rs11654749. This SNP was nominally associated with BDRABS in COPDGene NHW subjects, with the same effect direction (β=0.011, P=0.007). We performed the meta-analysis without the AA population (Supplementary Table1), with very similar results as that including all five study populations together.
Table 4.
Meta-analysis of top variants associated with BDR from 5 COPD populations
| SNP | Chr | Gene | Allele | Effect | Std Err | P Value |
|---|---|---|---|---|---|---|
| BDRABS | ||||||
| rs61824320 | 1 | KCNK1 | A | −0.0142 | 0.0027 | 2.02×10−7 |
| rs9898686 | 17 | KCNJ2 | T | 0.0138 | 0.0027 | 2.05×10−7 |
| rs68120025 | 17 | KCNJ2 | C | 0.0138 | 0.0027 | 2.05×10−7 |
| rs68193035 | 17 | KCNJ2 | A | 0.0138 | 0.0027 | 2.05×10−7 |
| rs9906150 | 17 | KCNJ2 | A | 0.0137 | 0.0026 | 2.06×10−7 |
| rs9899756 | 17 | KCNJ2 | C | 0.0138 | 0.0027 | 2.08×10−7 |
| rs67158616 | 17 | KCNJ2 | C | 0.014 | 0.0028 | 5.35×10−7 |
| rs12956045 | 18 | MC5R | A | −0.013 | 0.0026 | 8.30×10−7 |
| BDRPRED | ||||||
| rs68193035 | 17 | KCNJ2 | A | 1.26 | 0.24 | 1.79×10−7 |
| rs68120025 | 17 | KCNJ2 | C | 1.26 | 0.24 | 1.79×10−7 |
| rs9906150 | 17 | KCNJ2 | A | 1.25 | 0.24 | 1.82×10−7 |
| rs9898686 | 17 | KCNJ2 | T | 1.26 | 0.24 | 1.83×10−7 |
| rs9899756 | 17 | KCNJ2 | C | 1.25 | 0.24 | 1.85×10−7 |
| rs11871999 | 17 | KCNJ2 | T | 1.26 | 0.26 | 9.36×10−7 |
| BDRBASE | ||||||
| rs12956045 | 18 | MC5R | A | −0.45 | 0.09 | 4.69×10−7 |
| rs2278843 | 10 | PKD2L1 | G | 0.42 | 0.08 | 7.11×10−7 |
| rs7079679 | 10 | PKD2L1 | A | 0.42 | 0.08 | 7.26×10−7 |
| rs28650403 | 19 | NWD1 | G | −1.26 | 0.26 | 9.82×10−7 |
| rs28428860 | 19 | NWD1 | C | −1.26 | 0.26 | 9.94×10−7 |
| rs28668077 | 19 | NWD1 | C | −1.26 | 0.26 | 9.95×10−7 |
Most significant variants associated with BDR among the meta-analysis including COPDGene NHW and AA, ECLIPSE, GenKOLS, and NETT. Allele: reference allele.
Figure 1.

Manhattan plots for meta-analysis results for each bronchodilator responsiveness outcome
We examined SNPs previously associated with BDR in asthma GWAS (9, 11, 26, 27) (Supplementary Table 9). In asthma, the SNP rs4452682 in the gene SLC22A23 was associated with BDR(11). This SNP was nominally associated with BDRBASE in the COPD meta-analysis (β =0.63, P=2.5×10−3), although the effect size was in the opposite direction. A rare variant in the gene SLC24A4 was also previously associated with BDR in an asthma study(26). Although this SNP, rs77441273, was not present in our GWAS meta-analysis, several SNPs in this gene demonstrated nominal association with BDRABS (rs60243508, β=0.017, P=0.008). The gene SPATA13 was previously associated with BDR in a gene-based GWAS among African Americans with asthma(27). Although this gene did not replicate in our gene-based VEGAS analysis and the top reported SNP did not replicate in our meta-analysis, we tested all 464 of the genotyped and imputed SNPs available in the gene SPATA13 for their association with BDRABS. Twenty-eight of these SNPs were nominally associated with BDRABS in the COPD meta-analysis, including rs9511156 (β=0.02, P=0.007); however these associations were not significant after correction for multiple testing. In addition, among the COPDGene AA population, the top SNP rs9507294 from the asthma study showed nominal association with BDRBASE (β=1.94 , P=0.05). The remainder of the asthma GWAS SNPs were not significantly associated with BDR in the COPD analyses, and none of the asthma BDR genes were significant in the gene-based VEGAS analysis. The ADRB2 codon 16 and 27 SNPs (rs1042713 and rs1042714) that have been previously identified in asthmatic populations were not significantly associated with BDR in the COPD GWAS. None of the top COPD variants were associated with BDR when examined in two prior asthma GWAS(9, 11). The SNPs from candidate genes previously associated with BDR in COPD populations (6), including EPHX1 (rs3753661), SERPINE2 (rs6712954), and ADRB2 (rs1042717), were not associated with BDR in our analysis.
We tested the top SNPs from the BDR meta-analysis for association with lung function in four of the five COPD populations (COPDGene NHW and AA, GenKOLs, ECLIPSE). SNP rs61824320 in the gene KCNK1 was significantly associated with FEV1/FVC (β= −0.0042 , P=0.03) and FEV1 (β= −0.02 , P=0.04) (M. Cho and S. Lutz, personal communication). The remainder of top SNPs from the BDR meta-analysis were not significantly associated with lung function.
