

Abstract
Platelet-activating-Factor (PAF), a potent inflammatory mediator, is involved in endothelial permeability. This study was designed to characterize PAF receptor (PAF-R) expression and its specific contribution to the modifications of adherens junctions in mouse endothelial cells. We demonstrated that PAF-R was expressed in mouse endothelial cells and was functionally active in stimulating p42/p44 MAPK and phosphatidylinositol 3-kinase (PtdIns3′-kinase)/Akt activities. Treatment of cells with PAF induced a rapid, time- and dose-dependent (10−7 to 10−10M) increase in tyrosine phosphorylation of a subset of proteins ranging from 90 kDa to 220 kDa, including the VE-cadherin, the latter effect being prevented by the tyrosine kinase inhibitors, herbimycin A and bis-tyrphostin. Furthermore, we demonstrated that PAF promoted formation of multimeric aggregates of VE-cadherin with PtdIns3′-kinase which was also inhibited by herbimycin and bis-tyrphostin. Finally, we showed by immunostaining of endothelial cells VE-cadherin, that PAF dissociated adherens junctions. The present data provide the first evidence that the treatment of endothelial cells with PAF promoted activation of tyrosine kinases and the VE-cadherin tyrosine phosphorylation and PtdIns3′-kinase association, that ultimately lead to the dissociation of adherens junctions. Physical association between PtdIns3′-kinase, serving as a docking protein, and VE-cadherin may thus provide an efficient mechanism for amplification and perpetuation of PAF-induced cellular activation.
Keywords: 1-Phosphatidylinositol 3-Kinase; Adherens Junctions; Enzyme Inhibitors; Fluorescent Antibody Technique; Heart; Immunosorbent Techniques; Intracellular Signaling Peptides and Proteins; Lactams, Macrocyclic; Mice; Mitogen-Activated Protein Kinase 1; Mitogen-Activated Protein Kinase 3; Phosphorylation; Animals; Platelet Activating Factor; Platelet Membrane Glycoproteins; Protein-Tyrosine Kinases; Quinones; RNA, Messenger; Receptors, G-Protein-Coupled; Reverse Transcriptase Polymerase Chain Reaction; Tyrosine; Tyrphostins; Antigens, CD; Benzoquinones; Cadherins; Cell Line; Embryo, Mammalian; Endothelial Cells; Enzyme Activation
INTRODUCTION
Platelet-activating-Factor (PAF) plays a key role in allergic disorders and inflammation, and it has also been implicated in reproduction, cardiovascular, and in nervous and immune systems. PAF is synthesized by a variety of proinflammatory cells that participate in the development of inflammation, involving monocytes/macrophages, polymorphonuclear neutrophils, eosinophils, basophils, and platelets (1, 2). All these cells are targets of PAF bioactions as they bear PAF receptors (PAF-R) localized on their cell surface (3). The gene encoding human PAF-R possesses two 5′ noncoding exons, terminated by a tissue specific promoter, and each of these exons is spliced to a common acceptor site on a third exon that encodes an unique, functional PAF-R protein of 39 kDa (4). Upon binding to its receptor, PAF stimulates a number of signal transduction pathways, involving phospholipids turnover, through activation of phospholipase C (PLC) in platelets, macrophages, B cell lines, endothelial cells, and Kupffer cells (5, 6). PAF equally activates PLA2, phospholipase D, and phosphatidylinositol 3-kinase (PtdIns3′-kinase) in various cells and tissues (6) and is involved in activation of mitogen-activated protein kinase (MAPK) (7, 8) and induces an early tyrosine phosphorylation of numerous signaling proteins, such as focal adhesion kinase (p125FAK) in human endothelial cells (9) and pp60c-src (10) in platelets and PLC, Fyn, Syk, Lyn, and p85 regulatory subunit of PtdIns3′-kinase in human B cell lines (11). Recently it was shown that, PAF enhances the angiogenic activity of certain polypeptide mediators such as tumor necrosis factor and hepatocyte growth factor by promoting endothelial cell motility, suggesting a role for PAF in angiogenesis (12).
Endothelial adherens junctions regulate the transendothelial flux of liquid and plasma proteins (13). The endothelial cell-specific VE-cadherin is a component of endothelial adherens junctions involved in mediating cell-cell interactions (14). Endothelial cell adherens junctions disassemble in response to proinflammatory mediators such as thrombin (15), and histamine (16) resulting in increased transendothelial permeability. The endothelial junctional barrier is disrupted within 5 to 10 minutes, and VE-cadherin complex is redistributed to the membrane in association with increased endothelial permeability. Endothelial adherens junctions disappear and then reform within 2 hours to restore endothelial junctional integrity and normal vasopermeability (15). Tyrosine and serine/threonine kinases and phosphatases acting on catenins, the proteins linking VE-cadherin to the actin cytoskeleton, seem to play an important role in the disassembly of endothelial adherens junctions (17).
The cytoplasmic tail of the classical cadherins, including VE-cadherin, comprises two well-characterized domains. The juxtamembrane domain binds to the catenin p120, an armadillo family protein that is thought to regulate cadherin adhesive interactions by modulating the activity of Rho family GTPases (18). At the carboxyl-terminal region of the cadherin cytoplasmic tail, a domain termed the catenin binding domain interacts with β-catenin or plakoglobin (19). Accordingly, VE-cadherin cytoplasmic domain was shown to regulate endothelial protrusive activity in vitro, suggesting that VE-cadherin may be essential for the invasive process (20). In addition, gene ablation experiments strongly suggested that VE-cadherin might be involved in VEGF-induced survival pathway (21).
The present study focused on the signaling triggered by PAF through PAF-R, leading to activation of tyrosine kinase phosphorylation pathways, in endothelial cell adherens junctions. Our data demonstrate that PAF, induces activation of both MAPK p44/42 and PtdIns3′-kinase signaling pathways, and finally triggers the VE-cadherin tyrosine phosphorylation and dissociation of adherens junction. We showed for the first time a link between the PAF-R signaling, the tyrosine kinase phosphorylations, and the adherens junctions in the regulation of endothelial cell barrier integrity.
MATERIALS AND METHODS
Antibodies
Commercially available antibodies used were as follows: for immunoprecipitation, monoclonal antiphosphotyrosine mAb 4G10 (Upstate Biotechnology, Inc., Lake Placid, NY), mouse monoclonal anti-p85 subunit of PtdIns3′-kinase (Transduction Laboratories, Lexington, KY), and for western blotting, monoclonal antiphosphotyrosine mAb 4G10, polyclonal anti-phospho Akt, polyclonal anti-active MAPK (Promega, Madison), and horseradish peroxidase-conjugated goat anti–mouse IgG, goat anti–rabbit IgG, rabbit anti-rat (Bio-Rad Laboratories (Hercules, CA). For immunoflorescence, Cy3-conjugated affinipure goat anti-rat IgG and goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, Inc. (Westgrove, PA).
Reagents
PAF, phosphatidylinositol, phosphatidylinositol 3-kinase (PtdIns3′-kinase) inhibitor (wortmanin), tyrosine protein kinase inhibitor (herbimycin, bis-tyrphostin), benzamidine, leupeptin, pepstatin A, Triton X-100 were purchased from Sigma-Aldrich (Saint Louis, Missouri). [γ32P]-ATP (3000 Ci/mmol) and the enhanced chemiluminescence detection reagents were purchased from PerkinElmer (Lifesciences, Belgium). Nitrocellulose was obtained from Schleicher and Schuell (Ecquevilly, France). The micro-bicinchoninic acid protein assay reagent kit was from Pierce (Oud Beijerland, The Netherlands). Protein A-Sepharose was from Pharmacia (Netherland). Thin layer chromatography plates were from Merck.
