

Abstract
Nuclear receptor subfamily 4 group A member 2 (NR4A2) is an orphan nuclear receptor that is over-expressed in cancer and promotes cell proliferation, migration, transformation, and chemoresistance. Increased expression and function of NR4A2 have been attributed to various signaling pathways, but little is known about microRNA (miRNA) regulation of NR4A2 in cancer. To investigate the posttranscriptional regulation of NR4A2, we used a 3′ untranslated region (UTR) reporter screen and identified miR-34 as a putative regulator of NR4A2. By using computer predictions, we identified and confirmed an miRNA recognition element in the 3′ UTR of NR4A2 that was responsible for miR-34–mediated suppression. We next demonstrated that overexpression of exogenous miR-34 or activation of the p53 pathway, which regulates endogenous miR-34 expression, decreased NR4A2 expression. Consistent with previous reports, overexpression of NR4A2 blocked the induction of p53 target genes, including mir-34a. This was a phenotypic effect, as NR4A2 overexpression could rescue cells from p53-induced inhibition of proliferation. In summary, our results are the first characterization of a cancer-related miRNA capable of regulating NR4A2 and suggest a network and possible feedback mechanism involving p53, miR-34, and NR4A2.
The nuclear receptor (NR) superfamily is a group of ligand-regulated transcription factors that control specific gene activity. NRs are thus important drug targets1,2. The 48 members of the human NR family share a common modular structure that includes an N-terminal domain containing an activation function 1 (AF-1) region, a DNA-binding domain (DBD), a hinge region, and a C-terminal ligand-binding domain (LBD) that also harbors an AF-2 domain. NR subfamily 4 group A (NR4A) consists of three highly homologous NRs, including NR4A2 (also called Nurr1, NOT, TINUR, or NGFI-Bβ), that are characterized as immediate-early genes induced by mitogens, growth factors, and other stimuli3. The NR4A receptors have been implicated as having roles in multiple tissues and diseases, including cancer4, and their expression and function are associated with various oncogene and tumor suppressor pathways5.
NR4A2 is involved in cancer progression through a mechanism that has yet to be fully described. Most research indicates that NR4A2 has an oncogenic-like role, as it mediates cell proliferation, survival, transformation, invasion, and migration6,7,8,9,10. NR4A2 is highly expressed in squamous cell carcinoma (SCC), as compared to normal patient tissues, and prostaglandin-mediated induction of NR4A2 expression in SCC leads to increased resistance to 5-fluorouracil (5-FU)11. The effects of NR4A2 on chemoresistance are also seen in colorectal cancers12,13, and high expression of NR4A2 predicts a poor outcome for gastric cancer patients receiving 5-FU therapy14. NR4A2 may mediate these effects through a previously described NR4A2-p53 interaction that serves to suppress p53 transactivation, thereby protecting cells from p53-induced apoptosis15. Another NR4A family member, NR4A1 (also called Nur77, TR3, or NGFI-Bα), has also been implicated in p53 suppression16.
As the main arbiter of cell cycle progression, DNA repair, and apoptosis, p53 is a central hub for regulating tumor suppression. These effects are mediated through p53 target genes, which include the microRNA-34 (miR-34) family17,18,19,20. MicroRNAs (miRNAs) are small, endogenous, noncoding RNAs that help regulate target gene networks by binding complementarily to the 3′ untranslated regions (UTRs) of target genes to degrade or prevent their translation into proteins21. The miR-34 family, which consists of three isoforms (miR-34a, miR-34b, and miR-34c) encoded by two p53 direct transcriptional target genes (mir-34a and mir-34b/c), is considered to be partly responsible for carrying out p53’s tumor suppressive function by targeting the 3′ UTRs of genes that are critical to the cell cycle and survival, such as BCL2, cyclins, cyclin-dependent kinases (CDKs), and MYCN19,22. In mice, mir-34a is expressed at higher levels than mir-34b/c, except in lung tissues, where mir-34b/c is dominantly expressed19. Although miR-34 is dispensable for p53 tumor suppression23, it is critical for enhancing p53 stability and activity through miR-34–mediated suppression of negative regulators of p53, such as Sirt124 and Hdm425. The miR-34 isoforms have been clinically characterized as tumor suppressors in multiple cancer types, often independent of p53 mutation, as in neuroblastoma, where mir-34a is commonly deleted26,27, or in other cancers characterized by epigenetic silencing of miR-3428,29.
Recently, miR-132 was reported to target mouse Nr4a230,31; the first characterization of an miRNA targeting an NR4A member. In this study, to determine whether human NR4A2 is regulated by miRNAs and investigate its oncogenic-like role, we used an miRNA screening approach to identify cancer-relevant posttranscriptional regulatory networks of NR4A2, a subject that has not been fully explored. We identified miR-34 as a direct regulator of NR4A2 through a specific sequence in its 3′ UTR. Furthermore, we determined that elevated miR-34 levels, resulting from exogenous overexpression or endogenous induction in a p53-dependent manner, decreased the levels of NR4A2. Corroborating the reported NR4A2-mediated inhibition of p5315, we found that overexpression of NR4A2 inhibited the ability of p53 to activate its target genes, including mir-34a. Lastly, overexpression of NR4A2 attenuated the sensitivity of cells to the p53 activator Nutlin-3a. These data are consistent with previous findings and, for the first time, identify miR-34 as a direct negative regulator of NR4A2. Together, they reveal a novel regulatory network linking p53, miR-34, and NR4A2, in which p53 can overcome its inhibition by endogenous NR4A2 through upregulating miR-34.
Results
Identification of miRNAs directly targeting the 3′ UTR of NR4A2
Increased NR4A2 expression in cancer has been characterized as resulting from cell signaling events5, but miRNA-mediated regulation of NR4A2 in cancer has hitherto been unexplored. To identify putative miRNAs capable of regulating NR4A2 through its 3′ UTR, we assessed the effects of 75 cancer-relevant miRNAs by using a luciferase reporter system (Fig. 1a). Precursor miRNAs selected from an established library32 (see Supplementary Table S1) or an miRNA negative control (pSIF) were cotransfected with the NR4A2 3′ UTR reporter plasmid (WT 3UTR) in 293T cells. We observed that several miRNAs, including miR-34c, were capable of decreasing the luminescence reporter signal to a significantly greater extent than was miR-132, an miRNA that targets Nr4a230,31 (log2 fold changes of −0.47521 and −0.29685, respectively; P ≤ 0.0001 and P = 0.0086, respectively) (Fig. 1b and Table 1). This suggested that miR-34c was a modulator of NR4A2 through its 3′ UTR.
Figure 1. Screening for miRNAs that directly target the 3′ UTR of NR4A2.

