

Abstract
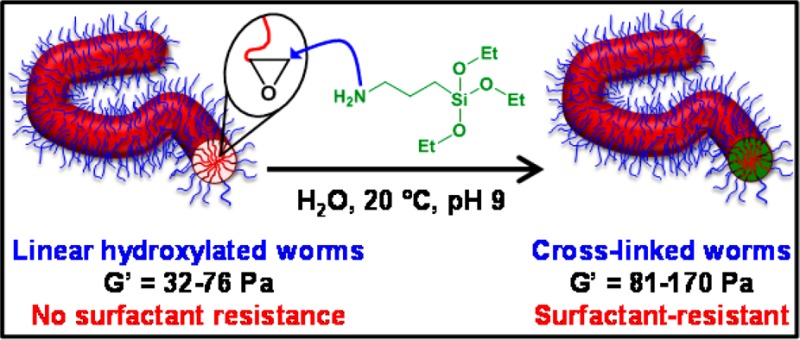
A poly(glycerol monomethacrylate) (PGMA) chain transfer agent is chain-extended by reversible addition–fragmentation chain transfer (RAFT) statistical copolymerization of 2-hydroxypropyl methacrylate (HPMA) with glycidyl methacrylate (GlyMA) in concentrated aqueous solution via polymerization-induced self-assembly (PISA). A series of five free-standing worm gels is prepared by fixing the overall degree of polymerization of the core-forming block at 144 while varying its GlyMA content from 0 to 20 mol %. 1H NMR kinetics indicated that GlyMA is consumed much faster than HPMA, producing a GlyMA-rich sequence close to the PGMA stabilizer block. Temperature-dependent oscillatory rheological studies indicate that increasing the GlyMA content leads to progressively less thermoresponsive worm gels, with no degelation on cooling being observed for worms containing 20 mol % GlyMA. The epoxy groups in the GlyMA residues can be ring-opened using 3-aminopropyltriethoxysilane (APTES) in order to prepare core cross-linked worms via hydrolysis-condensation with the siloxane groups and/or hydroxyl groups on the HPMA residues. Perhaps surprisingly, 1H NMR analysis indicates that the epoxy–amine reaction and the intermolecular cross-linking occur on similar time scales. Cross-linking leads to stiffer worm gels that do not undergo degelation upon cooling. Dynamic light scattering studies and TEM analyses conducted on linear worms exposed to either methanol (a good solvent for both blocks) or anionic surfactant result in immediate worm dissociation. In contrast, cross-linked worms remain intact under such conditions, provided that the worm cores comprise at least 10 mol % GlyMA.
Introduction
Over the past fifty years or so, block copolymer self-assembly has become a well-recognized and widely adopted route for the production of organic nanoparticles in a wide range of solvents. Many copolymer morphologies have been reported in the literature.1−5 However, there have been relatively few studies of block copolymer worms, cylinders, or rods via traditional post-polymerization processing routes, such as a solvent switch in dilute solution.2,6−13 This is presumably because such highly anisotropic morphologies typically occupy relatively little phase space, which means that the range of required block compositions tends to be rather narrow. In contrast, polymerization-induced self-assembly (PISA) has recently enabled the rational synthesis of block copolymer worms in the form of highly concentrated dispersions in a wide range of polar and non-polar solvents.14−32
The worm morphology is particularly interesting for various potential applications. Discher and co-workers have shown that poly(ethylene oxide)–poly(caprolactone) diblock copolymer worms exhibit substantially extended in vivo circulation times compared to the equivalent spherical morphology.9 Armes and co-workers have recently demonstrated the advantages offered by highly anisotropic worms when deployed as Pickering emulsifiers:33 they are much more strongly adsorbed at the oil–water interface compared to the equivalent spheres, yet retain a relatively high specific surface area.34,35 Several research groups have studied the rheological properties of block copolymer worms,31,32,36−40 with thermoreversible gelation being observed in aqueous solution,14,31 polar solvents such as ethanol,41 and non-polar solvents such as n-alkanes.19,32,42
Many strategies have been explored for the covalent stabilization of block copolymer nano-objects. Core cross-linked spherical micelles have been reported by various groups,43−46 while Wooley47−51 and Armes52−55 have worked extensively on shell cross-linked micelles. Both Antonietti et al.56 and Bates and co-workers7,37 have cross-linked polybutadiene-based block copolymer worms in dilute solution using γ radiation or redox chemistry, respectively. In contrast, Liu’s group has developed various photochemical strategies based on cinnamoyl side groups.57,58 In the context of PISA formulations, cross-linked block copolymer spheres, worms, and vesicles have been reported by copolymerizing small amounts of divinyl comonomers such as ethylene glycol di(meth)acrylate or poly(ethylene glycol) diacrylate.32,34,59−62 However, this strategy is somewhat problematic for the worm morphology, since relatively small perturbations in the block composition can result in the formation of mixed phases, rather than pure worms. An alternative post-polymerization approach was reported by Chambon et al. for cross-linked block copolymer vesicles, whereby pendent epoxy groups were reacted with small molecule or oligomeric diamines.63 Similarly An and co-workers prepared poly(poly(ethylene oxide)methyl ether methacrylate)–poly(2-(acetoacetoxy)ethyl methacrylate) (PPEOMA–PAEMA) diblock copolymer vesicles using PISA via RAFT dispersion polymerization in ethanol.64 These vesicles were subsequently cross-linked using O,O′-1,3-propanediylbisoxylamine dihydrochloride, which reacted with ketone groups in the PAEMA core-forming block. Very recently, the same team chain-extended a poly(N,N-dimethylacrylamide) (PDMA) macro-CTA using a binary mixture of diacetone acrylamide (DAAM) and an asymmetric cross-linker allyl acrylamide (ALAM) to prepare vesicles.65 The acrylamide groups in DAAM and ALAM have similar reactivities, whereas the allyl group in ALAM reacts significantly more slowly. This leads to in situ cross-linking of the vesicles toward the end of the copolymerization.
Generally speaking, there are relatively few literature reports describing the synthesis and cross-linking of diblock copolymer worms.6−8,57,58,66−69 Herein we describe the facile preparation of core cross-linked diblock copolymer worms. More specifically, a series of hydroxyl-functional methacrylic diblock copolymer worms containing varying amounts of glycidyl methacrylate (GlyMA) in the core-forming block are prepared in aqueous solution via PISA. Such worms are then covalently stabilized via cross-linking of the core-forming block using 3-aminopropyltriethoxysilane (APTES) (see Figure 1). The physical properties of aqueous dispersions of these cross-linked worms are compared to those of the linear worm precursors using various characterization techniques, including transmission electron microscopy (TEM), dynamic light scattering (DLS), and oscillatory rheology.
Figure 1.

Synthesis of a PGMA56 macro-CTA via RAFT solution polymerization of GMA in ethanol using a CPDB RAFT agent and its subsequent chain extension via statistical copolymerization of varying molar ratios of HPMA and GlyMA to form diblock copolymer worms in aqueous solution via polymerization-induced self-assembly (PISA). Such worms are then cross-linked using APTES in a two-step post-polymerization process involving (i) an epoxy–amine reaction with the GlyMA residues and (ii) hydrolysis–condensation reaction with the hydroxyl groups on the HPMA residues.
Experimental Section
Materials
Glycerol monomethacrylate (GMA; 99.8% purity) was kindly donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. 2-Hydroxypropyl methacrylate (HPMA) was purchased from Alfa Aesar and used as received. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) was purchased from Wako Pure Chemical Industries (Japan) and used as received. Glycidyl methacrylate (GlyMA), 2-cyano-2-propyl benzodithioate (CPDB), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; V-501; 99%), 3-aminopropyltriethoxysilane (APTES), d4-sodium trimethylsilyl propanoate (TMSP), sodium dodecyl sulfate (SDS), deuterated methanol-d4, ethanol (99%, anhydrous grade), methanol and dichloromethane were purchased from Sigma-Aldrich UK and were used as received. All solvents were of HPLC-grade quality.
Synthesis of Poly(glycerol monomethacrylate) (PGMA56) Macro-CTA via RAFT Solution Polymerization in Ethanol
A typical protocol for the synthesis of PGMA56 macro-CTA was as follows: GMA (203.0 g, 1.268 mol), CPDB (6.03 g, 0.020 mol; target DP = 63), ACVA (1.14 g, 4.07 mmol; CPDB/ACVA molar ratio = 5.0), and anhydrous ethanol (156.0 g, 3.38 mol) were added to a round-bottomed flask to afford a 55% w/w solution. The resulting pink solution was purged with N2 for 20 min, before the sealed flask was immersed into an oil bath set at 70 °C. After 140 min (69% conversion as judged by 1H NMR) the polymerization was quenched by immersion of the flask into an ice bath and exposing the reaction mixture to air. The crude polymer was then precipitated into a 10-fold excess of dichloromethane and washed three times using this nonsolvent to remove residual unreacted GMA monomer before being dried under high vacuum for 3 days at 40 °C. 1H NMR studies indicated a mean degree of polymerization of 56 via end-group analysis (the integrated aromatic RAFT end-group signals at 7.1–7.4 ppm were compared to those assigned to the two oxymethylene protons at 3.5–4.4 ppm). Taking into account the target DP of 63 and the GMA conversion of 69%, this indicated a CTA efficiency of 76%. GPC studies (DMF eluent, refractive index detector; calibrated against a series of near-monodisperse poly(methyl methacrylate) standards) indicated an Mn of 15 000 g mol–1 and an Mw/Mn of 1.11.
Synthesis of PGMA56–PHPMA144 Diblock Copolymer Worms via RAFT Aqueous Dispersion Polymerization
A typical protocol for the chain extension of PGMA56 macro-CTA with 144 units of HPMA via RAFT aqueous dispersion polymerization was as follows. PGMA56 macro-CTA (0.399 g, 0.043 mmol), HPMA monomer (0.90 g, 6.0 mmol), and VA-044 (3.50 mg, 0.011 mmol; PGMA56 macro-CTA/VA-044 molar ratio = 4.0) were added to a 25 mL round-bottomed flask, prior to addition of water to produce a 15% w/w aqueous solution. The reaction solution was purged under nitrogen for 30 min at 20 °C prior to immersion into an oil bath set at 50 °C. The reaction mixture was stirred for 105 min to ensure almost complete conversion of the HPMA monomer (>99% by 1H NMR analysis), and then the HPMA polymerization was quenched by exposure to air followed by cooling to ambient temperature. The resulting dispersion was diluted with deionized water to give a free-standing 7.5% w/w worm gel that was characterized by DLS, TEM, and rheology without further purification.
Synthesis of PGMA56–P(HPMAy-stat-GlyMAz) Diblock Copolymer Worms via RAFT Aqueous Emulsion/Dispersion Polymerization
A typical protocol for chain extension of PGMA56 macro-CTA with 122 units of HPMA and 22 units of GlyMA via RAFT aqueous dispersion/emulsion polymerization was as follows: PGMA56 macro-CTA (0.418 g, 0.046 mmol), HPMA monomer (0.800 g, 5.5 mmol), GlyMA monomer (0.140 g, 1.0 mmol), and VA-044 (3.70 mg, 0.011 mmol; PGMA56 macro-CTA/VA-044 molar ratio = 4.0) were added to a 25 mL round-bottomed flask, prior to addition of sufficient water to afford a 15% w/w aqueous solution. This reaction solution was purged under nitrogen for 30 min at 20 °C prior to immersion into an oil bath set at 50 °C. The reaction mixture was stirred for 105 min to ensure almost complete conversion of the HPMA and GlyMA comonomers (>99% by 1H NMR analysis). Then the copolymerization was quenched by exposure to air, followed by cooling to ambient temperature. The resulting dispersion was immediately diluted with deionized water to 7.5% w/w solids, yielding a free-standing worm gel that was characterized by DLS, TEM, and rheology without further purification.