Buffers
Buffer B was: 10mM Tris/HCl (pH7.4), 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% (v/v) Triton X-100 and 0.5% (v/v) Nonidet P-40.
Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
Total endothelial cell RNA was isolated using the RNAgents Total RNA Isolation System (22). The amplification parameters were as follows: 40 cycles (94°C for 1 min, 55°C for 1 min, 72°C for 1 min) for PAF-R, for VE-cadherin gene 5 min at 94°C, n (n=24, 26, 28, 30) cycles de: 94°C for 1 min, 57°C for 1 min, 72°C for 1 min; followed by 10 min at 72°C for final extension, using a PCR apparatus (Biometra Trio-Thermoblock). To ensure semi quantitative results, the number of PCR cycles for each set of primers was selected to be in the linear range of amplification. Hybridized filters were visualized and signals quantified using a Fluorimager (Molecular Dynamics, Sunnyvale, CA). Primers and probes used in these studies were for murine PAF-Receptor, sens: 5′ CAG TGT GCC CAT CCT TGT TG, and antisens 5′ CCT GAT GGA AGT TGG TCT GG, 1, for murine VE-cadherin gene, sens, 5′ ACG GAC AAG ATC AGC TCC TC, antisens 5′ TCT CTT CAT CGA TGT GCA TT. The RT-PCR products were analyzed by fractionation of 10 μL aliquots on a 2.5% agarose/TAE gel. Control samples analyzed in the absence of reverse transcriptase were free of genomic DNA.
Cell culture, extraction, and immunoprecipitation
Mouse embryonic heart endothelial cells (H5V) were obtained from Garlanda et al., 1994, and cultured in Dulbecco’s modified Eagle’s medium (DMEM), 4500 mg/L glucose, sodium pyruvate (Invitrogen corporation, Grand Island, NY), supplemented with 10% fetal bovine serum (FBS), 1% penicillin, 1% streptomycin, 200 mM L-glutamine (all Life Technologies, Inc).
Before PAF stimulation, endothelial cells, were pre-treated for 10 min at 37°C with 5 μl in 5 ml of a mix solution of Na3VO4, 250 mM, hydrogen peroxyde (H2O2) 5M, H2O 4:4:92). PAF stimulation was performed at 37°C for the indicated concentrations and time. The reactions were stopped by addition of 200 μl of ice-cold lysis buffer (buffer A).
Lysing cells in Triton lysis buffer
To avoid potential dephosphorylation of tyrosine residues, cells were rinsed twice with cold PBS and immediately lysed in cold buffer A: 20mM Tris/acetate (pH7.0), 0.27M sucrose, 1% (v/v) Triton X-100, 1mM dithiothreitol (DTT), 1mM EDTA, 1mM EGTA, 1mM Na3VO4, 1mM benzamidine, 4μg/ml leupeptin and 1μg/ml pepstatin A. Cell lysates were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE), or stored at −20°C.
Lysing cells in SDS lysis buffer
In some experiments, cells were rinsed twice with ice-cold PBS, and lysed in ice-cold SDS lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% (v/v) SDS) supplemented with phosphatase inhibitors (1 mM Na3VO4, 1 mM NaF), and protease inhibitors (4 μg/ml aprotinin, 4 μg/ml leupeptin). The cells were harvested and sonicated for 10s at 4°C, and lysates were cleared by centrifugation at 12,000 g for 10 min at 4°C. Protein concentration of each sample was determined using a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). Samples were boiled in a 1:5 ratio (v/v) with 5X Laemmli buffer containing 2.5% (v/v) β-mercaptoethanol, and subjected to SDS-PAGE or stored at −20°C.
Immunoprecipitation
Equal amounts of protein from each cell lysate were incubated with the appropriate antibody and 40 μl of a 50% (v/v) mixture of protein A-Sepharose beads (Amersham Biosciences) for 30 min at 4°C on a rotator. The supernatants were then incubated with the anti-phosphotyrosine antibody or the anti- PtdIns3′-kinase antibody (2 μg/ml) overnight at 4°C. The immunoprecipitates were then collected by incubation with 20 μl of ProteinA-Sepharose. Immunoprecipitates were washed four times in ice-cold NP-40 lysis buffer and collected by centrifugation for 10 min at 4°C. Samples were eluted from ProteinA-Sepharose beads by boiling in a 1:5 ratio (v/v) with 5X Laemmli buffer containing a final concentration of 2.5% (v/v) β–mercaptoethanol, and subjected to SDS-PAGE.
MAP Kinase Assays
Activation of p42/p44 MAP kinase was determined through the immunoblotting of endothelial cell lysates using antibodies that specifically recognize the phosphorylated amino acid residues on activated phospho-MAPK. Following the indicated treatment, H5V cells were washed twice with ice-cold phosphate-buffered saline and lysed with buffer A containing 10 mM sodium orthovanadate for 20 min on ice. The cell lysates were then harvested from the culture dishes, and the cellular debris were removed by centrifugation. Of the total cell lysates, 80 μg of proteins were separated on a 10% denaturing polyacrylamide gel, and the proteins were subsequently transferred to nitrocellulose membranes.
PtdIns3′-kinase activity
PtdIns3′-kinase activity was measured by adding 100 μg of sonicated phosphatidylinositol and 10 μCi of [γ-32P]ATP, 30 mM MgCl2 and 35 μM ATP in a total volume of 60 μl. Reactions were performed for 20 min at 30°C and stopped by the addition of 100 μl of 1 N HCl and 200 μl of chloroform/methanol (1:1 by vol). After centrifugation and removal of the upper layer, 100 μl of methanol/HCl (1:1) was added. After further centrifugation, lipids were separated on thin layer chromatography (TLC) plates with a solvent system of chloroform/methanol/NH4OH (45:35:10 by vol.). The TLC plates associated radioactivity was determined using Phosphorimager (Biorad) quantification. Immunoprecipitation with mouse monoclonal anti-p85 antibody was used as positive control for PtdIns3′-kinase activity.
Immunofluorescence studies
Endothelial cells (H5V) were plated onto coverslips at a density of 45,000 cells/ml and were grown to confluence. After stimulation, with PAF for 30 min at 37 °C, the cells were fixed with 3.5% paraformaldehyde in PBS for 20 min at room temperature and washed 3 times with PBS containing MgCl2 0.5 mM, CaCl2 1 mM. Permeabilization was performed with Triton X-100 (0.5 % in PBS) for 10 min. The cells were then washed 3 times in PBS, and non specific binding sites were saturated with PBS/BSA (1 mg/ml) for 30 min. Incubation with the primary antibody (phosphotyrosine and VE-cadherin 2μg/ml) was performed in PBS for 1 h at room temperature. After three washes with PBS, the cells were then incubated for 1 h with the cyanine-labeled secondary antibody. After 3 washes in PBS, the nuclei were labeled with Hoescht 1 μg/ml for 5 min and then washed 3 times with PBS. The coverslips were then rinsed, dried in ethanol and mounted on glass slides with Aquamount.