(a) A reporter plasmid containing the 3′ UTR of NR4A2 downstream from a firefly luciferase (Fluc) gene was used to identify miRNAs that putatively regulate NR4A2, relative to an internal Renilla luciferase (Rluc) control gene. (b) 293T cells were transfected with the NR4A2 3′ UTR (WT 3UTR) reporter construct for 24 h. Transfected cells were reseeded in 96-well plates and reverse transfected with 75 individual cancer-relevant miRNAs. After 48 h of transfection, a Dual-Glo luciferase assay was performed, the ratio of Fluc/Rluc was calculated, and the log2 fold change was determined for each miRNA (n = 3), relative to a transfection control (pSIF, n = 9), and presented as a waterfall plot. The Fluc/Rluc value for pSIF was set as 1. Statistical significance was calculated using a one-way ANOVA and Dunnett’s test for multiple comparisons between pSIF and the indicated miRNAs. (c) A schematic of the predicted miR-34 seed region in the NR4A2 3′ UTR (WT 3UTR). This predicted miR-34 binding site in the NR4A2 3′ UTR was mutated, and the resulting loss of complementarity is shown (34mut). (d) WT 3UTR or 34mut reporter constructs were cotransfected with pSIF or the indicated miR-34 isoforms into HCT116 colorectal carcinoma cells for 72 h, after which a Dual-Glo luciferase assay was performed. The fold change of the Fluc/Rluc ratio with respect to pSIF was calculated (the Fluc/Rluc value of pSIF for each reporter transfection group was set as 1), and the statistical significance of the relation between pSIF and miR-34a or miR-34c was determined using a two-way ANOVA and Tukey’s test for multiple comparisons (****P ≤ 0.0001; n.s.P > 0.05). Statistically significant changes in WT 3UTR and 34mut for each miRNA transfection are indicated by ####(P ≤ 0.0001).
Table 1. miRNAs that putatively regulate NR4A2 through its 3′ UTR.
| Rank | miRNA ID | Log2 fold change | P-value | TargetScan | miRanda | PicTar | Other predicted NR4A genes | Other targets |
|---|---|---|---|---|---|---|---|---|
| 1 | hsa-miR-335 | −0.47549 ± 0.0223 | <0.0001 | No | No | No | NR4A3 | BCL2L2, SOX4, RB1, RUNX2 |
| 2 | hsa-miR-34c | −0.4752 ± 0.1028 | <0.0001 | Yes | Yes | Yes | — | BCL2, CCND1, CDK4/6, FRA1, MET, MYC, MYCN, SIRT1, SNAI1 |
| 3 | hsa-miR-144 | −0.44443 ± 0.0696 | <0.0001 | No | No | No | NR4A3 | NOTCH1, PTEN, TGFB1 |
| 4 | hsa-miR-214 | −0.37908 ± 0.0942 | 0.0002 | No | Yes | Yes | NR4A1 | BCL2L2, EZH2, PTEN, TWIST1 |
| 5 | hsa-miR-191 | −0.37123 ± 0.1471 | 0.0002 | No | No | No | — | CDK6, SOX4 |
| 6 | hsa-miR-15a | −0.36165 ± 0.0343 | 0.0004 | No | No | No | NR4A1, NR4A3 | BCL2, CCND1, CCND2, CCNE1, CRKL, VEGFA |
| 7 | hsa-miR-155 | −0.36052 ± 0.0926 | 0.0004 | No | No | No | NR4A3 | APC, FOXO3, MLH1, RUNX2, SMAD1, SMAD2, SMAD5 |
| 8 | hsa-miR-20a | −0.35512 ± 0.0296 | 0.0005 | Yes | Yes | Yes | NR4A3 | CCND1, CDKN1A, E2F1, HIF1A, KIT, PTEN |
| 9 | hsa-miR-25 | −0.35227 ± 0.128 | 0.0006 | No | No | No | NR4A3 | BCL2L11, CDH1, EZH2, MDM2, TP53 |
| 17 | hsa-miR-132 | −0.296850 ± 0.1684 | 0.0086 | Yes | Yes | Yes | — | CDKN1A, NR4A2, RB1, SIRT1 |
The log2 fold changes from the reporter assay screen of the top nine miRNAs and miR-132 are listed. The significance of each miRNA in comparison to the transfection control (pSIF) was determined using a one-way ANOVA and Dunnett’s test for multiple comparisons. Three online prediction algorithms (TargetScanHuman, www.targetscan.org; miRanda, www.microrna.org; and PicTar, pictar.mdc-berlin.de) were used to predict if a seed region match was present in the NR4A2 3′ UTR. The same algorithms were used to predict if any of the listed miRNAs targeted other NR4A members. The other known targets of each miRNA are listed (taken from miRTarBase, mirtarbase.mbc.nctu.edu.tw).
By using three different miRNA prediction algorithm tools (TargetScan33, miRanda34, and PicTar35), we identified NR4A2 as a predicted target of the miR-34 family (Table 1), further suggesting that this family of miRNAs were regulators of NR4A2. As expected, miR-132 was also predicted to target NR4A2. To corroborate the findings from “first-generation” prediction tools, we used the CoMeTa interactive database36, which integrates thousands of publicly available gene-expression datasets with the assumption that the predicted targets of an miRNA will be coexpressed with each other. The authors noted that targets falling within the top 50th percentile of the CoMeTa analysis “co-rank” list for putative miRNA targets are highly predictive, based on previous dataset validation. CoMeTa analysis indicates that NR4A2 falls within the top 40.568 and 39.182 percentiles for miR-34a and miR-34c-5p, respectively. By using the prediction algorithms, we identified a predicted miRNA recognition element complementary to the miR-34 seed region in the 3′ UTR of NR4A2 and mutated this sequence to disrupt the complementarity in order to determine if there was direct regulation by miR-34 at this specific site (Fig. 1c). The wild-type 3′ UTR (WT 3UTR) reporter signal was effectively suppressed by cotransfection of miR-34a or miR-34c (52% and 62% of the control level, respectively; P ≤ 0.0001 for all) (Fig. 1d). The effect of miR-34a or miR-34c, compared to that of the control, was not significant when the miRNA was cotransfected with the 3′ UTR containing the seed-region mutation (34mut). Mutation of this predicted seed region was able to rescue the attenuation of the luminescence signal by miR-34a or miR-34c (Fig. 1d), increasing the signal from 52% to 99% and from 62% to 111% of the control level, respectively (P ≤ 0.0001 for all). These data indicate that miR-34 can target NR4A2 at a specific seed complementarity region within its 3′ UTR. Furthermore, by analyzing a dataset from 97 patient samples with rectum adenocarcinoma, we found that the expression of NR4A2 and miR-34a was inversely correlated to a similar extent as other miR-34 targets AXL37 and SIRT124 (see Supplementary Figs S1 and S2). The read counts for miR-34b and miR-34c in this dataset were very low (data not shown), which is consistent with the known expression patterns of mir-34b/c primarily in lung tissues19, making correlation analyses for the mir-34b/c isoforms not suitable.