In the VEGAS gene-based analysis (Supplementary Tables 10–12), using the P value results from the meta-analysis of all five studies, KCNK1 approached genome-wide significance for the outcome BDRABS (P=8.4×10−5) and BDRPRED (P=3.8×10−4). The gene SH2B adaptor protein 3, SH2B3, was a top gene for all three traits, approaching genome wide significance for BDRABS (P=3.20×10−5), BDRPRED (7.0 × 10−5) and BDRBASE (1.50 × 10−5). We examined the top genes from the GWAS in the VEGAS analysis. KCNJ2 and SGCD were not significant in the gene-based analysis; however, CDH13 showed nominal significance for all three traits. We additionally performed Gene Ontology analysis of the top 50 genes from the GWAS and from the VEGAS analysis using the functional annotation software, DAVID. Among the top genes in the GWAS, there was enrichment for ion channel and ion transport genes, including KCNK1, FXYD1, and PKD2L1. Among the top genes from the VEGAS analysis, there was enrichment for chemotaxis and lipid biosynthesis.
Discussion
This is the first genome-wide association study of the response to inhaled β2-agonists among COPD subjects. In a meta-analysis including five COPD populations and over 5000 subjects, we identified several genetic variants associated with the response to inhaled β2-agonist bronchodilators. In the African-Americans from COPDGene, several variants in the genes CDH13 (Cadherin-13), SGCD (Sarcoglycan delta), and GOLGA8B (golgin A8 family, member B) demonstrated genome-wide significance for their association with the response to β2-agonists. In the non-Hispanic white COPDGene population, SNPs upstream and within the potassium channel genes, KCNJ2 and KCNK1 respectively, approached genome-wide significant association with the response to inhaled bronchodilators. This association remained in a meta-analysis including all five COPD case populations, although not at the genome-wide significance level.
In the primary analysis among the COPDGene NHW population, the SNP rs17575208 on chromosome 6 upstream from the gene Ephrin-type A receptor 7, EPHA7, was genome-wide significant with a P value of 7.23×10−9 for the outcome BDRABS, 8.3×10−8 for BDRPRED and 4.15×10−6 for BDRBASE. Although mutations in this gene have been found in resected non-small cell lung cancer human specimens, little is known about a potential role for EPHA7 in COPD, asthma, or BDR. In our meta-analysis, this SNP had a P value of 2.32 × 10−5 for the outcome BDRABS and 1.30×10−4 for the outcome BDRPRED, however the association was not significant among the other populations tested (P>0.05). This is an imputed SNP with minor allele frequency of 1.4% but good imputation quality (R2 = 0.82), and therefore may suggest a promising gene for future studies.
We identified several SNPs on chromosome 17 upstream from KCNJ2, also known as KIR2.1 or Potassium inwardly-rectifying channel, subfamily J, member 2, that were suggestively associated with BDR. Inwardly-rectifying potassium channels were initially described in cardiac, skeletal, and brain tissue, and especially in smooth muscle of small arterioles. These channels play a role in potassium-mediated constriction in response to hypoxemia or ischemia(28). KCNJ2 encodes Kir2.1, a strong inwardly rectifying potassium channel, which has been identified in small and large bronchial smooth muscle cells and plays a role in membrane depolarization (29) (30). Although the exact role for these channels in smooth muscle relaxation remains to be determined, it is possible that these channels play a role in the response to increased extracellular potassium, such as that induced by hypoxemia or acidic environments, leading to membrane hyperpolarization and smooth muscle relaxation(28, 30).
Genotyped and imputed SNPs within the potassium channel gene, KCNK1, located on chromosome 1, were also associated with BDRABS. This gene is also known as TWIK-1, and encodes the 2-pore protein potassium channel subfamily K member 1, or inward rectifying potassium channel protein TWIK-1. Two-pore potassium channels have been identified in a lung epithelial cell line(31). TWIK-1 transcript has been identified on the apical membrane of human bronchial epithelial cells, and has been suggested to play a role in hyperpolarization of membrane action potential(32). The top variant in this gene was additionally associated with measures of lung function, suggesting a potential link to COPD severity. Further studies will be necessary to confirm the roles of these potassium channel genes in BDR and COPD severity.
The identification of multiple potassium channel genes in the single SNP and gene-based analyses suggests a potential role for these channels in moderating the response to inhaled bronchodilators. Further, both the GWAS and gene-based analysis were enriched for ion channel genes. Cellular potassium levels play a key role in maintaining membrane potential, and potassium channels have been demonstrated to mediate the effects of β-agonists(2). Other potassium channel genes, such as the KCNQ voltage activated channels, have been found to ameliorate methacholine bronchoconstriction in rat lung models(33), and these inwardly-rectifying potassium channels may play a similar role in bronchial smooth muscle relaxation. The potassium channel opening medication cromakalin has been tested in animal and human asthma subjects(34). However, limited knowledge is available about the role of potassium channels in mediating smooth muscle relaxation. The identification of potassium-channel genes suggests the importance of revisiting this class of medications for COPD and asthma therapeutics.
We noted a statistically significant, but clinically small, difference in the response to inhaled β2-agonists between the non-Hispanic white and African American subjects in COPDGene. To our knowledge, this is the first demonstration that NHW and AA subjects with COPD may respond differently to inhaled β2-agonists. Because of this difference, we examined the AA population alone for variants associated with BDR. In AA subjects, SNPs in the genes CDH13, SGCD, and GOLGA8B were associated with bronchodilator responsiveness. CDH13 encodes the protein T-cadherin, which functions as an adiponectin receptor(35) and is expressed in mouse lungs in response to allergen stimulation with ovalbumin (36). Adiponectin is protective against allergen-induced inflammatory cell response in mouse lungs and airway hyperresponsiveness(37) and T-cadherin knock-out mice demonstrate reduced immune response, airway hyperresponsiveness, and mucus hyperplasia compared to wild-type mice (36). Elevated adiponectin levels have been associated with increased radiographic measures of percent emphysema and lower response to inhaled bronchodilators among subjects with COPD(38).
