

Abstract
C3 glomerulopathy refers to a disease process in which abnormal control of complement activation, degradation or deposition results in predominant C3 fragment deposition within the glomerulus and glomerular damage. Recent studies have improved our understanding of its pathogenesis. The key abnormality is uncontrolled C3b amplification in the circulation and/or along the glomerular basement membrane. Family studies in which disease segregates with structurally abnormal complement factor H-related (CFHR) proteins demonstrate that abnormal CFHR proteins are important in some types of C3 glomerulopathy. This is currently thought to be due to the ability of these proteins to antagonize the major negative regulator of C3 activation, complement factor H (CFH), a process termed ‘CFH de-regulation’. Recent clinicopathological cohort studies have led to further refinements in case definition, culminating in a 2013 consensus report, which provides recommendations regarding investigation and treatment. Early clinical experience with complement-targeted therapeutics, notably C5 inhibitors, has also now been published. Here, we summarize the latest developments in C3 glomerulopathy.
Keywords: C3 glomerulopathy, complement, dense deposit, factor H
INTRODUCTION
C3 glomerulopathy designates ‘a disease process due to abnormal control of complement activation, deposition, or degradation and characterized by predominant glomerular C3 fragment deposition with electron-dense deposits on electron microscopy’ [1]. It often progresses to end-stage kidney disease (ESKD) and recurs after renal transplantation. The prototypical form of C3 glomerulopathy is dense deposit disease (DDD), which historically has been diagnosed based on linear, hyperosmiophilic electron-dense deposits occupying the middle layer of the glomerular basement membrane (GBM). ‘C3 glomerulonephritis' (C3GN) refers to those cases of C3 glomerulopathy in which the electron-dense deposits do not have this classic appearance.
One striking observation in patients with C3 glomerulopathy is how commonly clinical presentation is preceded by an infectious episode. This suggests that infection may initiate or exacerbate the disease process, for example by acting as an exogenous trigger of C3 activation. Once C3 is activated, abnormal glomerular accumulation of C3 fragments occurs due to uncontrolled C3b amplification via the alternative pathway (AP). In the majority of patients with C3 glomerulopathy, acquired or genetic defects in AP regulation can be demonstrated. In 2013, a consensus report in C3 glomerulopathy was published with recommendations regarding case definition, investigation and treatment [1]. At present, all patients in whom C3 glomerulopathy is diagnosed on either native or transplant kidney biopsy should be investigated for low plasma levels of C3, C4 and complement factor H (CFH), and the presence of paraprotein (i.e. monoclonal gammopathy), C3 nephritic factor and abnormal complement factor H-related protein 5 (CFHR5).
CFH is the main plasma regulator of the AP and consists of 20 protein subunits, termed short consensus repeat (SCR) domains, that are encoded within the regulators of complement (RCA) gene cluster on chromosome 1q32. The CFH–CFHR protein family (Figure 1A) comprises CFH and five structurally related CFHR proteins that are encoded downstream of the CFH gene. Frequent interspersed repeat elements across the CFH–CFHR locus predispose to genomic rearrangements, a number of which are associated with the renal pathologies atypical haemolytic uraemic syndrome (aHUS) and C3 glomerulopathy. CFH surface recognition domains at the carboxy (C)-terminus contain ligand-binding sites for glycosaminoglycans and C3b and are highly conserved on CFHR proteins. In contrast, the first four amino (N)-terminal SCR domains of CFH, which confer its complement regulatory functions, are absent from the CFHR proteins. As a consequence, CFHR proteins do not possess the physiological CFH regulatory activity mediated by these SCR domains. Distinct N-terminal SCR domains 1 and 2 are highly conserved on CFHR1, CFHR2 and CFHR5 (Figure 1B), and these are important in dimerization.
FIGURE 1:

The CFH–CFHR protein family. (A) CFH consists of 20 SCR domains. Those at the C-terminus (depicted in yellow) mediate surface recognition and are common to the five CFHR proteins (CFHR4 has two isoforms, CFHR4A and CFHR4B). In contrast, N-terminal SCR domains mediating CFH complement regulatory function (red) are not found on CFHR proteins. The N-terminal SCR domains 1 and 2 are highly conserved on CFHR1, CFHR2 and CFHR5 (blue) but not CFHR3 and CFHR4A/B, with very high sequence homology (B).