Post-Polymerization Cross-Linking of a 7.5% w/w Aqueous Dispersion of PGMA56-P(HPMAy-stat-GlyMAz) Worm Gel Using APTES
A typical protocol for the covalent cross-linking of PGMA56-P(HPMA122-stat-GlyMA22) diblock copolymer worm gel at 7.5% w/w solids using APTES was as follows. APTES (0.111 g, 0.5 mmol, APTES/GlyMA molar ratio = 1.0) was added to 9.1 g of a 7.5% w/w aqueous dispersion of PGMA56-P(HPMA122-stat-GlyMA22) diblock copolymer worms, and the epoxy–amine reaction was allowed to proceed for 24 h at 20 °C with continuous stirring of the shear-thinning worm gels.
Instrumentation
NMR Spectroscopy
1H NMR spectra were recorded using a 400 MHz Bruker Avance-500 spectrometer with 64 scans being averaged per spectrum.
Gel Permeation Chromatography (GPC)
Polymer molecular weights and polydispersities were determined using a DMF GPC setup operating at 60 °C and comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns connected in series to a Varian 390-LC multidetector suite (with only the refractive index detector being used) and a Varian 290-LC pump injection module. The GPC eluent was HPLC-grade DMF containing 10 mM LiBr at a flow rate of 1.0 mL min–1. DMSO was used as a flow-rate marker. Calibration was conducted using a series of ten near-monodisperse poly(methyl methacrylate) standards (Mn = 625–2 480 000 g mol–1). Chromatograms were analyzed using Varian Cirrus GPC software (version 3.3).
Dynamic Light Scattering (DLS)
Studies were conducted using a Malvern Zetasizer NanoZS instrument on 0.10% w/w copolymer dispersions in either water, methanol, or 1.0% w/w SDS aqueous solution at 25 °C before and after cross-linking in a glass cuvette at a fixed backscattering angle of 173°. Intensity-average hydrodynamic diameters were calculated via the Stokes–Einstein equation using a non-negative least-squares (NNLS) algorithm. All data were averaged over three consecutive runs.
Aqueous Electrophoresis
Aqueous electrophoresis studies were performed on 0.10% w/w aqueous copolymer dispersions (containing 10–3 mol dm–3 NaCl as background electrolyte) using a Malvern Zetasizer NanoZS instrument at 25 °C. The pH of the copolymer dispersion was initially basic and was adjusted using HCl. Zeta potentials were calculated from the Henry equation using the Smoluchowski approximation. All data were averaged over three consecutive runs.
Transmission Electron Microscopy (TEM)
As-prepared copolymer dispersions were diluted 150-fold at 20 °C in either methanol or water to generate 0.10% w/w dispersions. Copper/palladium TEM grids (Agar Scientific, UK) were surface-coated in-house to yield a thin film of amorphous carbon. The grids were then plasma glow-discharged for 30 s to create a hydrophilic surface. A micropipet was used to place droplets (12 μL) of aqueous copolymer dispersions onto freshly glow-discharged grids for 1 min, followed by careful blotting with filter paper to remove excess sample. To stain the aggregates, a 0.75% w/w uranyl formate solution (9 μL) was soaked on the sample-loaded grid for 20 s and then carefully blotted to remove excess stain. Each grid was then carefully dried using a vacuum hose. Imaging was performed using a FEI Tecnai Spirit microscope fitted with a Gatan 1kMS600CW CCD camera operating at 80 kV.
Rheology Studies
Storage (G′) and loss (G″) moduli were determined between 4 and 25 °C for diblock copolymer worm gels both before and after covalent cross-linking using a TA Instruments AR-G2 rheometer at a fixed strain of 1.0% and an angular frequency of 1.0 rad s–1. The copolymer concentration was fixed at 7.5% w/w for all experiments. A cone-and-plate geometry (40 mm 2° aluminum cone) was used for these measurements.
Results and Discussion
Block Copolymer Worm Syntheses
A well-defined near-monodisperse poly(glycerol monomethacrylate) (PGMA) macro-CTA was prepared via reversible addition–fragmentation chain transfer (RAFT) solution polymerization of GMA in ethanol at 70 °C using 2-cyano-2-propylbenzodithioate (CPDB) as the chain transfer agent (CTA).70,711H NMR spectroscopy studies suggested a mean degree of polymerization (DP) of 56, as judged by end-group analysis. Moreover, gel permeation chromatography (GPC) studies conducted in DMF against near-monodisperse poly(methyl methacrylate) (PMMA) standards indicated that the PGMA56 macro-CTA possessed a relatively narrow molecular weight distribution (Mn = 15 000 g mol–1 and Mw/Mn = 1.11). This homopolymer precursor was then chain-extended via statistical copolymerization of 0, 5, 10, 15, or 20 mol % of glycidyl methacrylate (GlyMA) with 2-hydroxypropyl methacrylate (HPMA) at 15% w/w solids using a PISA formulation (see Figure 1). This protocol produced a series of free-standing copolymer worm gels after cooling to room temperature. For each PGMA56-P(HPMAy-stat-GlyMAz) diblock copolymer synthesis, a mean DP of 144 was targeted for the core-forming block (i.e., y + z = 144). High monomer conversions (>99%) were achieved in all cases, as judged by the disappearance of monomer vinyl signals between 5.9 and 6.1 ppm in the 1H NMR spectra.
Covalent Stabilization of Block Copolymer Worms
The as-prepared 15% w/w aqueous dispersions of block copolymer worms described above were diluted to 7.5% w/w solids to allow efficient stirring when conducting post-polymerization derivatization reactions using 3-aminopropyltriethoxysilane (APTES) (see Figure 1). The primary amine group on this siloxane reagent reacts with the pendent epoxide groups72 located within the core-forming blocks, with APTES ingress aided by the partially hydrated nature of the HPMA-rich worm cores.14 In principle, the triethoxysilane component of the grafted APTES molecules should then undergo hydrolysis-condensation reactions, both with each other and also with the pendent secondary (and primary) hydroxyl groups on the HPMA residues, resulting in core cross-linked worms.73 Post-polymerization cross-linking was undertaken in order to minimize the possibility of in situ cross-linking during PISA, which might otherwise prevent the formation of worms or perhaps cause inter-worm aggregation. After cross-linking at 7.5% w/w solids, the diblock copolymer worm gels were expected to be dispersible in a good solvent for both blocks (e.g., methanol) and also possess enhanced resistance toward the presence of ionic surfactants, which are known to cause rapid dissociation of closely related PGMA–PHPMA linear diblock copolymer nano-objects.63 Furthermore, the rheological properties of the worm gels were investigated both before and after cross-linking. Recently, Lovett and co-workers reported that similar PGMA–PHPMA diblock copolymer worms prepared using a carboxylic acid-based RAFT CTA undergo worm-to-sphere transitions upon a pH switch as a result of end-group ionization.74 Thus, it was important to employ a non-ionic CTA in the present study in order to prevent such order–order morphological transitions on addition of the strongly basic APTES reagent.
Synthesis and Characterization of PGMA–P(HPMA-stat-GlyMA) Diblock Copolymer Worms
Previous syntheses of similar PGMA55–P(HPMA247-stat-GlyMA82) diblock copolymer vesicles were conducted by Chambon et al., with full conversion being attained after 4 h at 70 °C.63 According to 1H NMR studies, around 90% of the epoxide groups on the GlyMA residues survived these conditions, with 10% undergoing hydrolysis with water (to afford GMA residues) and/or pendent hydroxyl groups in HPMA resulting in partial in situ cross-linking. In the present study, diblock copolymer syntheses were conducted at 50 °C for 105 min in order to minimize such side reactions. 1H NMR studies confirmed the success of this modified protocol, with approximately 98% of epoxide groups surviving at full comonomer conversion (see Figure S1). The (co)polymerization kinetics for the synthesis of PGMA56–PHPMA144, PGMA56–P(HPMA130-stat-GlyMA14), and PGMA56–P(HPMA115-stat-GlyMA29) at 50 °C were monitored by 1H NMR (see Figure 2). Aliquots of reaction mixtures were extracted at regular time intervals and diluted prior to NMR analysis using CD3OD, which is a good solvent for all monomeric and copolymer species. Kinetic studies of the PGMA56–PHPMA144 diblock copolymer formulation indicated that full conversion was achieved after 90 min. After a brief induction period, consumption of the water-miscible HPMA monomer was relatively slow for 35 min. This may be the result of mild retardation, which is not fully understood.75 After 65 min (or 62% conversion, which corresponds to a PHPMA DP of 89), the rate of polymerization increases by an order of magnitude (see Figure S2). This is the result of micellar nucleation, which heralds a switch from RAFT solution polymerization to RAFT aqueous dispersion polymerization, as judged by both visual inspection and dynamic light scattering (DLS) (see Figure S2). According to Blanazs and co-workers, unreacted HPMA migrates into the micelle cores, increasing the local monomer concentration and hence leading to a faster rate of polymerization.76 A similar rate enhancement is also observed when HPMA is partially replaced by GlyMA (see Figure S2). However, in this case the water-immiscible GlyMA comonomer is consumed via aqueous emulsion polymerization. Interestingly, 1H NMR studies indicate significantly faster initial consumption of GlyMA compared to HPMA. For example, in the case of PGMA56–P(HPMA115-stat-GlyMA29), 13% GlyMA was consumed after 10 min whereas only 5% HPMA had reacted on the same time scale. Similar observations have been recently reported by Ratcliffe and co-workers for the RAFT statistical copolymerization of water-immiscible 4-hydroxybutyl methacrylate (HBMA) with water-miscible 2-hydroxyethyl methacrylate (HEMA) in aqueous solution.77 In the present study, this leads to a GlyMA-rich sequence for the core-forming block close to its junction with the PGMA stabilizer block. This is important because it has a significant effect on the physical properties of the resulting worm gel, as discussed in more detail below. Visual inspection of the reaction mixture indicates that a homogeneous solution is obtained within 10 min, which suggests that the remaining GlyMA concentration becomes sufficiently low for the statistical copolymerization to proceed as an aqueous dispersion polymerization before micellar nucleation occurs. This is consistent with the temperature-dependent water solubility of GlyMA reported by Ratcliffe and co-workers.78
Figure 2.
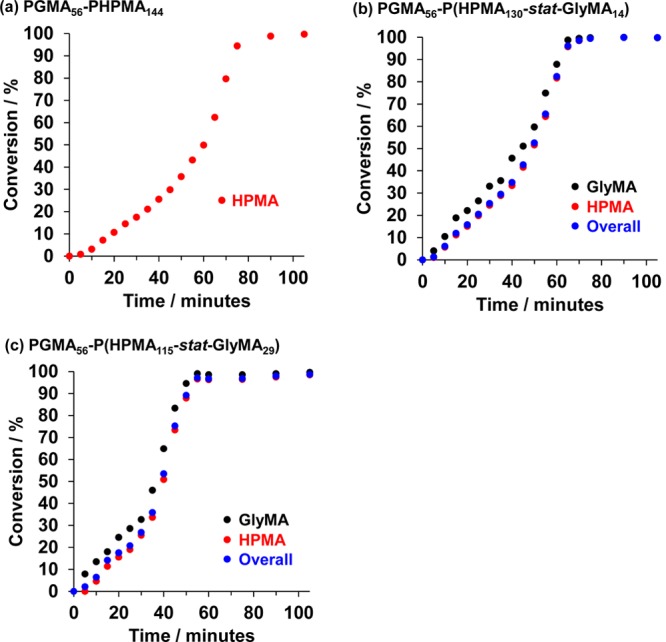
Conversion vs time curves obtained by 1H NMR for the (co)polymerization of HPMA (red circles), GlyMA (black circles), and the overall comonomer mixture (blue circles) at 50 °C using a PGMA56 macro-CTA when targeting diblock copolymer compositions of (a) PGMA56–PHPMA144, (b) PGMA56–P(HPMA130-stat-GlyMA14), and (c) PGMA56–P(HPMA115-stat-GlyMA29). All syntheses were conducted at 15% w/w solids.
As expected, partial replacement of HPMA with increasing amounts of GlyMA within the core-forming block induces micellar nucleation at shorter reaction times. For example, nucleation occurs after approximately 55 min when targeting PGMA56–P(HPMA130-stat-GlyMA14) but after only 40 min when targeting PGMA56–P(HPMA115-stat-GlyMA29).