miR-34 regulates endogenous NR4A2 levels
We next determined the in vitro effect of miR-34 on endogenous NR4A2 mRNA and protein levels through the use of miRNA mimics. Mature miR-34a-5p or miR-34c-5p mimics or a control (CmiR) were overexpressed in HCT116TP53−/− (KO) or HCT116 wild-type (WT) isogenic cell lines to assess the direct effects of forced miRNA expression on NR4A2. Transfection of mature miR-34 mimics increased the expression of each miRNA (Fig. 2a,b). Transfection of miR-34a-5p slightly increased miR-34c-5p expression, probably because of specificity differences in the stem loop primers caused by the approximately 80% sequence homology of the two miR-34 isoforms. Overexpression of the miR-34a-5p mimic reduced the endogenous NR4A2 expression to 59% and 64% of the control level in KO and WT cells, respectively, by 12 h post-transfection, whereas overexpression of the miR-34c-5p mimic reduced the NR4A2 expression to 71% and 66% of the control level in KO and WT cells, respectively (Fig. 2c), and this effect was independent of p53 status. At 24 h post-transfection, the effect of the miR-34 mimics on NR4A2 levels in HCT116 with wild-type p53 was no longer obvious, by comparison to the control. Additionally, the transfection of miR-34 mimics led to increased expression of the p53 target gene CDKN1A/p21 (Fig. 2d), increased p53 protein expression and acetylation, and subsequent p21 protein levels (see Supplementary Fig. S3) in wild-type HCT116 cells. The observation that increased levels of miR-34 led to enhanced p53 protein expression and activity probably reflects the known suppressive function of miR-34 on negative regulators of p53 protein stability (summarized in Supplementary Fig. S3)24,25, though TP53 gene expression (in terms of mRNA levels) was also slightly increased by the miR-34 mimics (see Supplementary Fig. S3). The slight but significant increase in TP53 expression induced by the miR-34 mimics is consistent with the observation that the levels of NR4A2 correlate inversely with that of TP53 gene expression, as discussed later. After 48 h of miR-34 mimic overexpression in HCT116 cells, the protein levels of NR4A2 (Fig. 2e) and another miR-34 target, Sirt1 (see Supplementary Fig. S3), were reduced in both KO and WT cells. Additionally, the effect of mature miR-34 mimics on NR4A2 expression was assessed in another colorectal cancer cell line, RKO, with similar results observed on CDKN1A and NR4A2 expression (see Supplementary Fig. S4). These data demonstrate that exogenous overexpression of a mature miR-34 decreases the levels of endogenous NR4A2 at both mRNA and protein level, regardless of the p53 status. The differential effect of exogenous miR-34 mimic on endogenous NR4A2 may be attributable to the different levels of endogenous NR4A2 mRNA in TP53+/+ and TP53−/− cells, although NR4A2 protein expression was substantially decreased in both TP53-isogenic backgrounds in response to exogenous miR-34 mimic. Additionally, we found that the protein level of NR4A2 was enhanced in RKO cells by a miR-34a inhibitor (see Supplementary Fig. S4), further supporting the regulation of NR4A2 by miR-34.
Figure 2. miRNA-34 downregulates endogenous NR4A2.

Mature miR-34a and miR-34c mimics (10 nM) were transfected into HCT116TP53−/− (KO) or HCT116 wild-type (WT) cells for 12 or 24 h. Expression of hsa-miR-34a–5p (a), hsa-miR-34c–5p (b), NR4A2 (c), and CDKN1A (d) was determined using TaqMan qPCR probes. Mature miRNA expression was normalized to RNU6B, and NR4A2 and CDKN1A were normalized to GAPDH. The value for the CmiR-transfected KO cells at each time point was set as 1. The statistical significance of the results of miR-34 transfections, compared to those of the control, in each cell line for each time point was calculated using a two-way ANOVA and Dunnett’s test for multiple comparisons. ****P ≤ 0.0001; *P ≤ 0.05. (e) Whole-cell lysates (45 μg) from HCT116TP53–/– (KO) and HCT116 wild-type (WT) cells transfected for 48 h with control (CmiR), miR-34a (34a), or miR-34c (34c) mimics (10 nM) were assessed for expression of NR4A2 and β-actin protein by performing Western blotting analysis.
p53 activation suppresses endogenous NR4A2 levels
As mir-34a is a direct transcriptional target of p5317,20, we next sought to determine if NR4A2 expression was regulated by endogenous miR-34a in a p53-dependent manner. We treated cells with a chemical activator of p53, Nutlin-3a38, and confirmed that the p53 protein level increased after the treatment in cells expressing wild-type p53 (WT) but not in p53-deficient (KO) cells (Fig. 3a). As expected, the expression of transcriptional targets of p53, CDKN1A/p21 and BBC3/Puma, was also induced in a p53-dependent manner (Fig. 3a). HCT116CDKN1A−/− and HCT116BBC3−/− cells were used to demonstrate that Nutlin-3a acts upstream of CDKN1A/p21 and BBC3/Puma through p53 (Fig. 3a). We also found increased expression of CDKN1A/p21 and mir-34a in cells expressing wild-type p53 (Fig. 3b,c), accompanied by decreased NR4A2 transcript levels after 48 h of treatment with Nutlin-3a (Fig. 3d). The NR4A family, including NR4A2, are immediate-early responsive genes that are inducible by many external stimuli3, and at an earlier time point (24 h), the levels of NR4A2 mRNA were actually increased by Nutlin-3a (Fig. 3d). In agreement with mRNA expression, Nutlin-3a treatment for 48 h also caused a decrease in the NR4A2 protein expression in WT cells, but not in KO cells (Fig. 3e), further demonstrating the p53 requirement for Nutlin-3a–induced NR4A2 decrease. In addition to NR4A2, Nutlin-3a was also capable of decreasing the protein expression of Sirt1 (Fig. 3e), a known target of miR-3424, suggesting that p53 activation can regulate other downstream factors, possibly through miR-34. We confirmed these results in other colorectal cell lines (RKO and SW48) that are isogenic for TP53 (see Supplementary Figs S5 and S6). These data indicate that p53 activation can induce the expression of mir-34a, while also decreasing the endogenous levels of NR4A2 and another known target of miR-34 (Sirt1). These results are consistent with the observation that the levels of miR-34 and NR4A2 are inversely correlated.
Figure 3. Nutlin-3a activation of p53 decreases endogenous NR4A2.

(a) HCT116 wild-type (WT), HCT116CDKN1A−/− (p21KO), HCT116TP53−/− (p53KO), and HCT116BBC3−/− (PumaKO) isogenic cell lines were treated for 48 h with 10 μM of Nutlin-3a or vehicle control (DMSO). Whole-cell lysates (40 μg) were assessed for expression of p53, p21, Puma, and β-actin by SDS-PAGE gel electrophoresis. (b–d) HCT116TP53−/− (KO) and HCT116 wild-type (WT) cells were treated with vehicle control (DMSO) or Nutlin-3a (5 or 10 μM) for 24 or 48 h. Expression of CDKN1A (b), mir-34a (c), and NR4A2 (d) was determined using TaqMan qPCR probes (normalized to GAPDH). The value for DMSO-treated KO cells at each time point was set as 1. The statistical significance of the results obtained with Nutlin-3a treatments, compared to those obtained with DMSO, in each cell line for each time point was calculated using a two-way ANOVA and Dunnett’s test for multiple comparisons. ****P ≤ 0.0001; ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05. (e) Whole-cell lysates (45 μg) from HCT116TP53−/− (p53KO) and HCT116 wild-type (WT) cells treated for 48 h with DMSO (0 μM) or Nutlin-3a (5 or 10 μM) were assessed for expression of indicated proteins by performing Western blotting analysis.
Overexpression of NR4A2 suppresses p53 activation
It was previously reported that NR4A2 suppressed the transcriptional activity of p53 in the presence or absence of doxorubicin15. We sought to determine the effects of NR4A2 overexpression on the induction of p53 activation by Nutlin-3a. HCT116 cells were transduced with lentivirus expressing empty vector control (EV) or 3xFlag-NR4A2, and overexpression of NR4A2 was confirmed (Fig. 4a). The EV- or 3xFlag-NR4A2–transduced cells were treated with Nutlin-3a to determine the effect on p53 downstream target genes. Upon Nutlin-3a treatment, overexpression of NR4A2 led to a significant attenuation of the p53-induced expression of the target genes mir-34a and CDKN1A/p21 (Fig. 4b,c). To assess the effect of NR4A2 on the binding of p53 to target gene promoters, we performed a chromatin immunoprecipitation (ChIP) assay. As shown in Supplementary Fig. S7, NR4A2 did not decrease p53 occupancy at the target gene promoters39,40, suggesting that another mechanism is responsible for the inhibitory effect of NR4A2 on p53. However, HCT116 cells overexpressing NR4A2 had diminished levels of p53 mRNA and protein (Fig. 4d,e and Supplementary Fig. S7). These data demonstrate that NR4A2 exerts inhibitory effects on the levels of p53 and its transcriptional target genes, which is consistent with previously reported results15 and suggests that the attenuation of p53 mRNA and protein levels, but not of p53 binding to target gene promoters, probably contributes to such inhibitory effects.