SGCD encodes the dystrophin-sarcoglycan complex protein subunit sarcoglycan-δ. This protein complex is expressed in skeletal and cardiac muscle and is thought to play a role in limb girdle muscular dystrophy(39). The delta-sarcoglycan complex has been identified in airway smooth muscle cells, and plays a role in mediating the transition of airway smooth muscle cells from contractile to proliferative phenotypes(40), suggesting a possible role in COPD pathogenesis. In addition, variants in the SGCD gene were recently associated with airway hyperresponsiveness in a GWAS among COPD subjects from the large multicenter Lung Health Study. These variants were also nominally associated with BDRBLINE in our population.
Variants in the gene GOLGA8B (Golgin A8 Family, Member B, GOLGA8B) were associated with BDRPRED and approached significant association with BDRABS. Although these were imputed SNPs with low minor allele frequency, the imputation was of good quality. GOLGA8B encodes a golgi system autoantigen, and this region has been associated with myopia in a large GWAS(41), although a potential role in bronchodilator responsiveness is unknown.
We examined the response to an inhaled bronchodilator as a quantitative variable using three measures that have been used in prior epidemiologic and genetic studies(5, 6). In a family-based study, Palmer and colleagues demonstrated that both BDRABS and BDRPRED have greater than 30% heritability, while BDRBASE is less heritable(5), suggesting that all three outcomes were suitable phenotypes to test for genetic association. The absolute change in FEV1 after β2-agonist administration is the most straightforward measure, but it does not account for baseline lung function, which is reduced in COPD. In contrast, BDRBASE has been shown to correlate with baseline lung function(42). These variables were all highly correlated. As no single measure appears to be the most comprehensive, we analyzed all three traits.
Quantitative measures of the response to inhaled bronchodilators differ from the binary definition used by the American Thoracic Society and European Respiratory Society(1). Prior studies have suggested that this binary outcome does not identify a phenotypically distinct subset of COPD patients, since the presence or absence of a bronchodilator response does not predict clinical outcomes and demonstrates intra-individual variability (42, 43). In contrast, linkage and candidate gene studies have previously identified genetic risk factors for quantitative measures of bronchodilator responsiveness(5, 6). In addition to our new GWAS results, these genetic associations suggest that there are distinct genetic risk factors that play a role in determining the quantitative response to inhaled β2-agonists. Identifying these markers may help to identify COPD patients who demonstrate a greater response to β2-agonists, or who may be unlikely to benefit and therefore should be prescribed alternative medications.
Several GWAS have identified SNPs and genes associated with BDR in asthma (8, 11, 26, 27, 44). We examined the top variants from the asthma studies for association with BDR in the COPD populations, as well as SNPs in the ADRB2 gene that have previously been associated with clinical response to long-acting β2-agonist administration in COPD(44–46). A variant in the SLC22A23 gene was nominally associated with BDR in COPD, although the effect size was in the opposite direction as originally reported. A previously identified intergenic SNP, rs11252394, on chromosome 5 also suggested significant association with BDRABS. However, none of the other asthma BDR variants demonstrated significant association. Although ADRB2 may be a candidate gene for bronchodilator responsiveness in asthma, variants from this gene have not demonstrated consistent association with BDR in COPD (6, 47, 48).
Our study has several limitations. It is common for investigators to replicate top GWAS findings in a replication population. However, in order to improve our power to detect an association, we used all available COPD cohorts in our meta-analysis to perform the largest GWAS of BDR to date. Our most significant findings are in biologically plausible genes, and the effect sizes are similar across all included cohorts. It is encouraging that we did find some cross-over between our top hits and those in asthma populations for both bronchodilator responsiveness and airway hyperresponsiveness despite the fact that these are different study populations. Although we identified several variants upstream from KCNJ2 as associated with BDRABS and BDRPRED, these results failed to meet genome-wide significance in the meta-analysis. An examination of each population demonstrates that these variants all had a similar effect size (Supplementary Figure 1, Supplementary Table 8), suggesting that the lack of significance may be related to sample size. In contrast, studies that have identified variants associated with lung function in the general population have had sample sizes up to ten times larger than the current study(49). This is the only genome-wide study of the response to inhaled bronchodilators in COPD performed to date. We were specifically interested in identifying genes associated with BDR in COPD populations, and therefore we are limited to available COPD cohorts for this analysis. Although the GWAS meta-analysis did not demonstrate genome-wide significance, the top SNP is upstream from the gene KCNJ2, and the protein-product potassium channel is relevant to the phenotype being studied. In addition, variants in this same region have previously been associated with lung function. We are additionally limited by the use of a one-time measurement of bronchodilator responsiveness. Although BDR as a binary trait is not necessarily stable in an individual COPD patient over serial measurements (43), we used quantitative outcomes in this analysis. Post-bronchodilator FEV1 is a stable phenotype over time(5), diminishing the noise in the quantitative measures. However, the presence of intra-individual variability may have diluted our ability to identify a significant genome-wide association. Future longitudinal studies in these populations that can account for intra-individual variability may better identify genetic risk factors for these outcomes.