SDS/PAGE and Western blotting
Endothelial cell lysates were analyzed by SDS/PAGE (12% acrylamide, 1% bis-acrylamide). Proteins were then transferred from the gel to nitrocellulose for 1 h and the residual binding sites were blocked by incubating the filters for 1 h in phosphate buffer saline containing 0.05% (v/v) Tween 20 and 5% (v/v) non-fat milk. The blots were subsequently incubated with the primary antibody for 1 h. After being washed, the blots were incubated for 1h with horseradish peroxidase-conjugated rabbit anti-mouse IgG diluted in phosphate buffer saline containing 0.05% (v/v) Tween 20. Immunoreactive proteins were visualized by chemiluminescence.
Data analysis
Each data point represents the mean +/−SD of the measures of three different wells or dishes in the same experiment. Each experiment has been reproduced at least three times in identical or similar configuration with similar results.
RESULTS
Detection of PAF-R mRNA in mouse endothelial cells by RT-PCR
Endothelial phenotype of H5V cells was confirmed by the VE-cadherin mRNA expression. As shown in Fig 1A, the amplification of VE-cadherin transcripts by RT-PCR produced a single product (154 bp) which corresponded in size to the mRNA expected for the murine VE-cadherin.
Figure 1. Expression of VE-cadherin and PAF-Receptor in mouse endothelial cells.
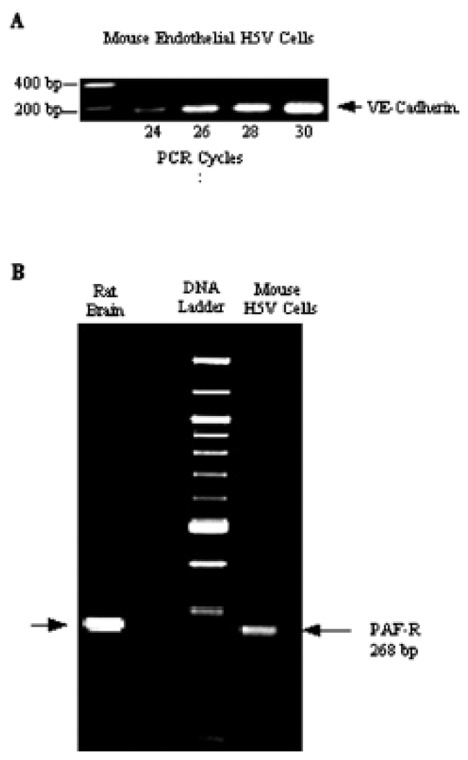
Total RNA (2 ng) was used for RT-PCR analysis. (A): After RT, VE-cadherin cDNA was amplified by 24 to 30-cycles PCR with primer set corresponding to with specific oligonucleotides for VE-cadherin. (B): After RT, PAF-R cDNA was amplified by 35-cycles PCR with primer set corresponding to PAF-R. PCR products were visualized ethidium bromide staining after agarose gel electrophoresis. M indicate 100-bp Promega size markers; lane 1 positive control from rat brain; lane 2 mRNA isolated from H5V cells.
PAF-receptor mRNA levels were determined by RT-PCR using the sets of primers corresponding to the coding region of the mouse PAF-R sequence (268 bp). Fig 1B shows a single band corresponding in size to the RT-PCR product expected for mouse PAF-R mRNA. Analysis of mRNA from rat brain displayed identical amplification product and served as a positive control for PAF-R expression.
Functionality of PAF-R in H5V cells
To determine whether the PAF-R was functional in H5V cells, we measured the activation of MAPK, tyrosine kinases, PtdIns3′-kinase and Akt which play a crucial role in angiogenesis and are known to be activated by PAF in other cell types (22). The dose-dependent phosphorylation of MAPK p44/p42 was used as a sensitive readout of PAF activity. The phospho-p44/42 MAPK antibody only detects p42 and p44 MAPK when catalytically activated by phosphorylation at Thr-202/Tyr-204 (23). The p44/42 MAPK phosphorylation, induced by PAF (10 nM), was observed as early as 2 min after its addition to the cells then reached maximum at 5 min and remained elevated up to 20 min (Figure 2A). Addition of PAF (1–100 nM) increased p44/42 MAPK phosphorylation in a dose-dependent manner (Figure 2B).
Figure 2. PAF signaling in mouse endothelial cells.
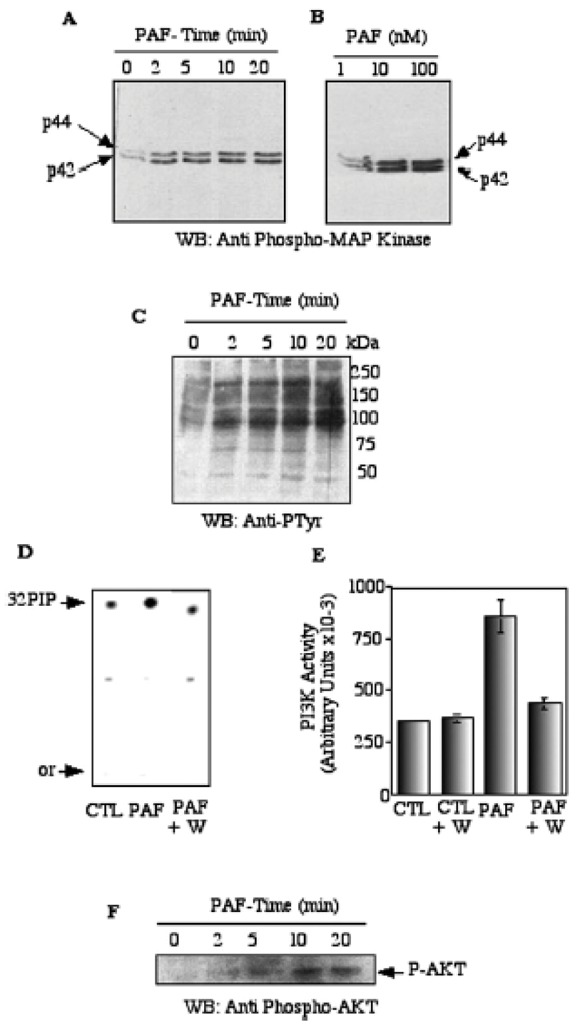
H5V cells were serum starved for 18 h followed by 10 nM PAF exposure for various time intervals. After each time point, cells were lysed and the lysates were were fractionated by SDS-PAGE, and transferred to nitrocellulose. Western blots were incubated with the anti-phospho-p44/42 MAPK (panel A, B). pTyr Ab (panel C), and the ECL system was used to visualize proteins. Panel D, PtdIns3′-kinase activity was assayed in endothelial cells in response to PAF stimulation: subconfluent H5V cells were serum-starved overnight, pretreated or not with 300 nM wortmannin for 30 min (lanes 1 and 7), and subsequently stimulated with PAF 10 nM for 15 min. Cells were lysed and 5 μg of total cellular extracts were incubated in the presence of PI and γ32P-ATP as substrates. The reaction was run for 20 min at 37°C and stopped by the addition of 100 μl HCL. The radioactive lipid products were analyzed by TLC and autoradiography. Panel E, Densitometric analysis of the radioactive phospholipids products separated by TLC. Triplicate assay were performed in each condition. Panel F, Activation of Akt was followed in PAF–treated cells by western blotting of cell samples with the anti-phospho-Akt.