Figure 4. NR4A2 overexpression suppresses p53 activation.

HCT116 cells were transduced for 16 h with lentivirus expressing empty vector (EV) or 3xFlag-NR4A2 (NR4A2). The cell medium was then changed and the cells remained in culture for a total of 48 h. The cells were then reseeded and treated with vehicle (DMSO) or Nutlin-3a (5 or 10 μM) for 24 or 48 h. Expression of NR4A2 (a) mir-34a (b) CDKN1A (c) and TP53 (d) was determined using TaqMan qPCR probes (normalized to GAPDH). The value for DMSO-treated EV at each time point was set as 1. The statistical significance of the difference between the results with EV and NR4A2 for each treatment was determined for each time point by using a two-way ANOVA with Sidak’s multiple comparisons test. ####P ≤ 0.0001; ###P ≤ 0.001; ##P ≤ 0.01; #P ≤ 0.05. (e) Whole-cell lysates from HCT116TP53−/− (KO) and HCT116 wild-type (WT) cells transduced with lentivirus expressing EV or 3xFlag-NR4A2 (F-NR4A2) were assessed for expression of Flag (indicating NR4A2), p53, and Gapdh protein by performing SDS-PAGE gel electrophoresis. Relative p53 expression was determined using Odyssey Image Studio to calculate the ratio of p53 to Gapdh protein band density (displayed at the bottom of the gel). The ratio for EV in WT cells was set as 1.0.
Knockdown of NR4A2 enhances p53 activation
To further confirm the inhibitory effect of NR4A2 on p53 transcriptional activity, we investigated the effects of reduced endogenous NR4A2 levels on Nutlin-3a–induced p53 transcriptional target gene expression. HCT116 cells were transfected with nontargeting control (NT) or siRNA targeting NR4A2 (siNR4A2), and knockdown of NR4A2 was confirmed (Fig. 5a). After 48 h of siRNA-mediated knockdown, the cells were treated with Nutlin-3a for an additional 24 h to determine the effects of NR4A2 knockdown on p53 target gene expression. We found that knocking down NR4A2 caused an enhancement of Nutlin-3a–induced expression of CDKN1A/p21, MDM2, and mir-34a (Fig. 5b–d). Consistent with the inhibitory effect of overexpressed NR4A2 on p53 gene expression (Fig. 4d), knocking down NR4A2 led to an increase in TP53/p53 gene expression for both the DMSO- and Nutlin-3a–treated groups (Fig. 5e). We also confirmed the reduced NR4A2 protein levels by the presence of siNR4A2 (Fig. 5f). Together, these data further suggest that NR4A2 can suppress p53 transcriptional activity, at least in part, by inhibiting the expression of p53 itself.
Figure 5. Knockdown of NR4A2 enhances p53 activation.

HCT116 cells were transfected for 24 h with nontargeting control (NT) or siRNA targeting NR4A2 (siNR4A2). The culture medium was then changed, and the cells remained in culture for a total of 48 h. The cells were then treated with vehicle (DMSO) or Nutlin-3a (10 μM) for 24 h. Expression of NR4A2 (a) CDKN1A (b) MDM2 (c) mir-34a (d) and TP53 (e) was determined using TaqMan qPCR probes (normalized to GAPDH). The value for DMSO-treated NT was set as 1. The statistical significance of the results was determined using a two-way ANOVA with Tukey’s multiple comparison test. The significance of the differences within each transfection group between transfection groups (####P ≤ 0.0001; ###P ≤ 0.001; ##P ≤ 0.01) is represented. (f) HCT116 cells were transfected with NT or siNR4A2 as indicated above, followed by treatment of DMSO or Nutlin-3a (10 μM) for indicated length of times. Whole-cell lysates (45 μg) were assessed for expression of indicated proteins by performing Western blotting analysis.
Overexpression of NR4A2 attenuates Nutlin-3a sensitivity
We next examined the cellular effects of NR4A2-mediated suppression of p53 after Nutlin-3a treatment. HCT116 cells transduced for 48 h with lentivirus expressing empty vector control (EV) or 3xFlag-NR4A2 were plated in normal growth medium, and the cell proliferation was monitored using live-cell imaging. After 14 h, the cell culture medium was replaced with fresh medium containing vehicle (DMSO) or Nutlin-3a (5 or 10 μM), and the monitoring of cell proliferation continued. In EV-expressing cells, the inhibitory effect of Nutlin-3a on cell proliferation was substantial (Fig. 6a). In cells overexpressing NR4A2, Nutlin-3a–induced inhibition of cell proliferation was attenuated at both concentrations of Nutlin-3a tested (Fig. 6b). At the end of the real-time monitoring period (108 h), the cells were collected and their mRNA and protein levels were examined to confirm that NR4A2 expression was increased in cells transduced with NR4A2-expressing lentivirus (Fig. 6c,d). These data indicate that overexpression of NR4A2 attenuates the inhibitory effect of Nutlin-3a on cell proliferation. Together, our data are consistent with the known oncogenic role of NR4A2 and, for the first time, identify miR-34 as a negative regulator of NR4A2 and reveal a novel functional network linking p53, miR-34, and p53.
Figure 6. Overexpression of NR4A2 attenuates Nutlin-3a sensitivity.

HCT116 cells were transduced for 16 h with lentivirus expressing empty vector (EV) or 3xFlag-NR4A2 (NR4A2). The cell medium was then changed, and the cells remained in culture for a total of 48 h. The cells were then reseeded into plates and the cell confluence was monitored using an IncuCyte ZOOM imaging system. After 14 h, the cell medium was replaced by medium containing vehicle (DMSO) or Nutlin-3a (5 or 10 μM) (indicated by Tx and arrow). The cell confluence for EV-expressing (a) or NR4A2-expressing (b) cells was monitored for a total of 108 h. (c) The cells were then collected and expression of NR4A2 was determined using TaqMan qPCR probes (normalized to ACTB). The value for DMSO-treated EV was set as 1. (d) Total protein lysates (35 μg) were assessed for expression of NR4A2 (low and high exposure), Flag (to indicate overexpressed NR4A2), and β-actin.
Discussion
Over the past decade, it has become increasingly evident that miRNA dysregulation plays an important role in human disease, including the development, progression, and therapeutic resistance of cancer. This process can be quite complex, as overlapping miRNA-mRNA networks can be formed, with a single miRNA having multiple targets or a single mRNA being a target for multiple miRNAs41. Research on miRNA regulation and the validation of biological targets has continued to increase in an effort to understand the multiple cellular pathways that are affected by specific miRNAs, and this area remains of particular interest as additional miRNA-based therapies are investigated and placed into clinical trials42,43.
The NR4A family of orphan nuclear receptors has been studied extensively in various cancer models, and its regulation and function have been connected to multiple oncogenic and tumor suppressive pathways5. However, the contribution of miRNAs to the expression of the NR4A family is unclear. In this report, we have presented a novel p53–miR-34 regulatory mechanism of the orphan nuclear receptor NR4A2, in which miR-34 directly and negatively regulates NR4A2, which is itself able to repress p53-induced gene expression to rescue the inhibition of cell proliferation (Fig. 7). This is the first miRNA to be characterized as targeting the human NR4A230,31.