The enhanced rate of copolymerization achieved under heterogeneous conditions leads to essentially full monomer conversion within relatively short time scales. More specifically, the synthesis of PGMA56–P(HPMA130-stat-GlyMA14) was complete after 75 min, while more than 99% conversion was observed for PGMA56–P(HPMA115-stat-GlyMA29) after only 60 min. In view of these kinetic data, it was decided to conduct these diblock copolymer syntheses for 105 min at 50 °C. These conditions were chosen to ensure very high (>99%) comonomer conversions while minimizing loss of pendent epoxide groups to side reactions, as discussed above.
At the end of each copolymerization, each of the five PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer dispersions were immediately diluted to 7.5% w/w solids to aid efficient mixing of the APTES cross-linker. Once fully dispersed, these 7.5% w/w dispersions were split into two batches. The first batch was used to determine the physical properties of the linear worms obtained prior to cross-linking, while the second batch was used to examine worm core cross-linking with APTES.
DMF GPC analysis of these five diblock copolymers prior to addition of the APTES cross-linker suggested minimal intrinsic cross-linking occurred during their synthesis, since no high molecular weight shoulder was observed at shorter retention times (see Figure 3). This was not unexpected, since the reaction of epoxy groups with the (mainly) secondary hydroxyl groups on the HPMA residues should be negligible at 50 °C. Furthermore, these GPC studies indicated relatively high blocking efficiencies, narrow molecular weight distributions, and similar number-average molecular weights for all five diblock copolymers. In striking contrast, DMF GPC analysis of PGMA55–P(HPMA247-stat-GlyMA82) vesicles prepared at 70 °C for 4 h performed by Chambon et al. indicated relatively high polydispersities and a prominent high molecular weight shoulder.63 This suggests that epoxide-based cross-linking occurs when such statistical copolymerizations are conducted over longer reaction times at elevated temperatures, although in principle differences in the levels of dimethacrylate impurity in the HPMA comonomer could be an alternative explanation.59,76
Figure 3.
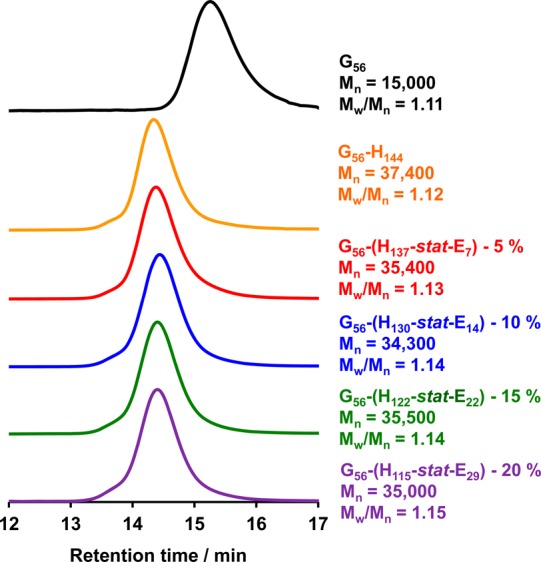
DMF GPC curves obtained for PGMA56 macro-CTA (black curve) and the corresponding traces for four PGMA56–P(HPMAy-stat-GlyMAz) (where y + z = 144; these copolymers are denoted as G56-(Hy-stat-Ez) for brevity) diblock copolymers prepared at 50 °C. Molecular weights are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards.
DLS and transmission electron microscopy (TEM) studies were conducted on dilute (0.1% w/w) dispersions of the five diblock copolymer worms prior to cross-linking in order to assess their colloidal stability in both water and methanol. DLS studies of dilute aqueous dispersions indicate that these worms possessed sphere-equivalent hydrodynamic diameters of 100–210 nm and relatively high polydispersities (>0.20), which compares well with literature data reported for such nano-objects.14,74 Moreover, relatively intense light scattering (derived count rates exceeding 30 000 kcps) was recorded in all cases, which is consistent with the presence of nano-objects (see Table 1). TEM images obtained for dried aqueous copolymer dispersions confirmed the presence of highly anisotropic worms in all cases. Image analysis of 50 worms per copolymer sample indicated well-defined worm widths of approximately 20–25 nm but highly variable worm lengths of 100–1200 nm (see Figure 4). In contrast, TEM studies of the same diblock copolymer dispersions diluted using methanol prior to drying confirmed the absence of any well-defined nano-objects (see Figure S3) while only very weak light scattering (<300 kcps) was observed by DLS. Both observations are consistent with molecular dissolution of copolymer chains in methanol, which is a good solvent for both blocks.73,79
Table 1. Summary of Dynamic Light Scattering (DLS) Data Obtained Both before and after APTES Cross-Linking for PGMA56–P(HPMAy-stat-GlyMAz) Diblock Copolymers (Where y + z = 144; These Copolymers Are Denoted as G56-(Hy-stat-Ez) for Brevity) in Pure Water, Methanol, and in a 1.0% w/w Aqueous SDS Solution.
| before
cross-linking |
after
cross-linking |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| water |
methanol |
water |
methanol |
SDS |
||||||
| copolymer composition | diam/nm (PDI) | derived count rate/kcps | diam/nm (PDI) | derived count rate/kcps | diam/nm (PDI) | derived count rate/kcps | diam/nm (PDI) | derived count rate/kcps | diam/nm (PDI) | derived count rate/kcps |
| G56-H144 | 102 (0.184) | 39 600 | 9 (0.216) | 140 | nd | nd | nd | nd | nd | nd |
| G56-(H137-stat-E7) | 150 (0.210) | 56 300 | 38 (0.252) | 250 | 152 (0.272) | 22 400 | 66 (0.169) | 3 600 | 48 (0.281) | 5 740 |
| G56-(H130-stat-E14) | 122 (0.206) | 51 400 | 14 (0.246) | 280 | 172 (0.345) | 36 300 | 266 (0.411) | 24 400 | 200 (0.255) | 31 700 |
| G56-(H122-stat-E22) | 128 (0.238) | 39 200 | 13 (0.354) | 210 | 235 (0.404) | 32 800 | 251 (0.295) | 30 300 | 231 (0.269) | 32 300 |
| G56-(H115-stat-E29) | 203 (0.286) | 41 500 | 61 (0.212) | 210 | 200 (0.412) | 28 900 | 220 (0.242) | 40 500 | 173 (0.238) | 34 000 |
Figure 4.
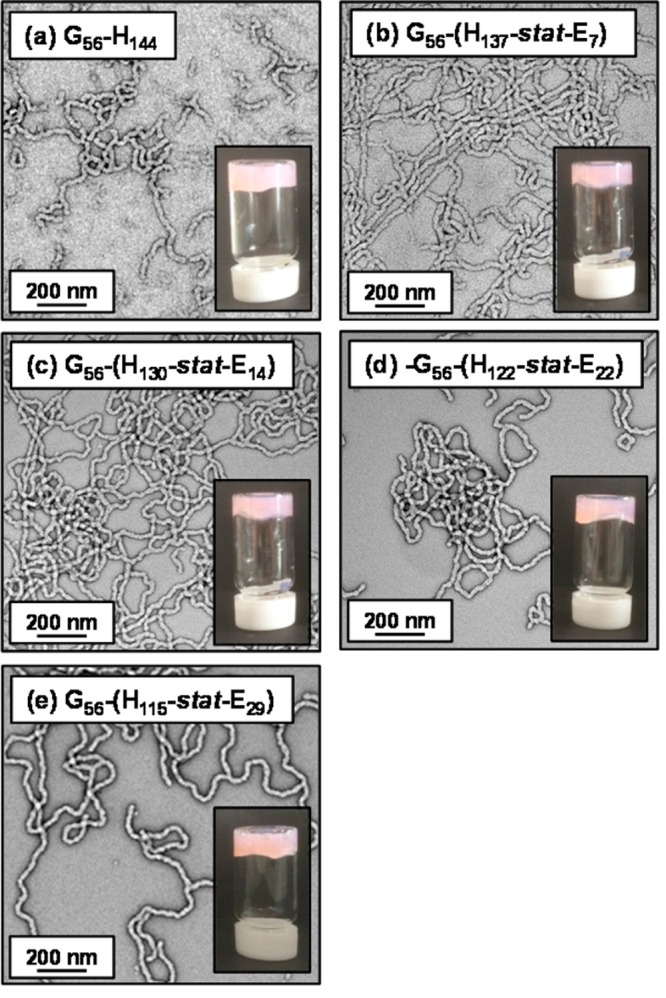
Representative TEM images obtained for dried 0.1% w/w aqueous dispersions of PGMA56–P(HPMAy-stat-GlyMAz) linear diblock copolymers prior to cross-linking (where y + z = 144; these copolymers are denoted as G56-(Hy-stat-Ez) for brevity). Digital photographic images of the corresponding free-standing gels recorded at 7.5% w/w solids are shown as insets.
Previous rheological studies on a similar PGMA54–PHPMA140 diblock copolymer worm gel have shown that degelation occurs on cooling to 5 °C.14,36 TEM and small-angle X-ray scattering (SAXS) studies confirmed that this is the result of a worm-to-sphere order–order morphological transition. This is driven by surface plasticization of the PHPMA core-forming block, which causes a shift in the packing parameter, P, from worm phase space (0.33 < P > 0.5) to spherical phase space (P < 0.33).3 As expected, the linear PGMA56–PHPMA144 diblock copolymer worm gel prepared in this study is similarly thermoresponsive. Its critical gelation temperature (CGT) was determined to be 13.5 °C on cooling to 5 °C, as judged by the point of cross-over of the storage modulus (G′) and loss modulus (G″) curves in temperature-dependent rheological studies (see Figure 5a). PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worm gels possess similar thermoresponsive degelation when up to 15 mol % GlyMA (z = 22) is incorporated into the core-forming block, as judged by rheology (see Figure 5 and Figure S4). However, increasing the GlyMA content suppresses the thermoresponsive behavior of the diblock copolymer worm gels, with lower CGTs being observed. As previously discussed, GlyMA is consumed faster than HPMA during the RAFT statistical copolymerization of these two comonomers. This results in a GlyMA-enriched block junction. However, as GlyMA residues are more hydrophobic than HPMA residues, progressively lower temperatures are required for the surface plasticization necessary to induce a worm-to-sphere transition (and hence degelation). Furthermore, more pronounced hysteresis is observed on returning to 25 °C. This is because the worm-to-sphere transition is relatively fast compared to the sphere-to-worm transition, since the latter process is highly cooperative. These rheological studies also indicate a reduction in storage modulus (G′) from 86 to 11 Pa at 25 °C on increasing the GlyMA content in the core-forming block from 0 to 20 mol %. Rheological studies of PGMA54–PHPMAy diblock copolymer worms reported by Verber and co-workers over a range of y values indicated that block compositions closer to the worm/sphere phase boundary formed weaker gels.36 Thus, it is possible that incorporating more GlyMA into the core-forming statistical block shifts the worm morphology toward this phase boundary.
Figure 5.