Within the COPDGene African American subjects, there were several SNPs that demonstrated genome-wide significance, but were of low minor allele frequency, including imputed SNPs in CDH13. However, all SNPs had a minor allele frequency greater than 1%, with excellent imputation quality (R2 ≥ 90%). Although the BDR outcomes were normally distributed, minimal skewing could result in false positive associations especially among variants with low minor allele frequency. In order to test our assumption of normality, we performed inverse normal transformation of the BDR outcomes and tested for variants for association with this transformed outcome. The order of SNPs was preserved with this transformed outcome, suggesting that our assumption of normality was correct. We confirmed imputation accuracy through direct genotyping. Furthermore, animal studies provide good evidence that CDH13 is biologically plausible as a gene potentially involved in the bronchodilator pathway. Although the top variants associated with BDR in the AA population were monomorphic in the Caucasian populations, several other CDH13 variants were nominally associated with BDR. In addition, the gene-based test identified this gene as nominally associated with bronchodilator responsiveness even though the top SNPs in the AA analysis were not included in the gene-based test.
In summary, in the largest COPD pharmacogenetics GWAS to date, we demonstrated that variants upstream from the gene KCNJ2 are associated with response to an inhaled short acting β2 -agonist bronchodilator. In addition, several SNPs in the genes CDH13 and SGCD were significantly associated with BDR in African Americans. These results may point to novel assessments or potential novel therapeutic pathways for COPD. Future studies will require larger COPD populations to identify genome-wide significant variants, and functional studies will help to identify a role for the SNPs and genes highlighted in the GWAS.
Supplementary Material
Figure 2.
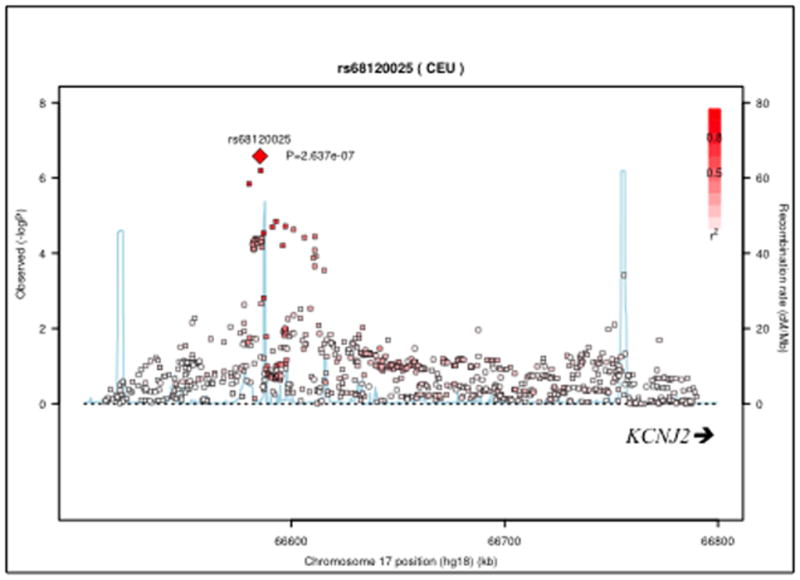
Regional association plot for KCNJ2 variants associated with the outcome BDRABS
Acknowledgments
Supported by: U.S. National Institutes of Health grants R01 HL089897, R01 HL089856, K12 HL089990, R01 HL094635, R01 NR013377, P01 HL105339, P01 HL083069; The Sheila J. Goodnight, MD, FCCP Clinical Research Grant in Women's Lung Health; COPDGene is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Pfizer, Novartis, Boehringer-Ingelheim, and Sunovion. ECLIPSE is supported by GlaxoSmithKline.
The authors would like to thank participants and field investigators in ECLIPSE, NETT, GenKOLS, and COPDGene for their willingness to contribute to medical research.
NIH Grant Support and Disclaimer
The project described was supported by Award Numbers R01 HL089897, R01 HL089856, K12 HL089990, R01 HL094635, P01 HL105339, and P01 HL083069from the National Heart, Lung, And Blood Institute and Award Number R01 NR013377 from the National Institute of Nursing Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, And Blood Institute, the National Institute of Nursing Research, or the National Institutes of Health.