Figure 7. Graphical summary.
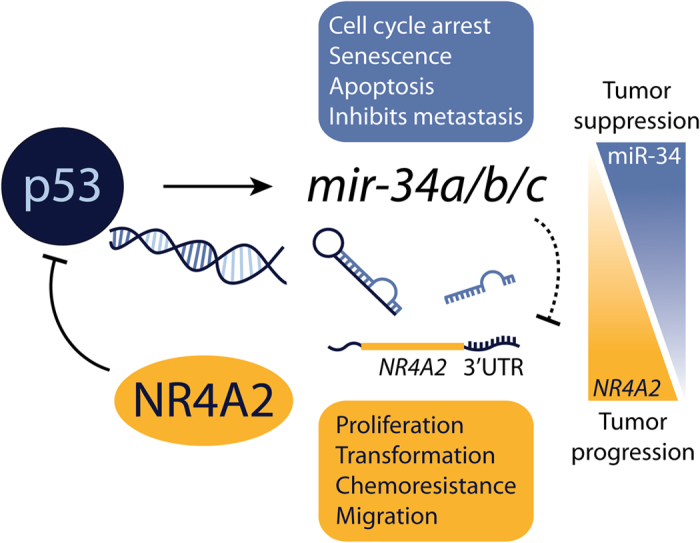
Here, we describe a novel miR-34 regulatory mechanism of NR4A2 that can act downstream of p53 activation. Additionally, our studies extend the finding that NR4A2 can suppress p53 expression and transcriptional targets, including Nutlin-3a–induced activation of mir-34a. The p53–miR-34 regulation of NR4A2 may serve as a protective mechanism to prevent p53 suppression by NR4A2, in addition to suppressing other tumorigenic properties of NR4A2. The long arrow indicates activation; the blunt arrows indicate inhibition; the blunt arrow with the dotted line highlights the inhibition of NR4A2 by miR-34 that indirectly activates p53. As tumors lose expression of mir-34, the expression and function of NR4A2 may become enhanced, shifting the balance from tumor suppression to tumor progression.
Based on the known roles of NR4A2 in oncogenic processes within cancer, we posited that a likely candidate miRNA would be one that harbored tumor-suppressive functions and was downregulated in cancer, consequently leading to upregulation of NR4A2. As presented here, we identified and described the direct regulation of NR4A2 by miR-34, a well-described tumor suppressor–like miRNA that targets genes involved in cancer progression and is increasingly being exploited for its therapeutic advantage22. Whereas other putative miRNAs were identified as targeting NR4A2, we focused on the miR-34 family because of the known role of NR4A members in inhibiting p5315,16 and the positive regulation of miR-34 by p5318, thereby framing our discovery of the regulation of NR4A2 by miR-34 as a possible feedback mechanism. The miR-34 isoforms, predominantly mir-34a, have tumor suppressor functions in multiple cancer types, which is sometimes attributed to their p53 status44,45, though the mechanisms of chromosomal deletion or epigenetic silencing are also major contributors26,27,28,29, and miR-34 expression is prognostic for patient outcome or relapse45,46,47,48. Likewise, several studies have demonstrated the ability of miR-34a restoration to sensitize cells to chemotherapeutic agents, including erlotinib49, Adriamycin/doxorubicin50, and 5-FU51. By using a colorectal cancer cell line pair that was sensitive or resistant to 5-FU, Akao et al. demonstrated that miR-34a was significantly downregulated in the resistant cells, and this was accompanied by increased expression of SIRT1, a target of miR-34. The expression of endogenous miR-34a remained low in 5-FU–resistant cells treated with 5-FU, and the restoration of miR-34a or knockdown of SIRT1 in the resistant cells overcame their resistant phenotype. In light of this observation, it would be interesting to study the relationship of miR-34 and NR4A2 with respect to chemoresistance, as increased NR4A2 expression is correlated with worse patient outcomes with regard to 5-FU therapy11,12,13,14.
By using an in vitro luminescence reporter-based screening assay and three computer-based prediction algorithms33,34,35,36, we identified miR-34c as a putative regulator of NR4A2 via its 3′ UTR region (Fig. 1 and Table 1). This regulatory effect was confirmed by using mutagenesis of the predicted miRNA recognition element that is complementary to the miR-34 seed region. Other miRNAs from this screen have been evaluated using computer-based and mutagenesis approaches (Table 1 and unpublished data), and the functional significance of these miRNA regulators of NR4A2 remains to be elucidated in future studies. Using publically available data, we found a weak negative (inverse) correlation of miR-34a with NR4A2 (see Supplementary Figs S1 and S2), which was similar to the correlations of miR-34a with other known miR-34 target genes, such as AXL37 and SIRT124, although NOTCH150 did not show a negative correlation as expected. Interestingly, the patient sample data demonstrated two populations of TP53-expressing cells, and when we considered only those patient tumor samples with normal levels of TP53, the correlation became stronger. As we would hypothesize, there was a positive correlation of NR4A2 with AXL and SIRT1 since these genes share a common miRNA regulator, miR-34. Consistent with our data in Figures 4d and 5e, the level of NR4A2 was negatively correlated to that of TP53 (see Supplementary Fig. S2).
The next objective was to determine whether miR-34 could regulate endogenous NR4A2, at both mRNA and protein levels. Indeed, overexpression of mature miR-34 isoforms reduced the endogenous levels of NR4A2 mRNA and protein in both wild-type and p53-deficient cells (Fig. 2 and Supplementary Fig. S4). This would be expected, as forced expression of exogenous miR-34 bypasses the requirement for p53. Additionally, miR-34 overexpression enhanced the p53 activity in wild-type cells, probably because of the known regulatory effect of miR-34 on negative regulators of p53 (Fig. 2 and Supplementary Fig. S3). This effect is largely the result of increased p53 protein levels rather than substantial changes in TP53 transcript levels (see Supplementary Fig. S3). The effect of miR-34 on NR4A2 was not as dramatic in the HCT116 p53 wild-type cells, especially at a later time point (24 h) (Fig. 2c), possibly because the lower levels of NR4A2 in these cells no longer respond to exogenous miR-34 expression after the initial transfection, although reduced protein expression was observed at 48 h post-transfection (Fig. 2e). This effect could also result from the delicate balance of NR4A2 expression being regulated directly by miR-34 or indirectly through cellular stress. Correspondingly, RKO p53 wild-type cells had higher levels of endogenous NR4A2 and responded better to miR-34 mimics than their p53-deficient counterpart (Supplementary Fig. S4), further supporting the notion that endogenous levels of NR4A2 affect its response to exogenous miR-34.
To further understand the importance of miR-34 regulation of NR4A2 in the cellular context, we examined the relationship between endogenous miR-34 and endogenous NR4A2 by using our isogenic cell line pairs that possess wild-type or deleted TP53. One interesting observation was that treating HCT116 cells with Nutlin-3a at an early time point (24 h) led to increased NR4A2 expression, but the opposite effect was observed at a later time point (48 h) (Fig. 3d). Consistent with this result, the NR4A2 protein expression was also reduced after 48 h of Nutlin-3a treatment in WT cells (Fig. 3e). The NR4A family are immediate-early genes that are inducible by many stimuli, including cell stress and cytotoxic agents3, whereas Nutlin-3a has a p53-independent role and can induce a DNA damage response leading to cell cycle arrest52. One of these properties might account for the earlier induction of NR4A2 by Nutlin-3a, as the NR4A family mediates DNA double-strand break repair53. This hypothesis could be further tested by using MDM2-deficient cells to determine if these effects are independent of the primary role of Nutlin-3a in Mdm2 inhibition.