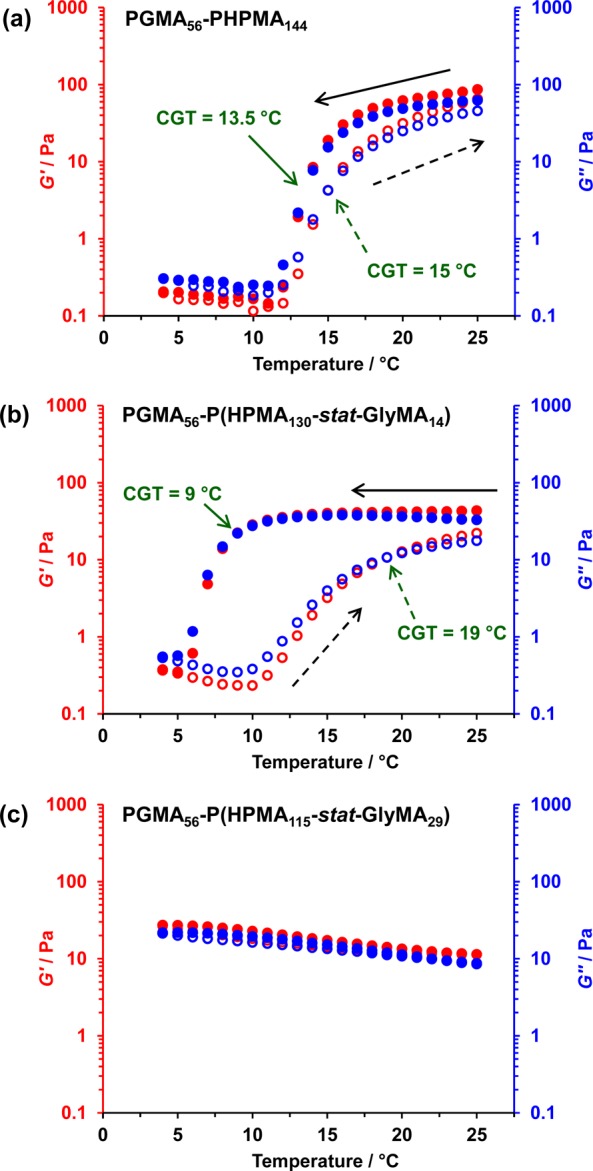
Variation of the storage modulus (G′; denoted by red data set) and the loss modulus (G″; denoted by blue data set) as a function of temperature (closed circles denote a 25 to 5 °C temperature sweep, and open circles denote a 5 to 25 °C temperature sweep) for a 7.5% w/w aqueous dispersion of (a) PGMA56–PHPMA144, (b) PGMA56–P(HPMA137-stat-GlyMA14), and (c) PGMA56–P(HPMA115-stat-GlyMA29) worms before cross-linking. Conditions: angular frequency = 1.0 rad s–1, applied strain = 1.0%, and rate of cooling/heating = 0.50 °C min–1.
It is worth considering why these worm gels are so soft at 25 °C. If the mean worm width and length for the linear PGMA56–PHPMA144 worms are 24 and 245 nm, respectively, and assuming a worm density of approximately 1.10 g cm–3, then the mean worm mass is estimated to be 1.22 × 10–16 g. Assuming a worm concentration of 7.5% w/w, then the worm number density is 6.2 × 1020 m–3. If we assume a mean copolymer molecular weight of 30 000 g mol–1, then the mean aggregation number (or average number of copolymer chains per worm) is estimated to be 2450. It is well-known that G′ = νekT.80 If G′ is 86 Pa (see Figure 5a), then the number density of elastically effective chains (νe) is estimated to be 2.1 × 1022 m–3. This means that each worm contributes 34 elastically effective chains. Thus, there is one just elastically effective chain per 72 copolymer chains. This explains why these worm gels are so soft: the copolymer chains are used rather inefficiently within the dissipative network. This is in part because the chains have some degree of mobility within the worms, especially when subjected to strain. In this context, it is noteworthy that variable temperature 1H NMR studies reported by Blanazs and co-workers provide direct experimental evidence for partial solvation of the PHPMA core-forming blocks.14 When the worm cores are covalently cross-linked using APTES, an increase in gel modulus is observed (see later). This means that the number of copolymer chains per elastically effective chain is reduced because core cross-linking links individual copolymer chains together, leading to the more effective distribution of stress within the worm gel.
Post-Polymerization Cross-Linking of PGMA–P(HPMA-stat-GlyMA) Diblock Copolymer Worms
On reaching full conversion, the aqueous worm gels were immediately diluted from 15 to 7.5% w/w to lower the gel viscosity. Once a homogeneous dispersion was achieved, APTES was added (APTES/GlyMA molar ratio = 1.0), and the shear-thinning gel was stirred overnight at 20 °C. As discussed earlier, the primary amine of the APTES reacts with the pendent epoxide groups in the GlyMA residues while the siloxane groups undergo multiple hydrolysis–condensation reactions that lead to highly cross-linked worm cores (see Figure 6). In reality, cross-linking is likely be even more complex because the secondary amines formed via ring-opening of the epoxide group can in principle react with a second epoxide. One interesting question here is the following: to what extent does the time scale for the epoxy–amine reaction differ from that of the hydrolysis-condensation reactions? To address this point, the rate of reaction of APTES with the epoxide groups and the rate of hydrolysis-condensation for the PGMA56–P(HPMA115-stat-GlyMA29) diblock copolymer worms were monitored by 1H NMR using d4-sodium trimethylsilylpropanoate (TMSP) as an internal standard (see Figure 7). Aliquots of the aqueous reaction mixture were extracted at regular intervals and diluted using CD3OD prior to NMR analysis. This choice of diluent enables chemical changes in the core-forming block to be monitored up to relatively high degrees of cross-linking. The rate of ring-opening by the nucleophilic APTES was determined by monitoring the disappearance of the characteristic epoxy proton signals at 3.0 ppm in the 1H NMR spectra relative to the internal standard (see blue data set shown in Figures 7b and 7a for the corresponding 1H NMR spectra). The integrated epoxy signal is reduced to 6% of its original value after 8 h (and to just 3% after 24 h). As the hydrolysis–condensation reaction proceeds, the chemical cross-links lead to worm core swelling in CD3OD, rather than worm dissolution. At higher degrees of cross-linking, the worm cores become solid-like and hence no longer solvated by the CD3OD; thus, signals associated with the P(HPMA-stat-GlyMA) core-forming block gradually become undetectable by 1H NMR. This can be used to infer the relative degree of cross-linking by determining either the normalized reduction in the methyl group signal assigned to the methacrylic backbone at 0.9 ppm (green data set in Figure 7) or that of the pendent methyl group assigned to the HPMA residues (see red data set in Figure 7 and Figure S5 for the corresponding 1H NMR traces). However, the latter method is preferred because in the former method data analysis is made more complicated by overlapping backbone methyl group signals arising from the PGMA stabilizer block, which remains soluble (and hence detectable) even after core cross-linking is complete. It was originally anticipated that the epoxy–amine reaction would occur prior to the hydrolysis–condensation reaction, leading to a sequential cross-linking process. However, the data shown in Figure 7 indicate that the relative integral of the HPMA methyl signal is attenuated at a comparable rate as that of the epoxy signals. This indicates that ring-opening of the epoxy groups and the hydrolysis–condensation reactions actually occur over similar time scales, suggesting that cross-linking does not proceed via a two-stage mechanism. However, the precise degree of cross-linking cannot be calculated because further cross-linking may occur that is no longer detectable by 1H NMR. It is perhaps noteworthy that the reaction times shown in Figure 7 correspond to the times at which each aliquot was taken from the reaction mixture—it does not include the time taken to run each 1H NMR spectrum. Diluting each aliquot with an equal volume of CD3OD may not adequately quench the reaction, so it was important to analyze each aliquot as soon as possible in order to minimize this “dead time”. (In practice, the time required for instrument setup and spectrum acquisition was around 15 min for each sample.) Notwithstanding such minor time domain errors, this spectroscopic study confirmed that an approximate time scale of 24 h is required for extensive cross-linking of each of the four GlyMA-containing diblock copolymer worms at 20 °C under the stated conditions.
Figure 6.
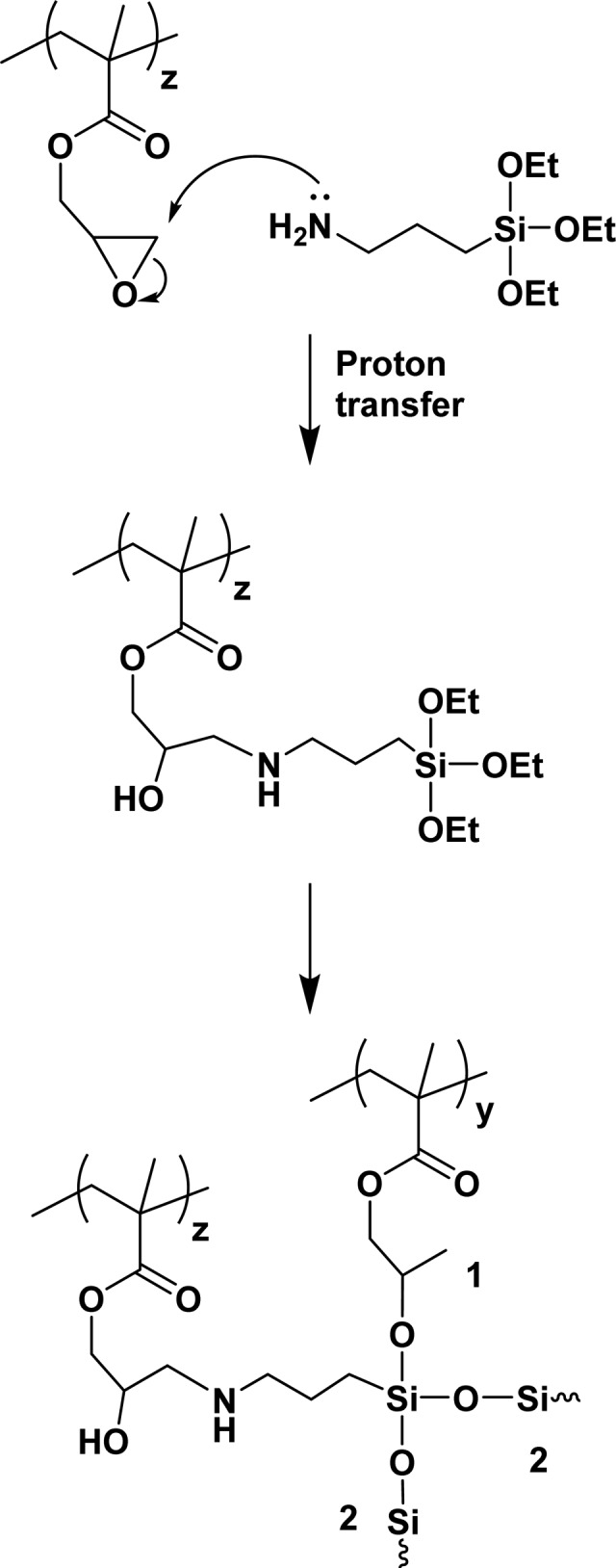
Reaction scheme illustrating worm core cross-linking chemistry by (i) epoxy ring-opening via nucleophilic attack with APTES and (ii) intermolecular cross-linking via hydrolysis-condensation. The latter step involves either reaction of the APTES with hydroxyl groups on HPMA residues on another copolymer chain (denoted as 1) and/or condensation with other APTES groups (denoted as 2). In reality, 1H NMR studies indicate that these two steps occur more or less simultaneously, rather than consecutively as shown (see main text for details). Moreover, the chemistry is likely to be more complex than that shown as the secondary amine species may react further.
Figure 7.