Tyrosine phosphorylation of membrane-associated proteins was determined by the stimulation of serum starved H5V cells and western blotting of cell lysates with an antiphosphotyrosine antibody. In PAF-treated cells, a dramatic time-dependent increase in the phosphotyrosine signal was observed especially for the bands of high molecular weight, as the proteins of 75 and 50 kDa remained unchanged (Figure 2C).
To evaluate the PtdIns3′-kinase activity, we measured the phosphorylation of the phosphatidylinositol. Total cellular extracts from control cells or cells treated with 10 nM PAF were incubated in the presence of PI and γ32P-ATP, as described in Materials and Methods. After extraction of phosphorylated lipids they were further separated by TLC. As shown in Figure 2D, addition of PAF (10 nM) induced a strong increase in cellular of PtdIns3′-kinase activity. In addition, pretreatment of H5V cells with a specific inhibitor of PtdIns3′-kinase, wortmannin (30 nM) for 20 min, completely abolished the PAF-induced PtdIns3′-kinase activity (Figure 2D, E). Figure 2E illustrates the quantification of the radioactive spots using Phosphor Imager.
To evaluate the Akt activity, we measured the phosphorylation of Akt (Ser 473). Cells were plated in the presence or in the absence of PAF and immunoblotted with an anti-phospho-Akt antibody. As shown in Figure 2F, PAF (10 nM) increased Akt phosphorylation as early as 2 min after addition and produced a maximal effect at 10 min. Collectively, these results indicated to us that the PAF signaling pathways were functional in mouse endothelial cells.
Characterization of tyrosine phosphorylation of VE-cadherin in mouse endothelial cells
In an attempt to identify the proteins which were phosphorylated after PAF treatment of the cells, we examined the phosphorylation of VE-cadherin as it has been proposed to be a point of convergence of signaling by endothelial specific growth factors including VEGF (21). Endothelial cells were exposed to 10 nM PAF for 20 min. Cell lysates were immunoprecipitated with the anti phosphotyrosine antibody and the immunoprecipitates were analyzed by SDS-PAGE and westernblotted with the anti-VE-cadherin antibody. PAF treatment of endothelial cells, resulted in a time-dependent phosphorylation of VE-cadherin protein. As shown in Figure 3A–B, the phosphotyrosine content of the 125 kDa protein increased rapidly within 5 min and was sustained up to 20 min of PAF treatment. The effect of PAF on VE-cadherin tyrosine phosphorylation was dose-dependent, already detectable at 0.1 nM (Figure 3C–D). Upon PAF challenge, the amount of VE-cadherin was not significantly modified in control cells, at any time of stimulation (data not shown). These data demonstrate that VE-cadherin is regulated by tyrosine phosphorylation in endothelial cells following PAF stimulation.
Figure 3. PAF induces tyrosine phosphorylation of VE-Cadherin.
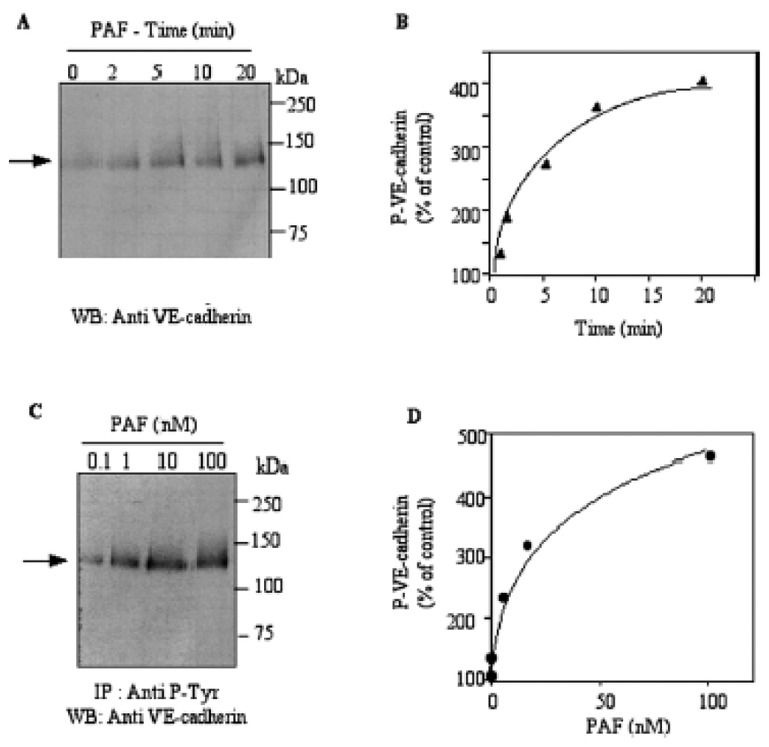
H5V cells were serum starved for 18 h followed by 10 nM PAF exposure for various time intervals. After each time point, cells were lysed and the lysates were immunoprecipitated from equal amounts of total cellular protein from control cells and PAF treated cells (500 μg) with an antiphosphotyrosine antibody. The immmunoprecipitates were separated using 12% SDS-PAGE and western-blotted (Panel A). The phosphorylated protein was revealed by incubating with the anti-VE-cadherin antibody followed by chemiluminescent detection. Panel B, Densitometric analysis of phospho-VE-cadherin, represented as percentage of control. Panel C, H5V cells were serum starved for 18 h followed by exposure to increasing concentrations of PAF for 10 min. After stimulation, cells were processed as in A Panel D, Densitometric analysis of phospho-VE-cadherin, represented as percentage of control (control being set at 100%). Typical results of one of three independent experiments are shown.
Effect of tyrosine kinase inhibitors
In order to elucidate the role of protein tyrosine kinases (PTK) in PAF-induced activation of VE-cadherin tyrosine phosphorylation, we utilized herbimycin, bis-tyrphostin AG126, as potent inhibitors of PTK. As shown on Figure 4 A, when the cells were pre-treated with PTK inhibitors, before the addition of PAF, the pattern of P-tyrosine was reduced to control levels by both inhibitors. Treatment of the cells with wortmanin, the PtdIns3′-kinase inhibitor, before PAF treatment, gave a pattern of tyrosine phosphorylation similar to PAF alone. In these conditions, analysis of VE-cadherin tyrosine phosphorylation (Figure 4 B–C) by immunoprecipitation showed that tyrosine phosphorylation of VE-cadherin was strongly inhibited by herbimycin and tyrphostin but not by wortmanin. Thus, it can be suggested that PAF stimulated tyrosine phosphorylation of VE-cadherin independently from the PtdIns3′-kinase pathway.
Figure 4. PAF-promoted tyrosine phosphorylations of VE-cadherin is blocked by tyrosine kinase inhibitors but not by wortmanin.