Finally, we investigated the previously reported inhibitory effect of NR4A2 on p53 activation15 to determine if this regulation had an effect in the cellular context. We confirmed that overexpression of exogenous NR4A2 can inhibit p53 induction of target genes in response to Nutlin-3a treatment (Fig. 4) and that the opposite effect is seen when NR4A2 expression is reduced by using siRNA (Fig. 5). Our data indicate that this effect might be achieved at least partially through NR4A2 suppression of TP53/p53 gene expression. Further investigation is warranted to determine if this suppression results from direct or indirect repression of the TP53 promoter. The suppressive effect of NR4A2 on the expression of p53 and its transcriptional targets was not reflected in changes in the binding of p53 to its target gene promoters. However, this finding may reflect the sensitivity of the assay, as even in the presence of NR4A2, enough p53 may be present to saturate the promoter response elements during Nutlin-3a treatment. It is also possible that NR4A2 is not inhibitory to p53 binding but is repressive of p53 transactivation at its target gene promoters.
NR4A2 inhibition of the p53 response has previously been investigated only with respect to the genotoxic agent doxorubicin; here, we have demonstrated a similar effect using a more targeted p53 activator. When cells overexpressing NR4A2 were subjected to prolonged treatment with Nutlin-3a, we observed a substantial rescue of cells from Nutlin-3a–induced inhibition of proliferation (Fig. 6), suggesting that the inhibitory effect of NR4A2 on p53 activation is responsible for phenotypic observations that are indicative of p53-induced tumor suppression. Interestingly, in cells overexpressing NR4A2, NR4A2 expression was increased in Nutlin-3a–treated cells after prolonged treatment (Fig. 6c). This effect may be attributable to the ability of NR4A2 to repress the inhibition of cell proliferation, and as the selective pressure of Nutlin-3a is applied to all the cells, those cells expressing the highest levels of NR4A2 persist in culture.
In summary, our study identified and confirmed a novel regulation of NR4A2 by miR-34. This regulatory effect is observed in the context of p53 activation, and NR4A2 itself is able to repress the p53 response. These events can be regarded as a positive feedback loop for p53 (Fig. 7), much like other miR-34 targets that also suppress p5324,25. In tumors expressing wild-type p53, this can serve as a means by which p53 can release itself from repression by NR4A2, further enhancing the p53 response. Alternatively, cells that contain p53 mutations or that overexpress NR4A2 through other means may lack the p53-mediated suppression of NR4A2, allowing NR4A2 to perform other oncogenic activities through its role as a transcription factor. As tumors lose expression of mir-34, through promoter methylation or the loss of p53, the expression and function of NR4A2 may become enhanced, shifting the balance from tumor suppression to tumor progression (Fig. 7), though this may not be directly traceable because of the complexity of miRNA-mRNA regulatory networks.
Materials and Methods
Cell culture
All cell lines were grown in culture at 37 °C in 5% CO2. Human embryonic kidney 293 T cells (ATCC, Manassas, VA) were grown according to the manufacturer’s protocol. Human colorectal carcinoma HCT116 wild-type and TP53−/− isogenic cell lines, which were derived from an adult male harboring a mutation in codon 13 of the ras proto-oncogene, were obtained from the Genetic Resources Core Facility at Johns Hopkins University School of Medicine (Baltimore, MD) and grown in McCoy’s 5A medium (ATCC) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific Inc., Rockford, IL) and 1% penicillin-streptomycin (Life Technologies, Carlsbad, CA). RKO human colon carcinoma (derived from a female harboring mutations in BRAF, NF1 and PIK3CA) and SW48 human colorectal adenocarcinoma (derived from a female harboring mutations in CTNNB1, FBXW7 and EGFR) (wild-type and TP53−/−) and HCT116 (CDKN1A−/− and BBC3−/−) isogenic cell line pairs54,55,56 were obtained from Horizon Discovery (Cambridge, UK) and grown according to the manufacturer’s protocol. Cells collected for RNA and protein extraction were detached with 0.05% trypsin-EDTA (Life Technologies). For all luminescence-based assays, the cells were plated using phenol-red–free DMEM or RPMI-1640 medium (Life Technologies).
3′ UTR reporter plasmid and microRNA screen
The NR4A2 3′ UTR was cloned by GeneCopoeia, Inc. (Rockville, MD), directly downstream from a firefly luciferase (Fluc) gene under the control of an SV40 promoter in the pEZX-MT01 vector, which also contains a Renilla luciferase (Rluc) gene under the control of a CMV promoter (as a transfection control). This reporter construct (WT 3UTR) was used to identify miRNAs that regulate Fluc activity through binding to the NR4A2 3′ UTR and degradation or translational inhibition of fused Fluc mRNA. The Rluc activity was used to normalize the Fluc. The 293T cells were cotransfected for 48 h with the 3′ UTR reporter plasmid and 75 cancer-related miRNAs selected from a previously described library32 (see Supplementary Table S1) by using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Fugene 6 (Promega, Madison, WI) was used for transient cotransfection of reporter gene plasmids and miRNAs into HCT116 cells according to the manufacturer’s instructions. Dual-Glo luciferase assays (Promega, Madison, WI) were performed to measure and calculate the ratios of firefly and Renilla luciferase activity. Luciferase activity was measured with an EnVision 2102 Multilabel Plate Reader (Perkin Elmer Inc., Waltham, MA).
miRNA target prediction and mutagenesis
Three miRNA target-prediction algorithms were used to identify putative miRNA regulators of NR4A2: TargetScanHuman, http://www.targetscan.org33; miRanda, http://www.microrna.org34; and PicTar, http://pictar.mdc-berlin.de/35. By using these algorithms, a putative seed region was determined and mutated using site-directed mutagenesis (Mutagenex, Inc., Piscataway, NJ). Reporter constructs containing either the wild-type (WT 3UTR) or mutated (34mut) 3′ UTR were used to demonstrate miR-34 specificity in the NR4A2 3′ UTR.
Molecular cloning
NR4A2 cDNA was cloned into the pEXM12-3xFLAG (N-terminal) vector (GeneCopoeia, Inc.). We used the primers listed in Supplementary Table S2 to amplify 3xFLAG-NR4A2 cDNA from pEX-3xFLAG-NR4A2 by PCR then subcloned it into a pSin-EF2-IRES-Blast lentiviral expression vector (kindly provided by Dr. Mark E. Hatley). All DNA constructs were confirmed by Sanger sequencing. Lentiviral expression plasmids were packaged into viral particles using the psPAX2 packaging (Addgene plasmid #12260) and pMD2.G envelope (Addgene plasmid #12259) vectors.
miRNA mimics and inhibitor, RNAi, and chemical treatments
Exogenous expression or inhibition of mature miRNAs was performed with mirVana mimics (CmiR: cat# 446058; miR-34a: cat# 446066; miR-34c: cat# 446066) or inhibitors (Cntrl. inhib.: cat# 446076; miR-34a inhib.: cat# 446084; used at 10 nM) transfected using Lipofectamine RNAiMAX reagent (Ambion, Life Technologies). All chemical treatments were performed in full growth medium containing 0.1% dimethyl sulfoxide (DMSO; as a negative control) (Thermo Fisher Scientific) or Nutlin-3a (Sigma Aldrich) as specified by the experimental design. Small interfering RNA (siRNA) was obtained from Dharmacon (Lafayette, CO) and transfected at a concentration of 20 nM using Lipofectamine RNAiMAX reagent.