COPD Foundation Funding
The COPDGene® project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, Novartis, Pfizer, Siemens, Sunovion, and GlaxoSmithKline
COPDGene® Investigators – Core Units
Administrative Core: James Crapo, MD (PI), Edwin Silverman, MD, PhD (PI), Barry Make, MD, Elizabeth Regan, MD, PhD
Genetic Analysis Core: Terri Beaty, PhD, Nan Laird, PhD, Christoph Lange, PhD, Michael Cho, MD, Stephanie Santorico, PhD, John Hokanson, MPH, PhD, Dawn DeMeo, MD, MPH, Nadia Hansel, MD, MPH, Craig Hersh, MD, MPH, Peter Castaldi, MD, MSc, Merry-Lynn McDonald, PhD, Emily Wan, MD, Megan Hardin, MD, Jacqueline Hetmanski, MS, Margaret Parker, MS, Marilyn Foreman, MD, Brian Hobbs, MD, Robert Busch, MD, Adel El-Bouiez, MD, Peter Castaldi, MD, Megan Hardin, MD, Dandi Qiao, PhD, Elizabeth Regan, MD, Eitan Halper-Stromberg, Ferdouse Begum, Sungho Won, Brittney Fredericksen, Sharon Lutz, PhD
Imaging Core: David A Lynch, MB, Harvey O Coxson, PhD, MeiLan K Han, MD, MS, MD, Eric A Hoffman, PhD, Stephen Humphries MS, Francine L Jacobson, MD, Philip F Judy, PhD, Ella A Kazerooni, MD, John D Newell, Jr., MD, Elizabeth Regan, MD, James C Ross, PhD, Raul San Jose Estepar, PhD, Berend C Stoel, PhD, Juerg Tschirren, PhD, Eva van Rikxoort, PhD, Bram van Ginneken, PhD, George Washko, MD, Carla G Wilson, MS, Mustafa Al Qaisi, MD, Teresa Gray, Alex Kluiber, Tanya Mann, Jered Sieren, Douglas Stinson, Joyce Schroeder, MD, Edwin Van Beek, MD, PhD
PFT QA Core, Salt Lake City, UT: Robert Jensen, PhD
Data Coordinating Center and Biostatistics, National Jewish Health, Denver, CO: Douglas Everett, PhD, Anna Faino, MS, Matt Strand, PhD, Carla Wilson, MS Epidemiology Core, University of Colorado Anschutz Medical Campus, Aurora, CO: John E. Hokanson, MPH, PhD, Jennifer Black-Shinn, MPH, PhD, Gregory Kinney, MPH, PhD, Sharon Lutz, PhD, Katherine Pratte, MSPH
COPDGene® Investigators – Clinical Centers
Ann Arbor VA: Jeffrey Curtis, MD, Carlos Martinez, MD, MPH, Perry G. Pernicano, MD
Baylor College of Medicine, Houston, TX: Nicola Hanania, MD, MS, Philip Alapat, MD, Venkata Bandi, MD, Mustafa Atik, MD, Aladin Boriek, PhD, Kalpatha Guntupalli, MD, Elizabeth Guy, MD, Amit Parulekar, MD, Arun Nachiappan, MD
Brigham and Women’s Hospital, Boston, MA: Dawn DeMeo, MD, MPH, Craig Hersh, MD, MPH, George Washko, MD, Francine Jacobson, MD, MPH
Columbia University, New York, NY: R. Graham Barr, MD, DrPH, Byron Thomashow, MD, John Austin, MD, Belinda D’Souza, MD, Gregory D.N. Pearson, MD, Anna Rozenshtein, MD, MPH, FACR
Duke University Medical Center, Durham, NC: Neil MacIntyre, Jr., MD, Lacey Washington, MD, H. Page McAdams, MD
Health Partners Research Foundation, Minneapolis, MN: Charlene McEvoy, MD, MPH, Joseph Tashjian, MD
Johns Hopkins University, Baltimore, MD: Robert Wise, MD, Nadia Hansel, MD, MPH, Robert Brown, MD, Karen Horton, MD, Nirupama Putcha, MD, MHS,
Los Angeles Biomedical Research Institute at Harbor UCLA Medical Center, Los Angeles, CA: Richard Casaburi, MD, Alessandra Adami, PhD, Janos Porszasz, MD, PhD, Hans Fischer, MD, PhD, Matthew Budoff, MD, Dan Cannon, PhD, Harry Rossiter, PhD
Michael E. DeBakey VAMC, Houston, TX: Amir Sharafkhaneh, MD, PhD, Charlie Lan, DO
Minneapolis VA: Christine Wendt, MD, Brian Bell, MD
Morehouse School of Medicine, Atlanta, GA: Marilyn Foreman, MD, MS, Gloria Westney, MD, MS, Eugene Berkowitz, MD, PhD
National Jewish Health, Denver, CO: Russell Bowler, MD, PhD, David Lynch, MD
Reliant Medical Group, Worcester, MA: Richard Rosiello, MD, David Pace, MD
Temple University, Philadelphia, PA: Gerard Criner, MD, David Ciccolella, MD, Francis Cordova, MD, Chandra Dass, MD, Robert D’Alonzo, DO, Parag Desai, MD, Michael Jacobs, PharmD, Steven Kelsen, MD, PhD, Victor Kim, MD, A. James Mamary, MD, Nathaniel Marchetti, DO, Aditti Satti, MD, Kartik Shenoy, MD, Robert M. Steiner, MD, Alex Swift, MD, Irene Swift, MD, Gloria Vega-Sanchez, MD
University of Alabama, Birmingham, AL: Mark Dransfield, MD, William Bailey, MD, J. Michael Wells, MD, Surya Bhatt, MD, Hrudaya Nath, MD
University of California, San Diego, CA: Joe Ramsdell, MD, Paul Friedman, MD, Xavier Soler, MD, PhD, Andrew Yen, MD
University of Iowa, Iowa City, IA: Alejandro Cornellas, MD, John Newell, Jr., MD, Brad Thompson, MD
University of Michigan, Ann Arbor, MI: MeiLan Han, MD, Ella Kazerooni, MD, Fernando Martinez, MD,
University of Minnesota, Minneapolis, MN: Joanne Billings, MD, Tadashi Allen, MD
University of Pittsburgh, Pittsburgh, PA: Frank Sciurba, MD, Divay Chandra, MD, MSc, Joel Weissfeld, MD, MPH, Carl Fuhrman, MD, Jessica Bon, MD
University of Texas Health Science Center at San Antonio, San Antonio, TX: Antonio Anzueto, MD, Sandra Adams, MD, Diego Maselli-Caceres, MD, Mario E. Ruiz, MD
ECLIPSE Principal investigators and participating centers include: Bulgaria: Y. Ivanov, Pleven; K. Kostov, Sofia. Canada: J. Bourbeau, Montreal; M. Fitzgerald, Vancouver; P. Hernández, Halifax; K. Killian, Hamilton; R. Levy, Vancouver; F. Maltais, Montreal; D. O'Donnell, Kingston. Czech Republic: J. Krepelka, Praha. Denmark: J. Vestbo, Hvidovre. The Netherlands: E. Wouters, Horn. New Zealand: D. Quinn, Wellington. Norway: P. Bakke, Bergen, Slovenia: M. Kosnik, Golnik. Spain: A. Agusti, Jaume Sauleda, Palma de Mallorca. Ukraine: Y. Feschenko, Kiev; V. Gavrisyuk, Kiev; L. Yashina, Kiev. UK: L. Yashina, W. MacNee, Edinburgh; D. Singh, Manchester; J. Wedzicha, London. USA: A. Anzueto, San Antonio, TX; S. Braman, Providence. RI; R. Casaburi, Torrance CA; B. Celli, Boston, MA; G. Giessel, Richmond, VA; M. Gotfried, Phoenix, AZ; G. Greenwald, Rancho Mirage, CA; N. Hanania, Houston, TX; D. Mahler, Lebanon, NH; B. Make, Denver, CO; S. Rennard, Omaha, NE; C. Rochester, New Haven, CT; P. Scanlon, Rochester, MN; D. Schuller, Omaha, NE; F. Sciurba, Pittsburgh, PA; A. Sharafkhaneh, Houston, TX; T. Siler, St Charles, MO; E. Silverman, Boston, MA; A. Wanner, Miami, FL; R. Wise, Baltimore, MD; R. ZuWallack, Hartford, CT.