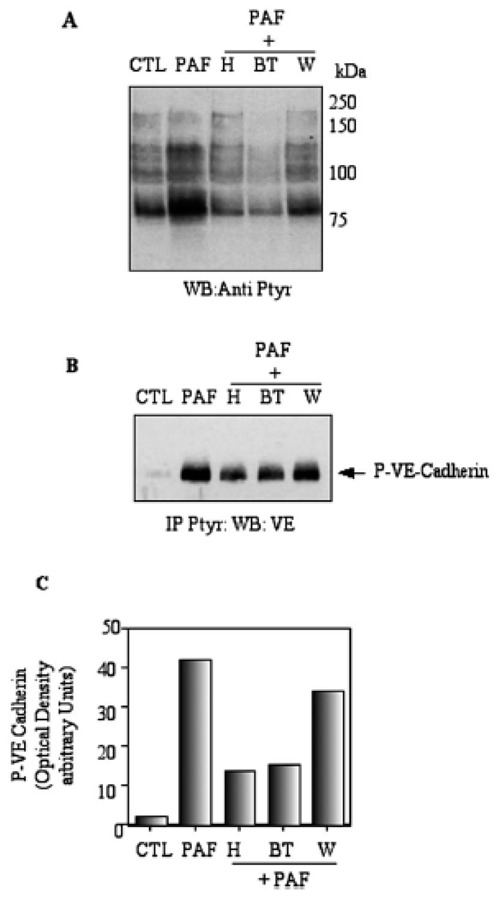
Panel A, H5Vcells were treated with 1 nM PAF for 15 min at 37 °C in the presence or in the absence of herbimycin (H) or Bis-tyrphostin (BT) or Wortmanin (W). Phosphorylated proteins were fractionated by SDS-PAGE, and transferred to nitrocellulose. Western blots were incubated with pTyr Ab, and the ECL system was used to visualize proteins. Panel B, proteins in lysates from control- and PAF-treated cells were immunoprecipitated with pTyr Ab and then treated as described under A. Western blots were incubated with VE-cadherin Ab, and the ECL system was used to visualize proteins. Panel C, Densitometric analysis of phospho-VE-cadherin VE-cadherin. Typical results of one out of three independent experiments are shown.
PAF stimulates complex formation between PtdIns3′-kinase and VE-Cadherin in endothelial cells
As the cytoplasmic tail of VE-cadherin contains several highly conserved tyrosine residues, we hypothesized that some of them are phosphorylated and could serve as recognition sites for SH2 domain proteins involved in intracellular signal transduction. As the prototypic p85 regulatory subunit of the PtdIns3′-kinase has two SH2 domains (24), and because PAF activated PtdIns3′-kinase (Fig 1D) in endothelial cells, we examined whether association between p85 subunit of PtdIns3′-kinase and VE-cadherin was detectable. Serum-starved cells were incubated with or without PAF (10 nM) and the cell lysates were immunoprecipitated with the antibody against VE-cadherin. Immune complexes were separated on SDS/PAGE, transferred to nitrocellulose membrane and immunoblotted with the antibody against the p85 subunit of PtdIns3′-kinase. As shown in Figure 5A, the level of PtdIns3′-kinase associated with VE-cadherin was barely detectable in untreated cells, while the stimulation with PAF strongly induced the association of PtdIns3′-kinase with VE-cadherin (Figure 5A). Immunoprecipitation with the anti- PtdIns3′-kinase antibody and immunoblotting with the anti-VE-cadherin led to similar conclusion ( data not shown).
Figure 5. PAF induced PtdIns3′-kinase association with VE-Cadherin.
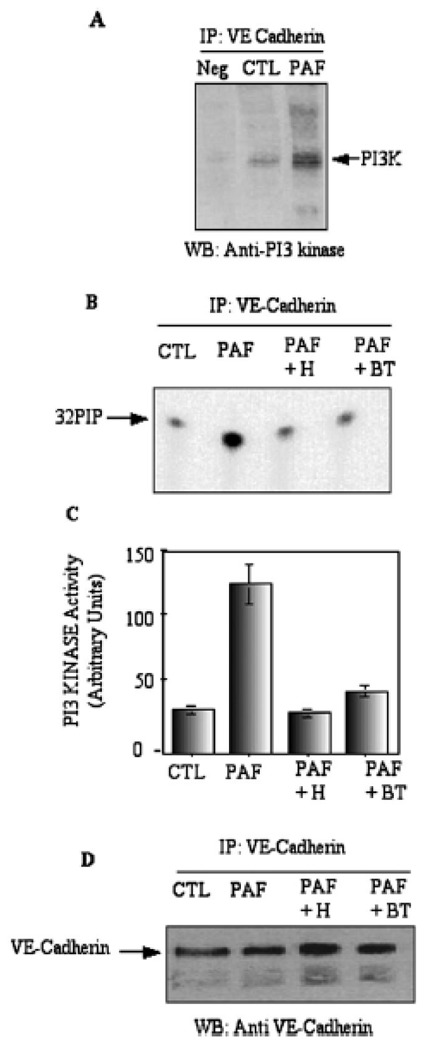
Panel A, H5Vcells were treated with 1 nM PAF for 15 min at 37 °C. Proteins in lysates from control- and PAF-treated cells were immunoprecipitated with anti-VE-cadherin antibody and then treated as described under Fig 1. Western blots were incubated with the anti PI3 kinase antibody: Neg: control with non-immune immunoglobulins. The arrow shows the p85 subunit of PtdIns3′-kinase. Panel B, Measurement of PtdIns3′-kinase activity associated with VE-cadherin: H5Vcells were treated with 1 nM PAF for 15 min at 37°C in the presence or in the absence of herbimycin (H) or Bis-tyrphostin (BT) or Wortmanin (W). The immunoprecipitates were incubated in the presence of PI and [γ32P]-ATP as substrate. The reaction was run for 20 min at 37°C and stopped by the addition of 100 μl HCL. The radioactive lipid products were extracted and analyzed by TLC and autoradiography. Panel C, the radioactive spots of 32P-phospholipids were quantitated using phosphoimager. Panel D, the VE-cadherin immmunoprecipitates were separated using 12% SDS-PAGE and western-blotted with the anti-VE-cadherin antibody. The arrow shows the mature form of VE-cadherin protein. Typical results of one out of three independent experiments are shown.
To confirm the association of PtdIns3′-kinase with VE-cadherin, we performed the PtdIns3′-kinase assay using the VE-cadherin immunoprecipitates. As shown in Fig 5 B–C, PtdIns3′-kinase activity was barely detectable in VE-cadherin immunoprecipitates from untreated cells. In contrast, in PAF-treated cells, the PtdIns3′-kinase activity associated with VE-cadherin increased 4-fold. When the cells were treated with the specific tyrosine kinase inhibitors, the PtdIns3′-kinase activity associated with VE-cadherin remained similar to control level. The levels of VE-cadherin, as determined by Western blotting, were not affected under these conditions (Fig 5D). These data strongly suggest that the PtdIns3′-kinase was associated with VE-cadherin in PAF-treated cells and that this association was dependent upon VE-cadherin tyrosine phosphorylation, as its was inhibited by the tyrosine kinase inhibitor.
PAF induced endothelial cell morphological changes and dissociates adherens junctions
The phase-contrast micrographs of intact endothelial cells grown for 4 days revealed their coble stone aspect (Figure 6, left upper panel). Following the exposure to 10 nM PAF for 10 min, the pronounced morphological changes were observed (Figure 6, right upper panel). In order to determine the effects of PAF exposure on the endothelial adherens junctions, we performed the immunofluorescent staining with the anti-VE-cadherin antibody. The immunostaining of VE-cadherin in control cells was typical for adherens junctions in confluent cells, distributed mainly at the periphery of the cells (Figure 6, middle panel). When the cells were stimulated with 10 nM PAF, the majority of VE-cadherin staining was redistributed throughout the cytoplasm and only a few adherens junctions were detected. In similar way, the tyrosine phosphorylated proteins were reveled with the anti-phosphotyrosine antibody in PAF-treated cells and showed similar pattern as compared with the VE-cadherin staining. Control, untreated cells exhibited similar staining with anti VE-cadherin and anti-phoshotyrosine antibodies (Figure 6, lower panel).