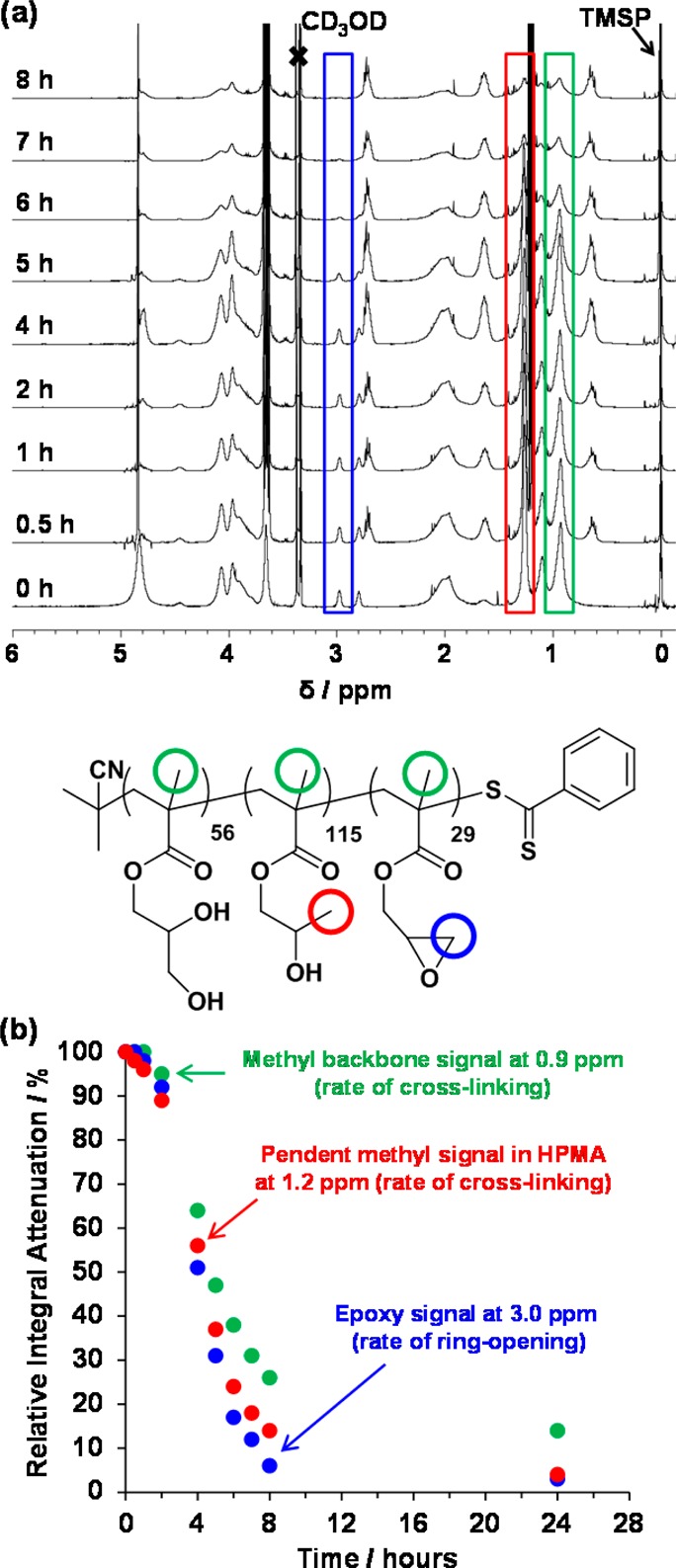
(a) 1H NMR spectra obtained at various time points following the reaction of APTES with PGMA56–P(HPMA115-stat-GlyMA29) after dilution into CD3OD. The amine reacts with GlyMA as judged by the reduction in the epoxy signal peak at 3.0 ppm compared to the internal standard TMSP. (b) Kinetics of the ring-opening epoxy–amine reaction (blue data set) as judged by the attenuation in the relative integral of the epoxide signal at 3.0 ppm compared to an internal standard by 1H NMR spectroscopy. Kinetics of worm core cross-linking as judged by the relative attenuation in the integrated pendent methyl group signal at 0.9 ppm assigned to the HPMA residues (red data set) and the relative attenuation in the integrated methyl signal at 1.2 ppm assigned to the methacrylate backbone (green data set) compared to the same internal standard at 0 ppm.
In principle, core cross-linking should prevent worm dissolution on dilution in methanol (which is a good solvent for both blocks). DLS studies conducted on 0.1% w/w aqueous worm dispersions (see Table 1) indicates that cross-linking causes a significant increase in the apparent hydrodynamic diameters [from 122 to 172 nm for the PGMA56–P(HPMA130-stat-GlyMA14) worms and from 128 to 235 nm for PGMA56–P(HPMA122-stat-GlyMA22) worms]. However, it is emphasized that only sphere-equivalent dimeters are reported by DLS, so it is difficult to interpret such observations in terms of changes in either worm contour lengths or worm widths. Moreover, this apparent increase in particle dimensions could in principle simply be a result of some degree of inter-particle cross-linking. Nevertheless, TEM images obtained for the four core cross-linked diblock copolymer worms containing 5, 10, 15, or 20 mol % GlyMA (see Figure 8) after dilution to 0.1% w/w aqueous dispersions do not indicate any discernible change in the original worm morphology. Furthermore, all four core cross-linked worm dispersions still form free-standing gels at 7.5% w/w solids, as judged by a tube inversion test (see Figure 8). However, DLS studies conducted on the same four worm dispersions after dilution to 0.1% w/w in methanol suggest that only worms comprising at least 10 mol % GlyMA are fully resistant to the presence of methanol (see Table 1). In contrast, the PGMA56–P(HPMA137-stat-GlyMA7) diblock copolymer (5 mol % GlyMA) shows a dramatic reduction in apparent hydrodynamic diameter from 152 nm in water to 66 nm in methanol, with a relatively low derived count rate (3600 kcps) being observed in the latter solvent. This suggests that the worms undergo a morphological transition to spheres and/or short worms. However, TEM images obtained (see Figure 9a) for this latter diblock copolymer dried as a 0.1% w/w dispersion in methanol suggest that no well-defined particles are present (i.e., worm dissolution most likely occurs under these conditions). Indeed, 1H NMR studies of this copolymer in CD3OD confirm a strong signal at around 1.25 ppm corresponding to the pendent methyl groups on the HPMA residues (data not shown). In contrast, DLS studies of the other three diblock copolymer worms (containing 10, 15, or 20 mol % GlyMA) in methanol indicate a much higher derived count rate of at least 22 000 kcps (see Table 1). Moreover, these diblock copolymer worms exhibit an increase in hydrodynamic diameter when dispersed in methanol as opposed to water. This is the result of swelling of the cross-linked worm cores because methanol is a good solvent for both blocks, but the relatively low degree of cross-linking is sufficiently high to prevent worm dissolution. TEM images obtained for PGMA56–P(HPMA130-stat-GlyMA14), PGMA56–P(HPMA122-stat-GlyMA22), and PGMA56–P(HPMA115-stat-GlyMA29) diblock copolymers dried from 0.1% w/w methanolic dispersions confirmed the persistence of the pure worm morphology in each case (Figure 9b–d).
Figure 8.

Representative TEM images obtained for dried 0.1% w/w aqueous dispersions of PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymers after APTES cross-linking of 7.5% w/w worm dispersions at 20 °C. Inset digital photographic images were recorded for the same aqueous copolymer dispersions at 7.5% w/w solids; free-standing gels are observed in each case.
Figure 9.

Representative TEM images obtained for core cross-linked PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymers (abbreviated G56-(Hy-stat-Ez) for the sake of brevity) after drying 0.1% w/w methanolic dispersions at 20 °C. (a) No well-defined nano-objects were observed at 5 mol % GlyMA, whereas the original worm morphology persists when core cross-linked worms contain higher proportions of GlyMA; see images (b), (c), and (d).
When APTES is reacted with the epoxy groups on the GlyMA residues, a secondary amine is generated (see Figure 6). Thus, the resulting core cross-linked worms might be expected to possess weakly cationic character below neutral pH (where the secondary amine groups become protonated). In a control experiment, aqueous electrophoresis studies conducted on a 0.1% w/w aqueous dispersion of linear PGMA56–PHPMA144 diblock copolymer worms indicated no cationic character from pH 10 to 3 (see Figure 10). In contrast, APTES-cross-linked PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms displayed cationic character below pH 7–9 (see Figure 10). However, these particles exhibit only relatively weak cationic character (+5 to +10 mV) at pH 5. In contrast, Penfold et al. have recently reported that linear PGMA50–PHPMA140 worms prepared using a morpholine-based RAFT agent exhibit zeta potentials of around +15 mV, even though there is only one terminal morpholine group per stabilizer block in this case.81 This discrepancy most likely arises because the cationic charge is located within the cores of the cross-linked worms in the present study, rather than in the stabilizer block. Above pH 9, all worms exhibit weakly anionic character (−5 to −10 mV). This most likely indicates the presence of carboxylic acid end-groups on some of the PGMA stabilizer chains resulting from use of ACVA initiator in their RAFT synthesis.74
Figure 10.
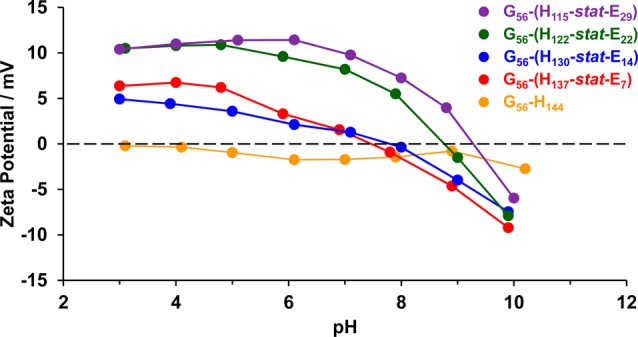
Zeta potential versus pH curves obtained at 25 °C for 0.1% w/w aqueous dispersions of linear PGMA56–PHPMA144 diblock copolymer worms and four examples of APTES cross-linked PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms in the presence of 10–3 M KCl.
It is noteworthy that the characteristic pink color of the worm gels that arises from the dithiobenzoate-based RAFT CTA is removed during the APTES cross-linking reaction (see Figure 8). This is the result of nucleophilic attack on the dithioester by the strongly basic primary amine groups (after APTES addition, the solution pH increases to pH 9–10).82,83 However, as the dithioester chain-ends are located within the worm cores, this side reaction is unlikely to adversely affect the physical properties of these copolymer worm dispersions.
Cross-linking also causes the PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms to form stronger gels, as judged by comparing the storage moduli (G′) of 7.5% w/w worm gels before and after cross-linking by oscillatory rheology (see Table 2). For example, cross-linking the PGMA56–P(HPMA130-stat-GlyMA14) worms leads to an increase in G′ from 43 to 81 Pa at 25 °C (see Figure 11). Previous work by Bates and co-workers suggest that this is due to worm stiffening, which leads to a longer worm persistence length.7 Moreover, temperature-dependent rheological studies indicate that the degelation that is observed on cooling linear diblock copolymer worm gels no longer occurs after worm core cross-linking (see Figure 11 and Figure S6). Clearly, covalent stabilization of the PGMA56–P(HPMAy-stat-GlyMAz) worms prevents their dissociation into spheres at around 5 °C. Moreover, even the relatively lightly cross-linked PGMA56–P(HPMA137-stat-GlyMA7) worm gel is no longer thermoresponsive (see Figure S6a), although DLS and TEM studies indicate that the same APTES-treated worms undergo dissolution when diluted in methanol (see Table 1).
Table 2. Summary of Data Obtained for PGMA56–P(HPMAy-stat-GlyMAz) Diblock Copolymer Worm Gels in the Presence and Absence of Methanol or 1.0% Aqueous SDS Solution before and after APTES Cross-Linking at 20 °C.
| before
cross-linking |
after
cross-linking |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| copolymer composition | Mn/g mol–1 a | Mw/Mna | G′ at 25 °C/Pab | thermoresponsive degelation?b | stable in the presence of methanol?c | stable in the presence of SDS?d | G′ at 25 °C/Pab | thermoresponsive degelation?b | stable in the presence of methanol?c | stable in the presence of SDS?d |
| G56-H144 | 37 400 | 1.12 | 86 | no | no | |||||
| G56-(H137-stat-E7) | 35 400 | 1.13 | 76 | yes | no | no | 170 | no | no | partial* |
| G56-(H130-stat-E14) | 34 300 | 1.14 | 43 | yes | no | no | 81 | no | yes | yes |
| G56-(H122-stat-E22) | 35 500 | 1.14 | 32 | yes | no | no | 119 | no | yes | yes |
| G56-(H115-stat-E29) | 35 000 | 1.15 | 11 | no | no | no | 13 | no | yes | yes |
Calculated using DMF GPC against a series of near-monodisperse PMMA calibration standards using a refractive index detector.
Determined for 7.5% copolymer worm gels using oscillatory rheology at an angular frequency of 1.0 rad s–1 and an applied strain of 1.0%.
As judged by DLS and TEM studies conducted on PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms diluted to 0.1% w/w in methanol.
As judged by DLS and TEM studies conducted on PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms diluted to 0.1% w/w in the presence of 1.0% w/w SDS aqueous solution (i.e., SDS/copolymer mass ratio = 10).
Figure 11.