RNA extraction and quantitative real-time PCR
RNA was extracted using Maxwell simplyRNA kits and a Maxwell 16 Instrument (Promega). For the experiments that used miRNA mimics, the total RNA (including small RNAs) was extracted from the collected cells using miRNeasy Mini kits (Qiagen) in accordance with the manufacturer’s instructions. RNA concentrations were measured using a NanoDrop 8000 UV-Vis Spectrophotometer (Thermo Fisher Scientific). All cDNA used in mRNA and miRNA quantitative real-time PCR (qPCR) analyses was synthesized from extracted RNA by using the SuperScript VILO cDNA synthesis kit (Life Technologies) according to the manufacturer’s protocol. The mRNA and miRNA expression data were generated using Applied Biosystems TaqMan assays (20×) and Fast Advanced Master Mix (Life Technologies). Thermal cycling for qPCR was performed with an Applied Biosystems 7900HT Fast Real-Time PCR system (Life Technologies) in accordance with the TaqMan Fast protocol.
Protein extraction and Western blot analysis
Protein was isolated by incubating cells in Pierce RIPA lysis buffer with added Halt Protease Inhibitor cocktail (Thermo Fisher Scientific) for 30 min on ice then sonicating the lysate for 10 s at 50% amplitude to shear the DNA. The protein concentration was measured using the Pierce BCA Protein Assay (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Protein lysates were resolved on NuPAGE 4–12% SDS-PAGE gradient gels (Life Technologies). After electrophoresis was completed, the proteins were transferred from the gels to nitrocellulose membranes with an iBlot dry transfer system (Life Technologies). Protein gels used for detection of NR4A2 protein were transferred to polyvinylidene fluoride (PVDF) membranes using wet transfer for 1 h at 100 V constant voltage. The specific antibodies used were rabbit anti-NR4A2 (Santa Cruz Biotechnology, sc-5568; M-196; 1:500 dilution)57,58 (see Supplementary Fig. S8), mouse anti-p53 (Santa Cruz Biotechnology, sc-65334; B-P3), rabbit anti-Sirt1 (Santa Cruz Biotechnology, sc-15404; H-300), goat anti-PUMAα (Santa Cruz Biotechnology, sc-19187; N-19); rabbit anti–acetyl-p53 (Lys382) (Cell Signaling, Danvers, MA, #2525); mouse anti-Gapdh (Ambion, Life Technologies); mouse anti-p21 (Oncogene Research Products, Boston, MA, OP64); mouse anti–β-actin (Sigma Aldrich, A5441; clone AC-15); and mouse anti-FLAG (Sigma Aldrich, F1804; clone M2). All antibodies were diluted in Odyssey Blocking Buffer (LI-COR Biotechnology, Lincoln, NE). The secondary antibodies were goat anti-rabbit and goat anti-mouse IRDye 800WC or IRDye 680LT (LI-COR). All Western blot imaging was conducted using a LI-COR Odyssey Infrared Imaging System.
Chromatin immunoprecipitation (ChIP)
HCT116 cells transduced with pSin-EF2-IRES-Blast empty vector (EV) or 3xFlag-NR4A2 lentivirus were grown in flasks and treated with vehicle (DMSO, 0.1%) or Nutlin-3a (10 μM) for 6 h. The chromatin was then crosslinked with 1% formaldehyde for 10 min. Cell extracts were digested for 10 min with 50 units of micrococcal nuclease (New England Biolabs, Ipswich, MA) at 37 °C and further sonicated to yield sheared DNA fragments with an average length of 200 to 1000 base pairs. The sonicated samples were centrifuged to pellet the cell debris, and the supernatant was diluted 7-fold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, and protease inhibitor cocktail). The samples were precleared with ChIP-grade Protein G agarose beads (Cell Signaling, #9007) in ChIP dilution buffer (1:1 ratio). Diluted supernatant (100 μL) was reserved as input (10%) for each treatment. Chromatin (1 mL) was used for each immunoprecipitation and was incubated overnight at 4 °C with mouse anti-p53 (Santa Cruz Biotechnology, sc-126; DO-1) or control mouse IgG. The antibody-protein-DNA complexes were precipitated by incubation with Protein G–agarose beads for 2 h at 4 °C. The protein-DNA complexes were eluted from the beads with elution buffer (1% SDS, 0.1 M NaHCO3). The crosslinks were reversed, and DNA was eluted from the protein-DNA complexes by adding 200 mM NaCl and incubating at least overnight at 65 °C. DNA was recovered and purified after protein digestion with Proteinase K at 45 °C for 2 h using the Qiagen DNeasy Blood & Tissue Kit (Qiagen Cat. No: 69506). Quantitative real-time PCR was performed to determine the changes in p53 occupancy at various known sites of p53 binding. The double-negative controls were nonspecific antibody (normal mouse IgG) and primers coding for regions that do not interact with p53. The thermal cycling conditions were 95 °C for 10 min followed by 45 cycles of 25 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. The primers used are listed in Supplementary Table S2.
Cell viability assays
Real-time cell growth in response to the various treatments was measured as the degree of cell confluence in culture plates and was determined using an IncuCyte ZOOM live-cell imaging system (Essen BioScience, Ann Arbor, MI). Cell proliferation curves were plotted using confluence values at specified time points for each treatment.
Analyses of expression data from online databases
The RNASeq and miRNASeq dataset of 164 patients with rectum adenocarcinoma was acquired from the TGCA Research Network: http://cancergenome.nih.gov/. After log2 transformation of reported normalized_count for mRNA expression and read_per_million_miRNA_mapped for mature miRNA expression, a subset of 97 samples with data generated from the HiSeq platform (for a consistent and reliable comparison) was investigated. The correlations between the expression of miR-34a and NR4A2, as well as other published miR-34 targets—AXL37, NOTCH150, and SIRT124—and TP53 were determined by the regression analysis using Stata software (College Station, TX).
Statistical analysis
All experiments were performed at least three times, and the independent replicates are represented as mean ± standard deviation. Data normalization, statistical tests used, and representations of P-value are indicated for each figure in the corresponding legend.
Additional Information
How to cite this article: Beard, J. A. et al. The orphan nuclear receptor NR4A2 is part of a p53–microRNA-34 network. Sci. Rep. 6, 25108; doi: 10.1038/srep25108 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Mark E. Hatley for kindly providing plasmids, Dr. R. Kiplin Guy for compound, Dr. Bert Vogelstein for cell lines, other members of the Chen research laboratory for valuable discussions, Dr. Keith A. Laycock (St. Jude Department of Scientific Editing) for editing the manuscript, and the Hartwell Center at St. Jude Children’s Research Hospital for DNA sequencing. This work was supported by ALSAC, St. Jude Children’s Research Hospital, and the National Institutes of Health [Grants R01GM086415, R01GM110034, P30-CA21765, and 5R25CA23944].
Footnotes
Author Contributions J.A.B. and T.C. proposed and organized the study. J.A.B. designed the experiments. J.A.B., A.T., J.H., J.D.H. and M.T.C. performed the experiments. Y.D.W. analyzed publically available expression data. J.A.B. wrote the main manuscript and prepared the figures. J.A.B. and T.C. revised and finalized the manuscript. All authors reviewed the final manuscript.