Figure 6. PAF dissociates adherens junctions.
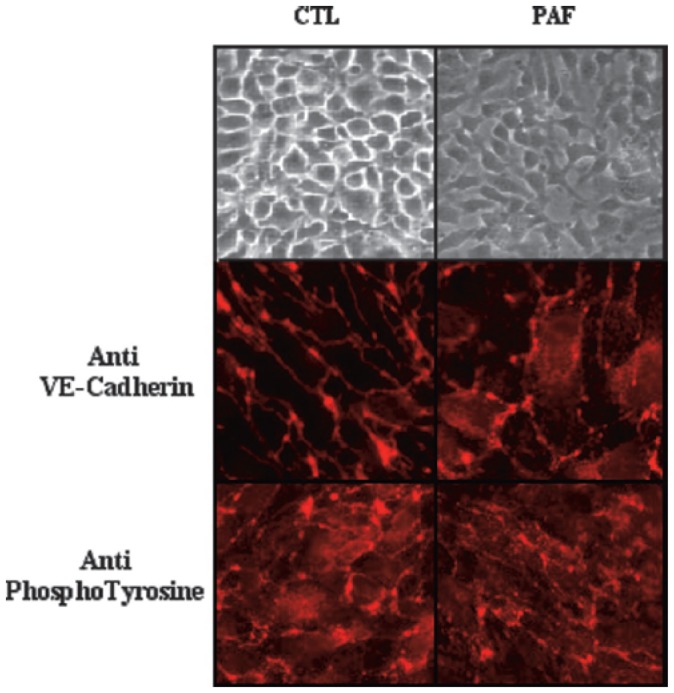
Endothelial cells were grown on glass slides for 4 days, followed by 10 nM PAF exposure for 30 min. The phase-contrast micrographs of intact endothelial cells grown for 4 days are presented in the upper panel. Control endothelial cells (CTL) and PAF-treated cells (PAF) were labelled with monoclonal anti-VE-cadherin (middle panels) and anti-phosphotyrosine (lower panels) antibodies, followed by rhodamine-conjugated secondary antibodies. Photographs were taken under 400 X magnification. This experiment is representative of three additional experiments.
DISCUSSION
PAF is one of the most potent phospholipid agonists that transmits outside-in signals to intracellular transduction systems and effector mechanisms in a variety of cell types, including endothelium (25). We show here, for the first time, that upon activation of PAF-R by PAF in mouse endothelial cells, several signaling events lead to tyrosine phosphorylation of junctionnal protein, the VE-cadherin. Only few in situ studies have been reported on PAF-R expression in blood vessels, and they were mainly related to PAF-induced increases in vascular permeability, showing a widespread localization of PAF-R in microvascular beds and especially its ubiquitous presence on endothelia and in pericytes, fibroblasts, and macrophages associated with microvessels (26). In the present study, we demonstrated that PAF-R is expressed and functionally active in mouse endothelial cells as demonstrated by the rapid activation of a set of protein kinases and where the dose-dependent phosphorylation of MAPK p44/p42 was used as a sensitive readout of PAF-R activity. Our results are in agreement with previous studies showing that the PAF-R, associated with heterotrimeric guanine nucleotide-binding proteins, is responsible for transducing PAF signals into various intracellular cascades (27). It is now accepted that, in addition to a well characterized pathway leading to MAPK activation following ligand-induced tyrosine kinase receptor autophosphorylation, many GPCRs, which lack intrinsic kinase activity, are also able to effectively activate MAPK (28). These includes the receptors for such diverse ligands as bombesin, endothelin-1, somatostatin, interleukin-8, oxytocin, lysophosphatidic acid (29).
PAF induces tyrosine phosphorylation of numerous cellular proteins, such as p125fak in human endothelial cells and brain (30, 31), p85 regulatory subunit of phosphatidylinositol 3-kinase (32), pp60src (10), Fyn, Syk, and Lyn in a human B cell line (32, 33). In the present study we provide an evidence that PAF induced a detectable tyrosine phosphorylation in H5V cell membranes, however the precise identity of tyrosine kinases activated in this model remains uncover. The Janus kinase/signal transducers and activators of transcription (Jak/STAT) pathway is one of the major mechanisms by which cytokine receptors transduce intracellular signals. To date, four mammalian Jaks have been identified (Jak1, Jak2, Jak3, and Tyk2), and have been characterized (34). Recently, the Tyk2 kinase was shown to be activated in response to PAF in myeloid cells and to associate with PAF-R, independently of agonist binding (35). Thus it remains to determine whether Tyk2 could be a potential mediator of PAF signalling in endothelial cells since it was shown to be present and activated by an other GPCR agonist, bradykinin, in endothelial cells (36).
Using a traditional lipid kinase assays to measure PtdIns3′-kinase activity and a PtdIns3′-kinase inhibitor we found that PAF was able to stimulate PtdIns3′-kinase activity, and to promote PKB/Akt phosphorylation in mouse endothelial cells. PtdIns3′-kinase activation by PAF may have several functional implications in endothelial cells: (i) promotes proliferation (ii) induces angiogenesis (iii) regulates endothelial nitric oxide synthase activity and finally increases Akt activity [see (2)for review]. PKB/Akt activity protects against apoptosis through its phosphorylation and inhibition of pro-apoptotic mediators (37), and PKB/Akt-mediated control of cell-cycle progression is well established (38). Taken together, these data suggest that PAF may be a key player in endothelial cell survival and/or cell proliferation.
Tyrosine phosphorylation of VE-cadherin has been described in cells stimulated by VEGF, thrombin or histamine and it was correlated with a rapid dissociation of adherens junctions, a critical step in angiogenesis and in inflammatory processes (13). Interestingly, in the present study, we demonstrated for the first time that VE-cadherin is tyrosine phosphorylated in a time and a dose-dependent manner in PAF-treated cells. Further studies will be required to identify the specific tyrosine kinase activated through the PAF-R. The covalent modification of the specific tyrosyl residues in growth factor receptors and in signaling molecules is involved in cellular communication (39). A conserved domain of about 100 amino acids, the SH2 domain, specifically recognizes and binds to phosphotyrosine, thereby promoting interactions of activated receptors and signaling molecules together (39). Our data demonstrated that the p85 subunit of PtdIns3′-kinase may bind to the phosphorylated form of VE-cadherin in PAF-treated endothelial cells, as this binding was impaired by the specific tyrosine kinase inhibitors. It is possible that both direct and indirect interactions exist between PtdIns3′-kinase and VE-cadherin. As an example, mutational studies in the cytoplasmic domain of the type 2 VEGF receptor have suggested that the p85 subunit of PtdIns3′-kinase may bind directly to Tyr-799 and Tyr-1173 (40). The phosphorylation sites of the VE-cadherin in vivo need to be determined to support this hypothesis.