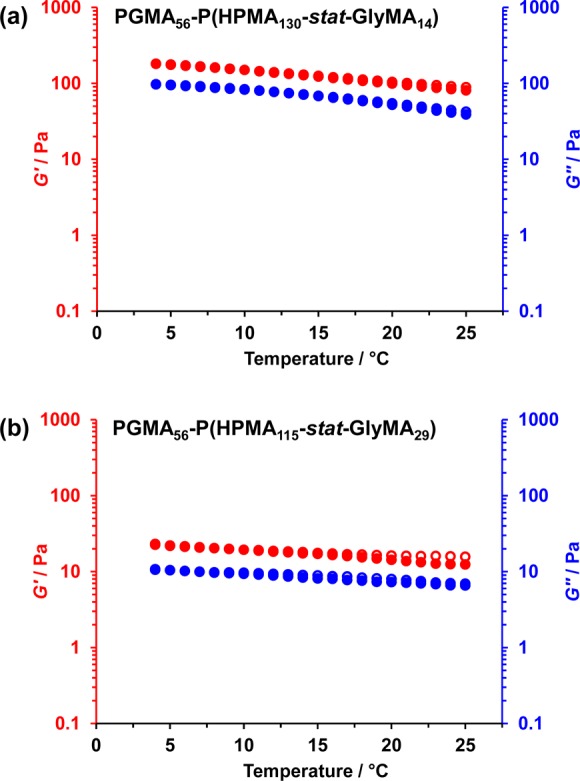
Variation in storage modulus (G′; red circles) and loss modulus (G″; blue circles) as a function of temperature (closed circles denote the cooling temperature sweep and open circles denote the heating temperature sweep) for 7.5% w/w aqueous dispersions of (a) PGMA56–P(HPMA130-stat-GlyMA14) and (b) PGMA56–P(HPMA115-stat-GlyMA29) after worm core cross-linking using APTES (final solution pH 9–10). Conditions: angular frequency = 1.0 rad s–1; applied strain = 1.0%; heating/cooling rate = 0.5 °C min–1.
G′ and G″ represent the energy per unit strain that is stored or dissipated, respectively. It is well-known that tan δ = G″/G′.84 If the latter parameter increases, then this indicates greater energy dissipation. For the linear worm gels reported herein, there are two likely energy dissipation mechanisms: (i) intra-worm interactions between the hydrophobic core-forming PHPMA blocks and (ii) inter-worm entanglements and/or multiple contacts. In each case energy is efficiently dissipated because such physical interactions are not fixed. Core cross-linking reduces tan δ, presumably because there is less dissipation via intra-worm interactions (see Figure 12). There may also be a contribution from the greater persistence length: the cross-linked worms acquire “rod-like” character compared to the highly flexible linear worms, which should reduce dissipation owing to entanglements or multiple contacts. However, tan δ values are still relatively high compared to other types of physical gels (e.g., peptide gels85−87), suggesting that cross-linking does not completely suppress dissipation.
Figure 12.
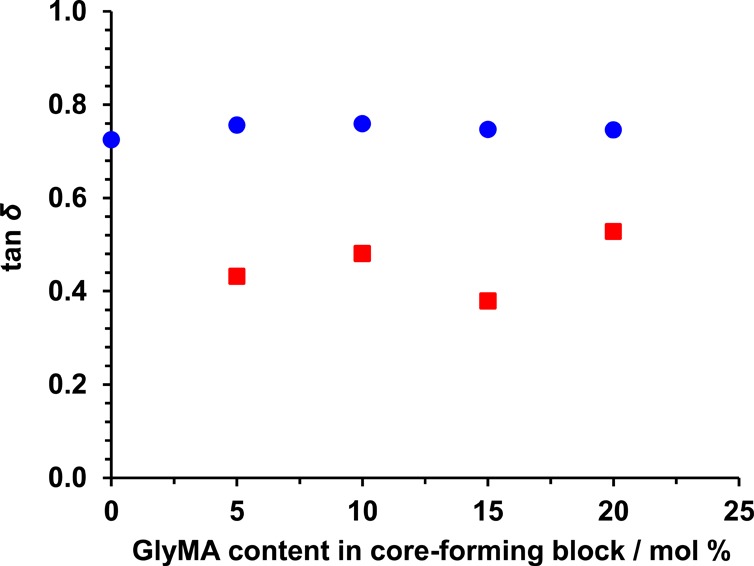
Values for tan δ at 25 °C for a series of PGMA56–P(HPMAY-stat-GlyMAZ) diblock copolymer worms before (blue data set) and after (red data set) after cross-linking. Covalent stabilization of the worms leads to a reduction in tan δ.
Chambon et al. demonstrated that PGMA55–PHPMA330 diblock copolymer vesicles fully dissociated to form individual copolymer chains when challenged with an anionic surfactant.63 In contrast, PGMA55–P(HPMA247-stat-GlyMA82) diblock copolymer vesicles that had been cross-linked using a small molecule (or polymeric) diamine proved to be surfactant-resistant. In principle, similar findings might be expected for the core cross-linked diblock copolymer worms described herein. Thus, core cross-linking is potentially useful because the resulting worms may be suitable as viscosity modifiers for various commercial surfactant-based home and personal care formulations. In this study, sodium dodecyl sulfate (SDS) was selected to assess the surfactant resistance of the worms, as this amphiphile was previously demonstrated to be particularly disruptive toward diblock copolymer vesicles.63 The surfactant resistance of all diblock copolymer worms was judged by TEM analysis of 0.1% w/w copolymer dispersions conducted in the absence and presence of 1.0% w/w SDS (i.e., a SDS/copolymer mass ratio of 10). As expected, when the linear PGMA56–PHPMA144 worm gels were subjected to an SDS challenge, there was an immediate reduction in turbidity, and DLS studies indicated a relatively low count rate of 260 kcps (see Table 1), suggesting rapid dissociation to form dissolved copolymer chains. This was corroborated by TEM, since no nano-objects could be observed (see Figure 13a). Similarly, linear PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms challenged with SDS also undergo immediate dissociation. In all cases no particles could be observed by TEM (see Figure S6). Interestingly, APTES cross-linked PGMA56–P(HPMA137-stat-GlyMA7) worms only exhibit partial resistance to this surfactant challenge. Rather than undergoing complete dissolution, a worm-to-sphere transition is instead observed by TEM (see Figure 13b), while DLS indicated a significant reduction in hydrodynamic diameter from 150 to 48 nm in the presence of SDS (see Table 1). However, on increasing the GlyMA content to 10, 15, or 20 mol % (and therefore the degree of core cross-linking), the worms became completely resistant to the presence of SDS. DLS studies of PGMA56–P(HPMA130-stat-GlyMA14), PGMA56–P(HPMA122-stat-GlyMA22), and PGMA56-P(HPMA115-stat-GlyMA29) diblock copolymer worms in the presence and absence of SDS revealed only minor changes in their apparent sphere-equivalent diameters (see Table 1). Furthermore, TEM images recorded after drying these diluted “worm plus surfactant” dispersions confirm that the original worm morphology is retained over time scales of months in each case (see Figure 13c–e).
Figure 13.

Representative TEM images obtained for dried dispersions of 0.1% w/w APTES cross-linked PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms exposed to the presence of 1.0% w/w SDS at 20 °C.
The colloidal stabilities of the five PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms (prepared targeting the same overall mean degree of polymerization; y + z = 144) before and after cross-linking are summarized in Table 2. Prior to cross-linking, none of the linear PGMA56–P(HPMAy-stat-GlyMAz) worms remained intact when challenged with either methanol or SDS. However, APTES treatment can significantly improve worm stability toward either reagent. In particular, for worm cores comprising at least 10 mol % GlyMA, TEM and DLS studies confirm that the worm morphology is preserved in the presence of either methanol or 1.0% w/w aqueous SDS solution. Furthermore, temperature-dependent oscillatory rheology studies demonstrate that worm core cross-linking results in stiffer gels that no longer exhibit thermoresponsive behavior.
Conclusions
In summary, a series of PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worm gels have been conveniently prepared by polymerization-induced self-assembly in concentrated aqueous solution by targeting a constant core-forming block DP of 144 in each case. Increasing the GlyMA content in such linear copolymers affords weaker gels, as judged by rheology studies. 1H NMR studies of the kinetics of statistical copolymerization of water-immiscible GlyMA with water-miscible HPMA indicate that the former comonomer is more reactive than the latter. Thus, the comonomer composition of the core-forming statistical block becomes GlyMA-rich at its junction with the PGMA stabilizer block. This explains why temperature-dependent rheological studies indicate that worms with higher GlyMA contents gradually become less thermoresponsive, since progressively lower temperatures are required to induce surface plasticization of the worms and hence degelation via a worm-to-sphere transition. Ultimately, thermally-induced degelation is no longer observed at a GlyMA content of 20 mol %. Such diblock copolymer worms can be core cross-linked by adding APTES. Perhaps surprisingly, 1H NMR studies indicate that the ring-opening and cross-linking reactions occur over similar time scales rather than via a two-stage reaction. The cross-linked worms are expected to be significantly stiffer than the linear worm precursors. Indeed, DLS provides some evidence for longer persistence lengths, and the cross-linked worms also form stronger gels, which in all cases no longer undergo thermally-induced degelation on cooling. TEM studies of dried diluted aqueous worm dispersions confirmed that core cross-linking produced no discernible change in the copolymer morphology. Furthermore, TEM studies conducted prior to cross-linking indicate that all of the linear PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymer worms are unstable with respect to the addition of either methanol (a good solvent for both blocks) or an anionic surfactant (SDS). In contrast, the corresponding cross-linked worms remain colloidally stable provided that the core-forming block contained at least 10 mol % GlyMA. Finally, it is noteworthy that the cross-linking chemistry described herein (i) utilizes cheap commercially available reagents, (ii) can be conveniently conducted at 20 °C in aqueous solution, and (iii) produces secondary amine groups within the worm cores, which results in weakly cationic worms below pH 7, as judged by aqueous electrophoresis.
Acknowledgments
S.P.A. and J.R.L. thank the University of Sheffield for a studentship and also GEO Specialty Chemicals for CASE support of this PhD project. S.P.A. and L.P.D.R. thank EPSRC for a postdoctoral grant (EP/K03071X). B.R.S. thanks EPSRC for a postdoctoral grant (EP/K030949/1). S.P.A. also thanks the ERC for a five-year Advanced Investigator grant (PISA 320372).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.6b00422.