Steering Committee
H. Coxson (Canada), C. Crim (GlaxoSmithKline, USA), L. Edwards (GlaxoSmithKline, USA), D. Lomas (UK), W. MacNee (UK), E. Silverman (USA), R. Tal Singer (Co-chair, GlaxoSmithKline, USA), J. Vestbo (Co-chair, Denmark), J. Yates (GlaxoSmithKline, USA).
Scientific Committee
A. Agusti (Spain), P. Calverley (UK), B. Celli (USA), C. Crim (GlaxoSmithKline, USA), B. Miller (GlaxoSmithKline, USA), W. MacNee (Chair, UK), S. Rennard (USA), R. Tal-Singer (GlaxoSmithKline, USA), E. Wouters (The Netherlands), J. Yates (GlaxoSmithKline, USA).
Footnotes
Supplementary information is available at The Pharmacogenomics Journal’s website.
Conflict of Interest:
The following authors report potential conflicts of interest: Dr Michael Cho receives funding from the NIH and the Alpha-1 Foundation; Dr David Lomas has received grant support, honoraria, and consultancy fees from GlaxoSmithKline. He chairs the GSK Respiratory Therapy Area Board; Dr Harvey Coxson has received $4800 in the years 2009 – 2012 for serving on the steering committee for the ECLIPSE project for GSK, he was the co-investigator on two multi-center studies sponsored by GSK and has received travel expenses to attend meetings related to the project. Dr Coxson has three contract service agreements with GSK (including the ECLIPSE study) to quantify the CT scans in subjects with COPD and a service agreement with Spiration Inc to measure changes in lung volume in subjects with severe emphysema. He has received a fee for speaking at a conference and related travel expenses from AstraZeneca (Australia). Dr Coxson was the recipient of a GSK Clinical Scientist Award in 2010; Dr Jørgen Vestbo has received honoraria for consulting and presenting from Almirall, AstraZeneca, Boehringer-Ingelheim, Chiesi, GSK, Novartis and Takeda; Julie Yates is an employee of and owns stock in GlaxoSmithKline; Dr Alvar Agusti has consulted and received honoraria for lecturing at meeting from different pharmaceutical companies commercializing bronchodilators, including GSK, Boheringer-Ingelheim, AstraZeneca, Almirall, Novartis and Chiesi. Dr Celli has worked as a researcher or consultant for the following companies: GSK, Almirall, Novartis, Forrest, Aeris, Boehringer-Ingelheim, Dey, Altana, Pfizer, and Rox. Dr Courtney Crim is an employee of GlaxoSmithKline LLC, the sponsor of the ECLIPSE trial. He holds stock/stock options in GSK as a portion of his compensation as an employee. As it relates to this manuscript, Dr Crim declares no potential conflict of interest; Dr Rennard has had or currently has a number of relationships with companies who provide products and/or services relevant to outpatient management of chronic obstructive pulmonary disease. These relationships include serving as a consultant, advising regarding clinical trials, speaking at continuing medical education programs and performing funded research both at basic and clinical levels. He does not own any stock in any pharmaceutical companies. These companies include: AARC, American Board of Internal Medicine, Able Associates, Align2 Acton, Almirall, APT, AstraZeneca, American Thoracic Society, Beilenson, Boehringer Ingelheim, Chiesi, CIPLA, Clarus Acuity, CME Incite, COPDFoundation, Cory Paeth, CSA, CSL Behring, CTS Carmel, Dailchi Sankyo, Decision Resources, Dunn Group, Easton Associates, Elevation Pharma, FirstWord, Forest, GLG Research, Gilead, Globe Life Sciences, GlaxoSmithKline, Guidepoint, Health Advance, HealthStar, HSC Medical Education, Johnson and Johnson, Leerink Swan, LEK, McKinsey, Medical Knowledge, Medimmune, Merck, Navigant, Novartis, Nycomed, Osterman, Pearl, PeerVoice, Penn Technology, Pennside, Pfizer, Prescott, Pro Ed Communications, PriMed, Pulmatrix, Quadrant, Regeneron, Saatchi and Saatchi, Sankyo, Schering, Schlesinger Associates, Shaw Science, Strategic North, Summer Street Research, Synapse, Takeda, Telecon SC, ThinkEquity; Dr Per Bakke has consulted for Boehringer- Ingelheim and received compensation; Professor Calverley has received funding from the UK MRC and holds an NIHR programme grant. He has been compensated for work on clinical trials steering committees for GSK, Boehringher Ingelheim and Takeda. He has spoken at meetings supported by these companies and by AstraZeneca, Novartis and Almirall. He holds no stock in any relevant concern and has no contacts with the tobacco industry. Dr Victor Kim has nothing to disclose in relationship to this manuscript but has served on an advisory committee for CSA and has participated in clinical trials sponsored by Boehringer Ingelheim, Glaxo-Smith-Kline, and Roche pharmaceuticals. VK is supported by NHLBI K23HL094696-03. Dr Craig Hersh has received consulting fees from Novartis and CSL Behring.