It has been shown that loss or truncation of VE-cadherin did not impair endothelial proliferation or differentiation, but increases endothelial apoptosis due to an inability of VE-cadherin deficient cells to respond to the survival activity of VEGF-A. The latter is known to mediate endothelial survival via the binding to VEGFR-2, thereby activating the PtdIns3′-kinase and Akt and also via increasing the levels of the antiapoptotic protein Bcl2 (21). Thus, VE-cadherin appears to be required for these VEGF-A-dependent survival signals through formation of a VE-cadherin/β-catenin/PtdIns3′-kinase/VEGFR-2 complex. Further studies will be required to identify the role of PtdIns3′-kinase–VE-cadherin association in PAF actions on endothelial cells.
It was demonstrated that PAF signaling is involved in angiogenesis (2). PAF is present in breast cancer tissues (41) and correlates with the tumor microvessel density (2). The adherens junctional molecule, VE-cadherin, functions to maintain the adherens junction stability and to suppress apoptosis of endothelial cells by forming a complex with VEGFR-2 and members of the armadillo family of cytoplasmic proteins. Our results demonstrate that PAF induced changes in cell shape endothelial monolayers at nanomolar concentrations which correlate with data from in vitro studies in cultured umbilical vein endothelial cells (42, 43). Disturbance of endothelial junctional protein distribution induced by PAF is not regulated at the transcriptional or translational level, as total cellular junctional protein expression and mRNA expression of VE-cadherin were not affected by PAF (44). Thus, the PAF-induced tyrosine phosphorylation of VE-cadherin, might be a likely mechanism involved in PAF-induced endothelial cell shape changes, since it has been suggested that PAF rapidly activates a cascade of phosphorylation steps that mediate cytoskeletal rearrangement (45).
In conclusion, we have demonstrated that PAF induced tyrosine phosphorylation of VE-cadherin and it’s association with PtdIns3′-kinase that lead to the dissociation of adherens junctions. Physical association between PtdIns3′-kinase, serving as a docking protein, and VE-cadherin may thus provide an efficient mechanism for amplification and perpetuation of PAF-induced endothelial cells activation.
Acknowledgments
We are indebted to Francine Cand for technical expertise. This work was supported by INSERM (EMI 02-19 and U525), Commissariat à l’Energie Atomique, Direction des Sciences du Vivant/Département Réponse Dynamique Cellulaire/Association pour la Recherche contre le Cancer (ARC#5588), Fédération Nationale des Centres de Lutte contre le Cancer, Fondation pour la Recherche Médicale, and Ligue Nationale contre le Cancer.
ABBREVIATIONS
The abbreviations used are
- ab
antibody
- bp
base pair(s)
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
Fetal bovine serum
- PAF
1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine or platelet-activating factor
- GPCR
G protein-coupled receptor
- MAP
mitogen-activated protein kinase
- PAF-R
PAF-receptor
- PtdIns3′-kinase
Phosphatidylinositol 3-kinase
- PI
phosphatidylinositol
- pTyr
Phosphotyrosine
References
- 1.Prescott SM, McIntyre TM, Zimmerman GA, Stafforini DM. Sol Sherry lecture in thrombosis: molecular events in acute inflammation. Arterioscler Thromb Vasc Biol. 2002;22:727–733. doi: 10.1161/01.atv.0000016153.47693.b2. [DOI] [PubMed] [Google Scholar]
- 2.Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80:1669–1699. doi: 10.1152/physrev.2000.80.4.1669. [DOI] [PubMed] [Google Scholar]
- 3.Honda Z, Nakamura M, Miki I, Minami M, Watanabe T, Seyama Y, Okado H, Toh H, Ito K, Miyamoto T, et al. Cloning by functional expression of platelet-activating factor receptor from guinea-pig lung. Nature. 1991;349:342–346. doi: 10.1038/349342a0. [DOI] [PubMed] [Google Scholar]
- 4.Mutoh H, Bito H, Minami M, Nakamura M, Honda Z, Izumi T, Nakata R, Kurachi Y, Terano A, Shimizu T. Two different promoters direct expression of two distinct forms of mRNAs of human platelet-activating factor receptor. FEBS Lett. 1993;322:129–134. doi: 10.1016/0014-5793(93)81552-b. [DOI] [PubMed] [Google Scholar]
- 5.Chao W, Liu H, Hanahan DJ, Olson MS. Platelet-activating factor-stimulated protein tyrosine phosphorylation and eicosanoid synthesis in rat Kupffer cells. Evidence for calcium-dependent and protein kinase C-dependent and in-dependent pathways. J Biol Chem. 1992;267:6725–6735. [PubMed] [Google Scholar]
- 6.Liu B, Nakashima S, Takano T, Shimizu T, Nozawa Y. Implication of protein kinase C alpha in PAF-stimulated phospholipase D activation in Chinese hamster ovary (CHO) cells expressing PAF receptor. Biochem Biophys Res Commun. 1995;214:418–423. doi: 10.1006/bbrc.1995.2303. [DOI] [PubMed] [Google Scholar]
- 7.Honda Z, Takano T, Gotoh Y, Nishida E, Ito K, Shimizu T. Transfected platelet-activating factor receptor activates mitogen-activated protein (MAP) kinase and MAP kinase kinase in Chinese hamster ovary cells. J Biol Chem. 1994;269:2307–2315. [PubMed] [Google Scholar]
- 8.Bazan HE, Varner L. A mitogen-activated protein kinase (MAP-kinase) cascade is stimulated by platelet activating factor (PAF) in corneal epithelium. Curr Eye Res. 1997;16:372–379. doi: 10.1076/ceyr.16.4.372.10699. [DOI] [PubMed] [Google Scholar]
- 9.Deo DD, Bazan NG, Hunt JD. Activation of platelet-activating factor receptor-coupled G alpha q leads to stimulation of Src and focal adhesion kinase via two separate pathways in human umbilical vein endothelial cells. J Biol Chem. 2004;279:3497–3508. doi: 10.1074/jbc.M304497200. [DOI] [PubMed] [Google Scholar]
- 10.Dhar A, Shukla SD. Electrotransjection of pp60v-src monoclonal antibody inhibits activation of phospholipase C in platelets. A new mechanism for platelet-activating factor responses. J Biol Chem. 1994;269:9123–9127. [PubMed] [Google Scholar]
- 11.Calcerrada MC, Catalan RE, Martinez AM. PAF-stimulated protein tyrosine phosphorylation in hippocampus: involvement of NO synthase. Neurochem Res. 2002;27:313–318. doi: 10.1023/a:1014911329489. [DOI] [PubMed] [Google Scholar]
- 12.Camussi G, Montrucchio G, Lupia E, De Martino A, Perona L, Arese M, Vercellone A, Toniolo A, Bussolino F. Platelet-activating factor directly stimulates in vitro migration of endothelial cells and promotes in vivo angiogenesis by a heparin-dependent mechanism. J Immunol. 1995;154:6492–6501. [PubMed] [Google Scholar]
- 13.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 14.Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani MG, Dejana E. Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler Thromb Vasc Biol. 1995;15:1229–1239. doi: 10.1161/01.atv.15.8.1229. [DOI] [PubMed] [Google Scholar]