Assigned 1H NMR spectra for PGMA56–P(HPMA115-stat-GlyMA29) diblock copolymer; conversion vs time curves, corresponding semilogarithmic plots and DLS data; TEM images for linear PGMA56–P(HPMAy-stat-GlyMAz) diblock copolymers in the presence of methanol or SDS; rheological data for PGMA56–P(HPMA137-stat-GlyMA7) and PGMA56–P(HPMA122-stat-GlyMA22) diblock copolymer worms before and after cross-linking; 1H NMR spectra obtained during cross-linking of PGMA56–P(HPMA115-stat-GlyMA29) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Discher D. E.; Eisenberg A. Polymer vesicles. Science 2002, 297, 967–973. 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- Jain S.; Bates F. S. On the Origins of Morphological Complexity in Block Copolymer Surfactants. Science 2003, 300, 460–464. 10.1126/science.1082193. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
- Foerster S.; Zisenis M.; Wenz E.; Antonietti M. Micellization of strongly segregated block copolymers. J. Chem. Phys. 1996, 104, 9956–9970. 10.1063/1.471723. [DOI] [Google Scholar]
- Bang J.; Jain S.; Li Z.; Lodge T. P.; Pedersen J. S.; Kesselman E.; Talmon Y. Sphere, Cylinder, and Vesicle Nanoaggregates in Poly(styrene-b-isoprene) Diblock Copolymer Solutions. Macromolecules 2006, 39, 1199–1208. 10.1021/ma052023+. [DOI] [Google Scholar]
- Wang X.; Guerin G.; Wang H.; Wang Y.; Manners I.; Winnik M. A. Cylindrical Block Copolymer Micelles and Co-Micelles of Controlled Length and Architecture. Science 2007, 317, 644–647. 10.1126/science.1141382. [DOI] [PubMed] [Google Scholar]
- Won Y.-Y.; Davis H. T.; Bates F. S. Giant Wormlike Rubber Micelles. Science 1999, 283, 960–963. 10.1126/science.283.5404.960. [DOI] [PubMed] [Google Scholar]
- Cheng C.; Qi K.; Germack D. S.; Khoshdel E.; Wooley K. L. Synthesis of Core-Crosslinked Nanoparticles with Controlled Cylindrical Shape and Narrowly-Dispersed Size via Core-Shell Brush Block Copolymer Templates. Adv. Mater. 2007, 19, 2830–2835. 10.1002/adma.200602986. [DOI] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava P.; Zheng J. X.; Li P.; Quirk R. P.; Harris F. W.; Cheng S. Z. D. Self-Assembled Polystyrene-block-poly(ethylene oxide) Micelle Morphologies in Solution. Macromolecules 2006, 39, 4880–4888. 10.1021/ma060677s. [DOI] [Google Scholar]
- Petzetakis N.; Dove A. P.; O’Reilly R. K. Cylindrical micelles from the living crystallization-driven self-assembly of poly(lactide)-containing block copolymers. Chem. Sci. 2011, 2, 955–960. 10.1039/C0SC00596G. [DOI] [Google Scholar]
- Dalhaimer P.; Bates F. S.; Discher D. E. Single Molecule Visualization of Stable, Stiffness-Tunable, Flow-Conforming Worm Micelles. Macromolecules 2003, 36, 6873–6877. 10.1021/ma034120d. [DOI] [Google Scholar]
- Groschel A. H.; Walther A.; Lobling T. I.; Schacher F. H.; Schmalz H.; Muller A. H. E. Guided hierarchical co-assembly of soft patchy nanoparticles. Nature 2013, 503, 247–251. 10.1038/nature12610. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Verber R.; Mykhaylyk O. O.; Ryan A. J.; Heath J. Z.; Douglas C. W. I.; Armes S. P. Sterilizable Gels from Thermoresponsive Block Copolymer Worms. J. Am. Chem. Soc. 2012, 134, 9741–9748. 10.1021/ja3024059. [DOI] [PubMed] [Google Scholar]
- Sugihara S.; Blanazs A.; Armes S. P.; Ryan A. J.; Lewis A. L. Aqueous Dispersion Polymerization: A New Paradigm for in Situ Block Copolymer Self-Assembly in Concentrated Solution. J. Am. Chem. Soc. 2011, 133, 15707–15713. 10.1021/ja205887v. [DOI] [PubMed] [Google Scholar]
- Fielding L. A.; Derry M. J.; Ladmiral V.; Rosselgong J.; Rodrigues A. M.; Ratcliffe L. P. D.; Sugihara S.; Armes S. P. RAFT dispersion polymerization in non-polar solvents: facile production of block copolymer spheres, worms and vesicles in n-alkanes. Chem. Sci. 2013, 4, 2081–2087. 10.1039/c3sc50305d. [DOI] [Google Scholar]
- Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Efficient Synthesis of Amine-Functional Diblock Copolymer Nanoparticles via RAFT Dispersion Polymerization of Benzyl Methacrylate in Alcoholic Media. Macromolecules 2012, 45, 5091–5098. 10.1021/ma300898e. [DOI] [Google Scholar]
- Yeow J.; Xu J.; Boyer C. Polymerization-Induced Self-Assembly Using Visible Light Mediated Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer Polymerization. ACS Macro Lett. 2015, 4, 984–990. 10.1021/acsmacrolett.5b00523. [DOI] [PubMed] [Google Scholar]
- Pei Y.; Thurairajah L.; Sugita O. R.; Lowe A. B. RAFT Dispersion Polymerization in Nonpolar Media: Polymerization of 3-Phenylpropyl Methacrylate in n-Tetradecane with Poly(stearyl methacrylate) Homopolymers as Macro Chain Transfer Agents. Macromolecules 2015, 48, 236–244. 10.1021/ma502230h. [DOI] [Google Scholar]
- Pei Y. W.; Lowe A. B. Polymerization-induced self-assembly: ethanolic RAFT dispersion polymerization of 2-phenylethyl methacrylate. Polym. Chem. 2014, 5, 2342–2351. 10.1039/C3PY01719B. [DOI] [Google Scholar]
- Boisse S.; Rieger J.; Belal K.; Di-Cicco A.; Beaunier P.; Li M.-H.; Charleux B. Amphiphilic block copolymer nano-fibers via RAFT-mediated polymerization in aqueous dispersed system. Chem. Commun. 2010, 46, 1950–1952. 10.1039/b923667h. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Boisse S.; Bui C.; Albouy P.-A.; Brulet A.; Li M.-H.; Rieger J.; Charleux B. Amphiphilic liquid-crystal block copolymer nanofibers via RAFT-mediated dispersion polymerization. Soft Matter 2012, 8, 1130–1141. 10.1039/C1SM06598J. [DOI] [Google Scholar]
- Zhang X.; Boissé S.; Zhang W.; Beaunier P.; D’Agosto F.; Rieger J.; Charleux B. Well-Defined Amphiphilic Block Copolymers and Nano-objects Formed in Situ via RAFT-Mediated Aqueous Emulsion Polymerization. Macromolecules 2011, 44, 4149–4158. 10.1021/ma2005926. [DOI] [Google Scholar]
- Wan W.-M.; Hong C.-Y.; Pan C.-Y. One-pot synthesis of nanomaterials via RAFT polymerization induced self-assembly and morphology transition. Chem. Commun. 2009, 5883–5885. 10.1039/b912804b. [DOI] [PubMed] [Google Scholar]
- Wan W.-M.; Pan C.-Y. One-pot synthesis of polymeric nanomaterials via RAFT dispersion polymerization induced self-assembly and re-organization. Polym. Chem. 2010, 1, 1475–1484. 10.1039/c0py00124d. [DOI] [Google Scholar]
- Zhao W.; Gody G.; Dong S.; Zetterlund P. B.; Perrier S. Optimization of the RAFT polymerization conditions for the in situ formation of nano-objects via dispersion polymerization in alcoholic medium. Polym. Chem. 2014, 5, 6990–7003. 10.1039/C4PY00855C. [DOI] [Google Scholar]
- Kang Y.; Pitto-Barry A.; Maitland A.; O’Reilly R. K. RAFT dispersion polymerization: a method to tune the morphology of thymine-containing self-assemblies. Polym. Chem. 2015, 6, 4984–4992. 10.1039/C5PY00617A. [DOI] [Google Scholar]
- Bauri K.; Narayanan A.; Haldar U.; De P. Polymerization-induced self-assembly driving chiral nanostructured materials. Polym. Chem. 2015, 6, 6152–6162. 10.1039/C5PY00919G. [DOI] [Google Scholar]
- Zhang W.; D’Agosto F.; Boyron O.; Rieger J.; Charleux B. Toward a Better Understanding of the Parameters that Lead to the Formation of Nonspherical Polystyrene Particles via RAFT-Mediated One-Pot Aqueous Emulsion Polymerization. Macromolecules 2012, 45, 4075–4084. 10.1021/ma300596f. [DOI] [Google Scholar]
- Tan J.; Sun H.; Yu M.; Sumerlin B. S.; Zhang L. Photo-PISA: Shedding Light on Polymerization-Induced Self-Assembly. ACS Macro Lett. 2015, 4, 1249–1253. 10.1021/acsmacrolett.5b00748. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Mahmood D.; Ryan A. J.; Armes S. P. RAFT Aqueous Dispersion Polymerization Yields Poly(ethylene glycol)-Based Diblock Copolymer Nano-Objects with Predictable Single Phase Morphologies. J. Am. Chem. Soc. 2014, 136, 1023–1033. 10.1021/ja410593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe L. P. D.; McKenzie B. E.; Le Bouëdec G. M. D.; Williams C. N.; Brown S. L.; Armes S. P. Polymerization-Induced Self-Assembly of All-Acrylic Diblock Copolymers via RAFT Dispersion Polymerization in Alkanes. Macromolecules 2015, 48, 8594–8607. 10.1021/acs.macromol.5b02119. [DOI] [Google Scholar]
- Binks B. P. Particles as surfactants - similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. 10.1016/S1359-0294(02)00008-0. [DOI] [Google Scholar]
- Thompson K. L.; Mable C. J.; Cockram A.; Warren N. J.; Cunningham V. J.; Jones E. R.; Verber R.; Armes S. P. Are block copolymer worms more effective Pickering emulsifiers than block copolymer spheres?. Soft Matter 2014, 10, 8615–8626. 10.1039/C4SM01724B. [DOI] [PubMed] [Google Scholar]
- Thompson K. L.; Mable C. J.; Lane J. A.; Derry M. J.; Fielding L. A.; Armes S. P. Preparation of Pickering Double Emulsions Using Block Copolymer Worms. Langmuir 2015, 31, 4137–4144. 10.1021/acs.langmuir.5b00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verber R.; Blanazs A.; Armes S. P. Rheological studies of thermo-responsive diblock copolymer worm gels. Soft Matter 2012, 8, 9915–9922. 10.1039/c2sm26156a. [DOI] [Google Scholar]
- Won Y.-Y.; Paso K.; Davis H. T.; Bates F. S. Comparison of Original and Cross-linked Wormlike Micelles of Poly(ethylene oxide-b-butadiene) in Water: Rheological Properties and Effects of Poly(ethylene oxide) Addition. J. Phys. Chem. B 2001, 105, 8302–8311. 10.1021/jp004078d. [DOI] [Google Scholar]
- Zhang W.; Charleux B.; Cassagnau P. Viscoelastic Properties of Water Suspensions of Polymer Nanofibers Synthesized via RAFT-Mediated Emulsion Polymerization. Macromolecules 2012, 45, 5273–5280. 10.1021/ma300529t. [DOI] [Google Scholar]
- Zhang W.; Charleux B.; Cassagnau P. Dynamic behavior of crosslinked amphiphilic block copolymer nanofibers dispersed in liquid poly(ethylene oxide) below and above their glass transition temperature. Soft Matter 2013, 9, 2197–2205. 10.1039/c2sm27602j. [DOI] [Google Scholar]
- Almgren M.; Brown W.; Hvidt S. Self-Aggregation and Phase-Behavior of Poly(Ethylene Oxide) Poly(Propylene Oxide) Poly(Ethylene Oxide) Block-Copolymers in Aqueous-Solution. Colloid Polym. Sci. 1995, 273, 2–15. 10.1007/BF00655668. [DOI] [Google Scholar]
- Pei Y. W.; Dharsana N. C.; Van Hensbergen J. A.; Burford R. P.; Roth P. J.; Lowe A. B. RAFT dispersion polymerization of 3-phenylpropyl methacrylate with poly 2-(dimethylamino)ethyl methacrylate macro-CTAs in ethanol and associated thermoreversible polymorphism. Soft Matter 2014, 10, 5787–5796. 10.1039/C4SM00729H. [DOI] [PubMed] [Google Scholar]
- Fielding L. A.; Lane J. A.; Derry M. J.; Mykhaylyk O. O.; Armes S. P. Thermo-responsive Diblock Copolymer Worm Gels in Non-polar Solvents. J. Am. Chem. Soc. 2014, 136, 5790–5798. 10.1021/ja501756h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A.; Liu G.; Tao J. Star Polymers and Nanospheres from Cross-Linkable Diblock Copolymers. Macromolecules 1996, 29, 2487–2493. 10.1021/ma951354r. [DOI] [Google Scholar]