References
- 1.Global initiative for chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2013 Revised 2013. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2013_Feb20.pdf.
- 2.Foreman MG, Campos M, Celedon JC. Genes and chronic obstructive pulmonary disease. Med Clin North Am. 2012;96(4):699–711. doi: 10.1016/j.mcna.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ram FS, Sestini P. Regular inhaled short acting beta2 agonists for the management of stable chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. Thorax. 2003;58(7):580–4. doi: 10.1136/thorax.58.7.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tashkin DP, Celli B, Decramer M, Liu D, Burkhart D, Cassino C, et al. Bronchodilator responsiveness in patients with COPD. Eur Respir J. 2008;31(4):742–50. doi: 10.1183/09031936.00129607. [DOI] [PubMed] [Google Scholar]
- 5.Palmer LJ, Celedon JC, Chapman HA, Speizer FE, Weiss ST, Silverman EK. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Hum Mol Genet. 2003;12(10):1199–210. doi: 10.1093/hmg/ddg125. [DOI] [PubMed] [Google Scholar]
- 6.Kim WJ, Hersh CP, DeMeo DL, Reilly JJ, Silverman EK. Genetic association analysis of COPD candidate genes with bronchodilator responsiveness. Respir Med. 2009;103(4):552–7. doi: 10.1016/j.rmed.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hizawa N, Makita H, Nasuhara Y, Betsuyaku T, Itoh Y, Nagai K, et al. Beta2-adrenergic receptor genetic polymorphisms and short-term bronchodilator responses in patients with COPD. Chest. 2007;132(5):1485–92. doi: 10.1378/chest.07-1103. [DOI] [PubMed] [Google Scholar]
- 8.Duan QL, Gaume BR, Hawkins GA, Himes BE, Bleecker ER, Klanderman B, et al. Regulatory haplotypes in ARG1 are associated with altered bronchodilator response. Am J Respir Crit Care Med. 2011;183(4):449–54. doi: 10.1164/rccm.201005-0758OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan QL, Lasky-Su J, Himes BE, Qiu W, Litonjua AA, Damask A, et al. A genome-wide association study of bronchodilator response in asthmatics. Pharmacogenomics J. 2013 doi: 10.1038/tpj.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan QL, Du R, Lasky-Su J, Klanderman BJ, Partch AB, Peters SP, et al. A polymorphism in the thyroid hormone receptor gene is associated with bronchodilator response in asthmatics. Pharmacogenomics J. 2013;13(2):130–6. doi: 10.1038/tpj.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Himes BE, Jiang X, Hu R, Wu AC, Lasky-Su JA, Klanderman BJ, et al. Genome-wide association analysis in asthma subjects identifies SPATS2L as a novel bronchodilator response gene. PLoS Genet. 2012;8(7):e1002824. doi: 10.1371/journal.pgen.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) Eur Respir J. 2008;31(4):869–73. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 13.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200–2. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, Demeo DL, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21(4):947–57. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7(1):32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Global Initiative for Chronic Obstructive Lung Disease; GOLD, editor. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Updated 2013. [Google Scholar]
- 18.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87(1):139–45. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansel NN, Pare PD, Rafaels N, Sin D, Sandford A, Daley D, et al. Genome Wide Association Study Identifies Novel Loci Associated with Airway Responsiveness in COPD. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hancock DB, Artigas MS, Gharib SA, Henry A, Manichaikul A, Ramasamy A, et al. Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function. PLoS Genet. 2012;8(12):e1003098. doi: 10.1371/journal.pgen.1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drake KA, Torgerson DG, Gignoux CR, Galanter JM, Roth LA, Huntsman S, et al. A genome-wide association study of bronchodilator response in Latinos implicates rare variants. J Allergy Clin Immunol. 2014;133(2):370–8. doi: 10.1016/j.jaci.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padhukasahasram B, Yang JJ, Levin AM, Yang M, Burchard EG, Kumar R, et al. Gene-based association identifies SPATA13-AS1 as a pharmacogenomic predictor of inhaled short-acting beta-agonist response in multiple population groups. Pharmacogenomics J. 2014;14(4):365–71. doi: 10.1038/tpj.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77(4):1165–232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 29.Snetkov VA, Ward JP. Ion currents in smooth muscle cells from human small bronchioles: presence of an inward rectifier K+ current and three types of large conductance K+ channel. Exp Physiol. 1999;84(5):835–46. [PubMed] [Google Scholar]