References
- Chen T. Nuclear receptor drug discovery. Curr Opin Chem Biol 12, 418–26 (2008). [DOI] [PubMed] [Google Scholar]
- Gronemeyer H., Gustafsson J. A. & Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3, 950–64 (2004). [DOI] [PubMed] [Google Scholar]
- Maxwell M. A. & Muscat G. E. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal 4, e002 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S., Kim K., Li X. & Lee S. O. NR4A orphan receptors and cancer. Nucl Recept Signal 9, e002 (2011). [Google Scholar]
- Beard J. A., Tenga A. & Chen T. The interplay of NR4A receptors and the oncogene-tumor suppressor networks in cancer. Cell Signal 27, 257–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke N. et al. Nuclear hormone receptor NR4A2 is involved in cell transformation and apoptosis. Cancer Res 64, 8208–12 (2004). [DOI] [PubMed] [Google Scholar]
- Llopis S. et al. Dichotomous roles for the orphan nuclear receptor NURR1 in breast cancer. BMC Cancer 13, 139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamoto T. et al. 1,1-Bis(3′-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol Cancer Ther 7, 3825–33 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamoto T., Czerniak B. A., Dinney C. P. & Kamat A. M. Cytoplasmic mislocalization of the orphan nuclear receptor Nurr1 is a prognostic factor in bladder cancer. Cancer 116, 340–6 (2010). [DOI] [PubMed] [Google Scholar]
- Smith A. G., Lim W., Pearen M., Muscat G. E. & Sturm R. A. Regulation of NR4A nuclear receptor expression by oncogenic BRAF in melanoma cells. Pigment Cell Melanoma Res 24, 551–63 (2011). [DOI] [PubMed] [Google Scholar]
- Shigeishi H. et al. PGE(2) targets squamous cell carcinoma cell with the activated epidermal growth factor receptor family for survival against 5-fluorouracil through NR4A2 induction. Cancer Lett 307, 227–36 (2011). [DOI] [PubMed] [Google Scholar]
- Han Y. et al. Nuclear orphan receptor NR4A2 confers chemoresistance and predicts unfavorable prognosis of colorectal carcinoma patients who received postoperative chemotherapy. Eur J Cancer 49, 3420–30 (2013). [DOI] [PubMed] [Google Scholar]
- Holla V. R., Mann J. R., Shi Q. & DuBois R. N. Prostaglandin E2 regulates the nuclear receptor NR4A2 in colorectal cancer. J Biol Chem 281, 2676–82 (2006). [DOI] [PubMed] [Google Scholar]
- Han Y. et al. Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer 119, 3436–45 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang T., Wang P., Ren H., Fan J. & Wang G. NGFI-B nuclear orphan receptor Nurr1 interacts with p53 and suppresses its transcriptional activity. Mol Cancer Res 7, 1408–15 (2009). [DOI] [PubMed] [Google Scholar]
- Zhao B. X. et al. p53 mediates the negative regulation of MDM2 by orphan receptor TR3. EMBO J 25, 5703–15 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T. C. et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26, 745–52 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L. et al. A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 17, 193–9 (2010). [DOI] [PubMed] [Google Scholar]
- Raver-Shapira N. et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 26, 731–43 (2007). [DOI] [PubMed] [Google Scholar]
- Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–33 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misso G. et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids 3, e194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion C. P. et al. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet 8, e1002797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M., Ferlito M. & Lowenstein C. J. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA 105, 13421–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada N. et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev 28, 438–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K. A. et al. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol Cancer Res 6, 735–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg-Gorenshtein G. et al. Reduced levels of miR-34a in neuroblastoma are not caused by mutations in the TP53 binding site. Genes Chromosomes Cancer 48, 539–43 (2009). [DOI] [PubMed] [Google Scholar]
- Lodygin D. et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–600 (2008). [DOI] [PubMed] [Google Scholar]
- Wang Z. et al. DNA hypermethylation of microRNA-34b/c has prognostic value for stage non-small cell lung cancer. Cancer Biol Ther 11, 490–6 (2011). [DOI] [PubMed] [Google Scholar]
- Yang D. et al. miR-132 regulates the differentiation of dopamine neurons by directly targeting Nurr1 expression. J Cell Sci 125, 1673–82 (2012). [DOI] [PubMed] [Google Scholar]
- Wu S. et al. MicroRNA-132 promotes estradiol synthesis in ovarian granulosa cells via translational repression of Nurr1. Reprod Biol Endocrinol 13, 94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takwi A. A. et al. miR-137 regulates the constitutive androstane receptor and modulates doxorubicin sensitivity in parental and doxorubicin-resistant neuroblastoma cells. Oncogene 33, 3717–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B. P., Burge C. B. & Bartel D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005). [DOI] [PubMed] [Google Scholar]
- John B. et al. Human MicroRNA targets. PLoS Biol 2, e363 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A. et al. Combinatorial microRNA target predictions. Nat Genet 37, 495–500 (2005). [DOI] [PubMed] [Google Scholar]
- Gennarino V. A. et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res 22, 1163–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudduluru G. et al. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 30, 2888–99 (2011). [DOI] [PubMed] [Google Scholar]
- Vassilev L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–8 (2004). [DOI] [PubMed] [Google Scholar]
- Millau J. F. et al. Formation of stress-specific p53 binding patterns is influenced by chromatin but not by modulation of p53 binding affinity to response elements. Nucleic Acids Res 39, 3053–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata H. et al. NuMA is required for the selective induction of p53 target genes. Mol Cell Biol 33, 2447–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M. E. Targeting of mRNAs by multiple miRNAs: the next step. Oncogene 29, 2161–4 (2010). [DOI] [PubMed] [Google Scholar]
- Bader A. G. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 3, 120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. & Rana T. M. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov 13, 622–38 (2014). [DOI] [PubMed] [Google Scholar]
- Corney D. C. et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res 16, 1119–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo E. et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 30, 1903–9 (2009). [DOI] [PubMed] [Google Scholar]
- Nakatani F. et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J Pathol 226, 796–805 (2012). [DOI] [PubMed] [Google Scholar]
- Svoboda M. et al. MiR-34b is associated with clinical outcome in triple-negative breast cancer patients. Diagn Pathol 7, 31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. The predictive effect of overexpressed miR-34a on good survival of cancer patients: a systematic review and meta-analysis. Onco Targets Ther 8, 2709–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Kelnar K. & Bader A. G. In-depth analysis shows synergy between erlotinib and miR-34a. PLoS One 9, e89105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. J. et al. MicroRNA-34a modulates chemosensitivity of breast cancer cells to adriamycin by targeting Notch1. Arch Med Res 43, 514–21 (2012). [DOI] [PubMed] [Google Scholar]
- Akao Y. et al. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett 300, 197–204 (2011). [DOI] [PubMed] [Google Scholar]
- Valentine J. M., Kumar S. & Moumen A. A p53-independent role for the MDM2 antagonist Nutlin-3 in DNA damage response initiation. BMC Cancer 11, 79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malewicz M. et al. Essential role for DNA-PK-mediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev 25, 2031–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S. et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA 106, 3964–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman T., Kinzler K. W. & Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 55, 5187–90 (1995). [PubMed] [Google Scholar]
- Yu J., Wang Z., Kinzler K. W., Vogelstein B. & Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 100, 1931–6 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. et al. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J 24, 2391–402 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. et al. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS One 3, e3794 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.