- O’Reilly R. K.; Hawker C. J.; Wooley K. L. Crosslinked block copolymer micelles: functional nanostructures of great potential and versatility. Chem. Soc. Rev. 2006, 35, 1068–1083. 10.1039/b514858h. [DOI] [PubMed] [Google Scholar]
- Blencowe A.; Tan J. F.; Goh T. K.; Qiao G. G. Core crosslinked star polymers via controlled radical polymerization. Polymer 2009, 50, 5–32. 10.1016/j.polymer.2008.09.049. [DOI] [Google Scholar]
- Kocak G.; Bütün V. Synthesis and stabilization of Pt nanoparticles in core cross-linked micelles prepared from an amphiphilic diblock copolymer. Colloid Polym. Sci. 2015, 293, 3563–3572. 10.1007/s00396-015-3727-0. [DOI] [Google Scholar]
- Thurmond K. B. II; Kowalewski T.; Wooley K. L. Water-Soluble Knedel-like Structures: The Preparation of Shell-Cross-Linked Small Particles. J. Am. Chem. Soc. 1996, 118, 7239–7240. 10.1021/ja961299h. [DOI] [Google Scholar]
- Huang H.; Kowalewski T.; Remsen E. E.; Gertzmann R.; Wooley K. L. Hydrogel-Coated Glassy Nanospheres: A Novel Method for the Synthesis of Shell Crosslinked Knedels. J. Am. Chem. Soc. 1997, 119, 11653–11659. 10.1021/ja9717469. [DOI] [Google Scholar]
- Joralemon M. J.; O’Reilly R. K.; Hawker C. J.; Wooley K. L. Shell click-crosslinked (SCC) nanoparticles: a new methodology for synthesis and orthogonal functionalization. J. Am. Chem. Soc. 2005, 127, 16892–16899. 10.1021/ja053919x. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Remsen E. E.; Wooley K. L. Shell Cross-Linked Nanoparticles Containing Hydrolytically Degradable, Crystalline Core Domains. J. Am. Chem. Soc. 2000, 122, 3642–3651. 10.1021/ja993941o. [DOI] [Google Scholar]
- Li Y.; Du W.; Sun G.; Wooley K. L. pH-Responsive Shell Cross-Linked Nanoparticles with Hydrolytically Labile Cross-Links. Macromolecules 2008, 41, 6605–6607. 10.1021/ma801737p. [DOI] [Google Scholar]
- Liu S. Y.; Weaver J. V. M; Tang Y. Q.; Billingham N. C.; Armes S. P.; Tribe K. Synthesis of shell cross-linked micelles with pH-responsive cores using ABC triblock copolymers. Macromolecules 2002, 35, 6121–6131. 10.1021/ma020447n. [DOI] [Google Scholar]
- Li Y.; Lokitz B. S.; Armes S. P.; McCormick C. L. Synthesis of Reversible Shell Cross-Linked Micelles for Controlled Release of Bioactive Agents. Macromolecules 2006, 39, 2726–2728. 10.1021/ma0604035. [DOI] [Google Scholar]
- Butun V.; Lowe A. B.; Billingham N. C.; Armes S. P. Synthesis of zwitterionic shell cross-linked micelles. J. Am. Chem. Soc. 1999, 121, 4288–4289. 10.1021/ja9840596. [DOI] [Google Scholar]
- Butun V.; Billingham N. C.; Armes S. P. Synthesis of shell cross-linked micelles with tunable hydrophilic/hydrophobic cores. J. Am. Chem. Soc. 1998, 120, 12135–12136. 10.1021/ja981932u. [DOI] [Google Scholar]
- Hentze H. P.; Krämer E.; Berton B.; Förster S.; Antonietti M.; Dreja M. Lyotropic Mesophases of Poly(ethylene oxide)-b-poly(butadiene) Diblock Copolymers and Their Cross-Linking To Generate Ordered Gels. Macromolecules 1999, 32, 5803–5809. 10.1021/ma9904058. [DOI] [Google Scholar]
- Tao J.; Stewart S.; Liu G.; Yang M. Star and Cylindrical Micelles of Polystyrene-block-poly(2-cinnamoylethyl methacrylate) in Cyclopentane. Macromolecules 1997, 30, 2738–2745. 10.1021/ma9612788. [DOI] [Google Scholar]
- Liu G. J.; Ding J. F.; Qiao L. J.; Guo A.; Dymov B. P.; Gleeson J. T.; Hashimoto T.; Saijo K. Polystyrene-block-poly (2-cinnamoylethyl methacrylate) nanofibers - Preparation, characterization, and liquid crystalline properties. Chem. - Eur. J. 1999, 5, 2740–2749. 10.1002/(SICI)1521-3765(19990903)5:9<2740::AID-CHEM2740>3.0.CO;2-V. [DOI] [Google Scholar]
- Li Y.; Armes S. P. RAFT synthesis of sterically stabilized methacrylic nanolatexes and vesicles by aqueous dispersion polymerization. Angew. Chem., Int. Ed. 2010, 49, 4042–4046. 10.1002/anie.201001461. [DOI] [PubMed] [Google Scholar]
- Shen W. Q.; Chang Y. L.; Liu G. Y.; Wang H. F.; Cao A. N.; An Z. S. Biocompatible, Antifouling, and Thermosensitive Core-Shell Nanogels Synthesized by RAFT Aqueous Dispersion Polymerization. Macromolecules 2011, 44, 2524–2530. 10.1021/ma200074n. [DOI] [Google Scholar]
- Liu G. Y.; Qiu Q.; Shen W. Q.; An Z. S. Aqueous Dispersion Polymerization of 2-Methoxyethyl Acrylate for the Synthesis of Biocompatible Nanoparticles Using a Hydrophilic RAFT Polymer and a Redox Initiator. Macromolecules 2011, 44, 5237–5245. 10.1021/ma200984h. [DOI] [Google Scholar]
- Liu G.; Qiu Q.; An Z. Development of thermosensitive copolymers of poly(2-methoxyethyl acrylate-co-poly(ethylene glycol) methyl ether acrylate) and their nanogels synthesized by RAFT dispersion polymerization in water. Polym. Chem. 2012, 3, 504–513. 10.1039/C2PY00533F. [DOI] [Google Scholar]
- Chambon P.; Blanazs A.; Battaglia G.; Armes S. P. How Does Cross-Linking Affect the Stability of Block Copolymer Vesicles in the Presence of Surfactant?. Langmuir 2012, 28, 1196–1205. 10.1021/la204539c. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Qu Q.; Yu W.; An Z. Single Monomer for Multiple Tasks: Polymerization Induced Self-Assembly, Functionalization and Cross-Linking, and Nanoparticle Loading. ACS Macro Lett. 2014, 3, 1220–1224. 10.1021/mz500650c. [DOI] [PubMed] [Google Scholar]
- Qu Q.; Liu G.; Lv X.; Zhang B.; An Z. In Situ Cross-Linking of Vesicles in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2016, 5, 316–320. 10.1021/acsmacrolett.6b00066. [DOI] [PubMed] [Google Scholar]
- Wang X.-S.; Arsenault A.; Ozin G. A.; Winnik M. A.; Manners I. Shell Cross-Linked Cylinders of Polyisoprene-b-ferrocenyldimethylsilane: Formation of Magnetic Ceramic Replicas and Microfluidic Channel Alignment and Patterning. J. Am. Chem. Soc. 2003, 125, 12686–12687. 10.1021/ja037521h. [DOI] [PubMed] [Google Scholar]
- Stewart S.; Liu G. Block Copolymer Nanotubes. Angew. Chem., Int. Ed. 2000, 39, 340–344. 10.1002/(SICI)1521-3773(20000117)39:2<340::AID-ANIE340>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Figg C. A.; Simula A.; Gebre K. A.; Tucker B. S.; Haddleton D. M.; Sumerlin B. S. Polymerization-induced thermal self-assembly (PITSA). Chem. Sci. 2015, 6, 1230–1236. 10.1039/C4SC03334E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Liu K.; Arsenault A. C.; Rider D. A.; Ozin G. A.; Winnik M. A.; Manners I. Shell-Crosslinked Cylindrical Polyisoprene-b-Polyferrocenylsilane (PI-b-PFS) Block Copolymer Micelles: One-Dimensional (1D) Organometallic Nanocylinders. J. Am. Chem. Soc. 2007, 129, 5630–5639. 10.1021/ja068730f. [DOI] [PubMed] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; Rizzardo E.; Thang S. H. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. 10.1021/ma9804951. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. 10.1071/CH05072. [DOI] [Google Scholar]
- Clayden J.; Greeves N.; Warren S.; Wothers P.. Organic Chemistry; Oxford University Press: New York, 2001. [Google Scholar]
- Save M.; Weaver J. V. M; Armes S. P.; McKenna P. Atom Transfer Radical Polymerization of Hydroxy-Functional Methacrylates at Ambient Temperature: Comparison of Glycerol Monomethacrylate with 2-Hydroxypropyl Methacrylate. Macromolecules 2002, 35, 1152–1159. 10.1021/ma011541r. [DOI] [Google Scholar]
- Lovett J. R.; Warren N. J.; Ratcliffe L. P.; Kocik M. K.; Armes S. P. pH-responsive non-ionic diblock copolymers: ionization of carboxylic acid end-groups induces an order-order morphological transition. Angew. Chem., Int. Ed. 2015, 54, 1279–1283. 10.1002/anie.201409799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barner-Kowollik C.; Buback M.; Charleux B.; Coote M. L.; Drache M.; Fukuda T.; Goto A.; Klumperman B.; Lowe A. B.; McLeary J. B.; Moad G.; Monteiro M. J.; Sanderson R. D.; Tonge M. P.; Vana P. Mechanism and kinetics of dithiobenzoate-mediated RAFT polymerization. I. The current situation. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5809–5831. 10.1002/pola.21589. [DOI] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. Mechanistic insights for block copolymer morphologies: how do worms form vesicles?. J. Am. Chem. Soc. 2011, 133, 16581–16587. 10.1021/ja206301a. [DOI] [PubMed] [Google Scholar]
- Ratcliffe L. P. D.; Blanazs A.; Williams C. N.; Brown S. L.; Armes S. P. RAFT polymerization of hydroxy-functional methacrylic monomers under heterogeneous conditions: effect of varying the core-forming block. Polym. Chem. 2014, 5, 3643–3655. 10.1039/c4py00203b. [DOI] [Google Scholar]
- Ratcliffe L. P. D.; Ryan A. J.; Armes S. P. From a Water-Immiscible Monomer to Block Copolymer Nano-Objects via a One-Pot RAFT Aqueous Dispersion Polymerization Formulation. Macromolecules 2013, 46, 769–777. 10.1021/ma301909w. [DOI] [Google Scholar]
- Zehm D.; Ratcliffe L. P. D.; Armes S. P. Synthesis of Diblock Copolymer Nanoparticles via RAFT Alcoholic Dispersion Polymerization: Effect of Block Copolymer Composition, Molecular Weight, Copolymer Concentration, and Solvent Type on the Final Particle Morphology. Macromolecules 2013, 46, 128–139. 10.1021/ma301459y. [DOI] [Google Scholar]
- Treloar L. R. G.The Physics of Rubber Elasticity; Oxford University Press: 2005. [Google Scholar]
- Penfold N. J. W.; Lovett J. R.; Warren N. J.; Verstraete P.; Smets J.; Armes S. P. pH-Responsive non-ionic diblock copolymers: protonation of a morpholine end-group induces an order-order transition. Polym. Chem. 2016, 7, 79–88. 10.1039/C5PY01510C. [DOI] [Google Scholar]
- Willcock H.; O’Reilly R. K. End group removal and modification of RAFT polymers. Polym. Chem. 2010, 1, 149–157. 10.1039/B9PY00340A. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. End-functional polymers, thiocarbonylthio group removal/transformation and reversible addition-fragmentation-chain transfer (RAFT) polymerization. Polym. Int. 2011, 60, 9–25. 10.1002/pi.2988. [DOI] [Google Scholar]
- Barnes H. A.A Handbook of Elementary Rheology; The University of Wales Institute of Non-Newtonian Fluid Mechanics: Aberystwyth, 2000. [Google Scholar]
- Jayawarna V.; Richardson S. M.; Hirst A. R.; Hodson N. W.; Saiani A.; Gough J. E.; Ulijn R. V. Introducing chemical functionality in Fmoc-peptide gels for cell culture. Acta Biomater. 2009, 5, 934–943. 10.1016/j.actbio.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Yan C.; Pochan D. J. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. 10.1039/b919449p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams D. J.; Topham P. D. Peptide conjugate hydrogelators. Soft Matter 2010, 6, 3707–3721. 10.1039/c000813c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.