- 30.Oonuma H, Iwasawa K, Iida H, Nagata T, Imuta H, Morita Y, et al. Inward rectifier K(+) current in human bronchial smooth muscle cells: inhibition with antisense oligonucleotides targeted to Kir2.1 mRNA. Am J Respir Cell Mol Biol. 2002;26(3):371–9. doi: 10.1165/ajrcmb.26.3.4542. [DOI] [PubMed] [Google Scholar]
- 31.Cook DI, Young JA. Effect of K+ channels in the apical plasma membrane on epithelial secretion based on secondary active Cl- transport. J Membr Biol. 1989;110(2):139–46. doi: 10.1007/BF01869469. [DOI] [PubMed] [Google Scholar]
- 32.Zhao KQ, Xiong G, Wilber M, Cohen NA, Kreindler JL. A role for two-pore K(+) channels in modulating Na(+) absorption and Cl(-) secretion in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L4–L12. doi: 10.1152/ajplung.00102.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brueggemann LI, Haick JM, Neuburg S, Tate S, Randhawa D, Cribbs LL, et al. KCNQ (Kv7) POTASSIUM CHANNEL ACTIVATORS AS BRONCHODILATORS: COMBINATION WITH A beta2-ADRENERGIC AGONIST ENHANCES RELAXATION OF RAT AIRWAYS. Am J Physiol Lung Cell Mol Physiol. 2014 doi: 10.1152/ajplung.00253.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelaia G, Gallelli L, Vatrella A, Grembiale RD, Maselli R, De Sarro GB, et al. Potential role of potassium channel openers in the treatment of asthma and chronic obstructive pulmonary disease. Life Sci. 2002;70(9):977–90. doi: 10.1016/s0024-3205(01)01487-4. [DOI] [PubMed] [Google Scholar]
- 35.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 2004;101(28):10308–13. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams AS, Kasahara DI, Verbout NG, Fedulov AV, Zhu M, Si H, et al. Role of the adiponectin binding protein, T-cadherin (Cdh13), in allergic airways responses in mice. PLoS One. 2012;7(7):e41088. doi: 10.1371/journal.pone.0041088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shore SA, Terry RD, Flynt L, Xu A, Hug C. Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol. 2006;118(2):389–95. doi: 10.1016/j.jaci.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 38.Carolan BJ, Kim YI, Williams AA, Kechris K, Lutz S, Reisdorph N, et al. The association of adiponectin with computed tomography phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(5):561–6. doi: 10.1164/rccm.201212-2299OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung D, Duclos F, Apostol B, Straub V, Lee JC, Allamand V, et al. Characterization of delta-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J Biol Chem. 1996;271(50):32321–9. doi: 10.1074/jbc.271.50.32321. [DOI] [PubMed] [Google Scholar]
- 40.Sharma P, Tran T, Stelmack GL, McNeill K, Gosens R, Mutawe MM, et al. Expression of the dystrophin-glycoprotein complex is a marker for human airway smooth muscle phenotype maturation. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L57–68. doi: 10.1152/ajplung.00378.2007. [DOI] [PubMed] [Google Scholar]
- 41.Solouki AM, Verhoeven VJ, van Duijn CM, Verkerk AJ, Ikram MK, Hysi PG, et al. A genome-wide association study identifies a susceptibility locus for refractive errors and myopia at 15q14. Nat Genet. 2010;42(10):897–901. doi: 10.1038/ng.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calverley PM, Burge PS, Spencer S, Anderson JA, Jones PW. Bronchodilator reversibility testing in chronic obstructive pulmonary disease. Thorax. 2003;58(8):659–64. doi: 10.1136/thorax.58.8.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albert P, Agusti A, Edwards L, Tal-Singer R, Yates J, Bakke P, et al. Bronchodilator responsiveness as a phenotypic characteristic of established chronic obstructive pulmonary disease. Thorax. 2012;67(8):701–8. doi: 10.1136/thoraxjnl-2011-201458. [DOI] [PubMed] [Google Scholar]
- 44.Rabe KF, Fabbri LM, Israel E, Kogler H, Riemann K, Schmidt H, et al. Effect of ADRB2 polymorphisms on the efficacy of salmeterol and tiotropium in preventing COPD exacerbations: a prespecified substudy of the POET-COPD trial. The Lancet Respiratory medicine. 2014;2(1):44–53. doi: 10.1016/S2213-2600(13)70248-0. [DOI] [PubMed] [Google Scholar]
- 45.Chung LP, Waterer G, Thompson PJ. Pharmacogenetics of beta2 adrenergic receptor gene polymorphisms, long-acting beta-agonists and asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2011;41(3):312–26. doi: 10.1111/j.1365-2222.2011.03696.x. [DOI] [PubMed] [Google Scholar]
- 46.Hawkins GA, Weiss ST, Bleecker ER. Clinical consequences of ADRbeta2 polymorphisms. Pharmacogenomics. 2008;9(3):349–58. doi: 10.2217/14622416.9.3.349. [DOI] [PubMed] [Google Scholar]
- 47.Bleecker ER, Meyers DA, Bailey WC, Sims AM, Bujac SR, Goldman M, et al. ADRB2 polymorphisms and budesonide/formoterol responses in COPD. Chest. 2012;142(2):320–8. doi: 10.1378/chest.11-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hersh CP. Pharmacogenetics of chronic obstructive pulmonary disease: challenges and opportunities. Pharmacogenomics. 2010;11(2):237–47. doi: 10.2217/pgs.09.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.