- 15.Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16:488–496. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- 16.Alexander JS, Alexander BC, Eppihimer LA, Goodyear N, Haque R, Davis CP, Kalogeris TJ, Carden DL, Zhu YN, Kevil CG. Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation. 2000;24:99–113. doi: 10.1023/a:1007025325451. [DOI] [PubMed] [Google Scholar]
- 17.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 18.Cascone I, Giraudo E, Caccavari F, Napione L, Bertotti E, Collard JG, Serini G, Bussolino F. Temporal and spatial modulation of Rho GTPases during in vitro formation of capillary vascular network. Adherens junctions and myosin light chain as targets of Rac1 and RhoA. J Biol Chem. 2003;278:50702–50713. doi: 10.1074/jbc.M307234200. [DOI] [PubMed] [Google Scholar]
- 19.Venkiteswaran K, Xiao K, Summers S, Calkins CC, Vincent PA, Pumiglia K, Kowalczyk AP. Regulation of endothelial barrier function and growth by VE-cadherin, plakoglobin, and beta-catenin. Am J Physiol Cell Physiol. 2002;283:C811–821. doi: 10.1152/ajpcell.00417.2001. [DOI] [PubMed] [Google Scholar]
- 20.Kouklis P, Konstantoulaki M, Malik AB. VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. J Biol Chem. 2003;278:16230–16236. doi: 10.1074/jbc.M212591200. [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 22.Izumi T, Shimizu T. Platelet-activating factor receptor: gene expression and signal transduction. Biochim Biophys Acta. 1995;1259:317–333. doi: 10.1016/0005-2760(95)00171-9. [DOI] [PubMed] [Google Scholar]
- 23.Pouyssegur J. Signal transduction. An arresting start for MAPK. Science. 2000;290:1515–1518. doi: 10.1126/science.290.5496.1515. [DOI] [PubMed] [Google Scholar]
- 24.Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling--which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 25.McManus LM, Pinckard RN. PAF, a putative mediator of oral inflammation. Crit Rev Oral Biol Med. 2000;11:240–258. doi: 10.1177/10454411000110020701. [DOI] [PubMed] [Google Scholar]
- 26.Brocheriou I, Stengel D, Mattsson-Hulten L, Stankova J, Rola-Pleszczynski M, Koskas F, Wiklund O, Le Charpentier Y, Ninio E. Expression of platelet-activating factor receptor in human carotid atherosclerotic plaques: relevance to progression of atherosclerosis. Circulation. 2000;102:2569–2575. doi: 10.1161/01.cir.102.21.2569. [DOI] [PubMed] [Google Scholar]
- 27.Ishii S, Nagase T, Shimizu T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 2002;68–69:599–609. doi: 10.1016/s0090-6980(02)00058-8. [DOI] [PubMed] [Google Scholar]
- 28.Chabre O, Cornillon F, Bottari SP, Chambaz EM, Vilgrain I. Hormonal regulation of mitogen-activated protein kinase activity in bovine adrenocortical cells: cross-talk between phosphoinositides, adenosine 3′,5′-monophosphate, and tyrosine kinase receptor pathways. Endocrinology. 1995;136:956–964. doi: 10.1210/endo.136.3.7867605. [DOI] [PubMed] [Google Scholar]
- 29.Schafer B, Gschwind A, Ullrich A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene. 2004;23:991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- 30.Soldi R, Sanavio F, Aglietta M, Primo L, Defilippi P, Marchisio PC, Bussolino F. Platelet-activating factor (PAF) induces the early tyrosine phosphorylation of focal adhesion kinase (p125FAK) in human endothelial cells. Oncogene. 1996;13:515–525. [PubMed] [Google Scholar]
- 31.Calcerrada MC, Catalan RE, Perez-Alvarez MJ, Miguel BG, Martinez AM. Platelet-activating factor stimulation of p125(FAK) and p130(Cas) tyrosine phosphorylation in brain. Brain Res. 1999;835:275–281. doi: 10.1016/s0006-8993(99)01612-1. [DOI] [PubMed] [Google Scholar]
- 32.Kuruvilla A, Pielop C, Shearer WT. Platelet-activating factor induces the tyrosine phosphorylation and activation of phospholipase C-gamma 1, Fyn and Lyn kinases, and phosphatidylinositol 3-kinase in a human B cell line. J Immunol. 1994;153:5433–5442. [PubMed] [Google Scholar]
- 33.Rezaul K, Sada K, Inazu T, Yamamura H. Platelet-activating factor stimulates calcium-dependent activation of protein-tyrosine kinase Syk in a human B cell line. Biochem Biophys Res Commun. 1997;239:23–27. doi: 10.1006/bbrc.1997.7419. [DOI] [PubMed] [Google Scholar]
- 34.Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Curr Opin Immunol. 1998;10:271–278. doi: 10.1016/s0952-7915(98)80165-9. [DOI] [PubMed] [Google Scholar]
- 35.Lukashova V, Asselin C, Krolewski JJ, Rola-Pleszczynski M, Stankova J. G-protein-independent activation of Tyk2 by the platelet-activating factor receptor. J Biol Chem. 2001;276:24113–24121. doi: 10.1074/jbc.M100720200. [DOI] [PubMed] [Google Scholar]
- 36.Ju H, Venema VJ, Liang H, Harris MB, Zou R, Venema RC. Bradykinin activates the Janus-activated kinase/signal transducers and activators of transcription (JAK/STAT) pathway in vascular endothelial cells: localization of JAK/STAT signalling proteins in plasmalemmal caveolae. Biochem J. 2000;351:257–264. doi: 10.1042/0264-6021:3510257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta SR. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 38.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 39.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 40.Dayanir V, Meyer RD, Lashkari K, Rahimi N. Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol 3-kinase and cell proliferation. J Biol Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 41.Pitton C, Lanson M, Besson P, Fetissoff F, Lansac J, Benveniste J, Bougnoux P. Presence of PAF-acether in human breast carcinoma: relation to axillary lymph node metastasis. J Natl Cancer Inst. 1989;81:1298–1302. doi: 10.1093/jnci/81.17.1298. [DOI] [PubMed] [Google Scholar]
- 42.Axelrad TW, Deo DD, Ottino P, Van Kirk J, Bazan NG, Bazan HE, Hunt JD. Platelet-activating factor (PAF) induces activation of matrix metalloproteinase 2 activity and vascular endothelial cell invasion and migration. Faseb J. 2004;18:568–570. doi: 10.1096/fj.03-0479fje. [DOI] [PubMed] [Google Scholar]
- 43.Bussolino F, Camussi G, Aglietta M, Braquet P, Bosia A, Pescarmona G, Sanavio F, D’Urso N, Marchisio PC. Human endothelial cells are target for platelet-activating factor. I. Platelet-activating factor induces changes in cytoskeleton structures. J Immunol. 1987;139:2439–2446. [PubMed] [Google Scholar]
- 44.Zhang Y, Gu Y, Lucas MJ, Wang Y. Antioxidant superoxide dismutase attenuates increased endothelial permeability induced by platelet-activating factor. J Soc Gynecol Investig. 2003;10:5–10. [PubMed] [Google Scholar]
- 45.Biancone L, Cantaluppi V, Boccellino M, Bussolati B, Del Sorbo L, Conaldi PG, Albini A, Toniolo A, Camussi G. Motility induced by human immunodeficiency virus-1 Tat on Kaposi’s sarcoma cells requires platelet-activating factor synthesis. Am J Pathol. 1999;155:1731–1739. doi: 10.1016/S0002-9440(10)65488-0. [DOI] [PMC free article] [PubMed] [Google Scholar]