RECENT FAMILY STUDIES
In 2010, Gale et al. demonstrated segregation of C3GN with a heterozygous mutation of CFHR5 involving internal duplication of exons 2 and 3 (which encode N-terminal SCR domains 1 and 2, respectively) in a large Greek Cypriot cohort [2]. Since the original description of Cypriot ‘CFHR5 nephropathy’, a number of families with autosomal dominant C3GN and distinct CFHR mutations have been reported. A striking common feature has been internal duplication of exons or formation of hybrid genes resulting in abnormal CFHR proteins with duplication of N-terminal SCR domains 1 and 2 (Figure 2). Recent family studies in C3 glomerulopathy and membranoproliferative glomerulonephritis (MPGN) are briefly summarized below (see also Table 1), followed by a discussion of the pathophysiological implications of abnormal CFHR proteins.
FIGURE 2:
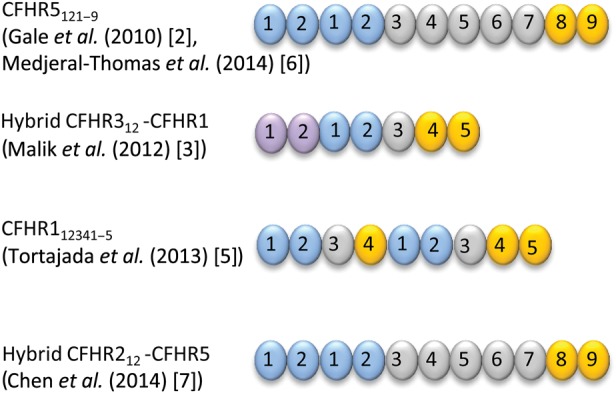
Family studies have identified abnormal CFHR proteins associated with C3 glomerulopathy. These include duplication of N-terminal SCR domains and formation of a hybrid CFHR protein containing two domains from CFHR3 (depicted in purple) linked to the whole of CFHR1.
Table 1.
Recent family studies in C3 glomerulopathy and MPGN
| Original report (year) | Reported diagnosis | Affected member (Case number) | Glomerular IF/IHC | EM deposits | C3 levels | C3NeF | CFH levels | Modified CFHR proteins |
Molecular defect(s) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gale et al. (2010) [2] | (Cypriot) CFHR5 nephropathy | Mother (Family 1 IV-5) Son (Family 1 V-4) Great uncle (Family 1 II.1) Aunt (Family 1 IV.1) Uncle (Family 2 III.2) |
C3 pos; C1q/C4/Ig neg | Subendothelial, mesangial > subepithelial | Normal | Neg | Normal | CFHR5121–9 | Heterozygous internal duplication of exons 2–3 (SCRs1–2) in CFHR5 | ||
| Malik et al. (2012) [3]/Neary (2002) | C3GN | Father (105) | C3 pos; Ig neg | Subendothelial, IM, subepithelial, mesangial | Normal | Neg | Normal | Hybrid CFHR312–CFHR1 (in addition to CFHR1, 3) | Heterozygous hybrid gene with deletion of exons 4–6 (SCRs 3–5) of CFHR3 and exon 1 of CFHR1 (in addition to normal CFHR3/1 genes) | ||
| Elder son (212) Younger son (213) |
Subendothelial, IM, subepithelial | Low–normal | |||||||||
| Daughter (214) | Normal | ||||||||||
| Paternal aunt (103) | — | — | |||||||||
| Paternal aunt's daughter (223) | — | Subendothelial, subepithelial, mesangial | — | — | — | ||||||
| Ozaltin et al. (2013) [4] | Glomerular microangiopathy with histologic signs of both MPGN and endothelial distress | Brother (Family UT-062 V-2) | — | — | Normal | — | — | — | Homozygous DGKE c.127C>T (p.Q43X) | ||
| Brother (Family UT-062 V-3) | C3 pos | Subendothelial | |||||||||
| Brother (Family UT-062 V-4) | |||||||||||
| Brother (Family UT-062 V-6) | |||||||||||
| Sister (Family HU-314 IV-1) | — | — | Homozygous DGKE c.610ΔA (p.T204Qframeshift210X) | ||||||||
| Sister (Family HU-314 IV-2) | — | — | |||||||||
| Sister (Family HU-500 V-1) | C3 neg; IgM, IgG pos | — | Homozygous DGKE c.889–2 (p.W350X) | ||||||||
| Twin brother (Family HU-500 V-2) | — | — | — | ||||||||
| Twin brother (Family HU-500 V-3) | — | — | — | ||||||||
| Tortajada et al. (2013) [5] | C3 glomerulopathy | Son (GN29) | Diffuse C3, mesangial IgM; IgG neg | Within GBM and mesangium | Slightly low | Neg | Normal | CFHR112341–5 | Heterozygous internal duplication of exons 2–5 (SCRs 1–4) of CFHR1 | ||
| Mother (GN29 M) | Diffuse C3; IgG/IgM neg | Subendothelial, subepithelial, mesangial | (with absent CFHR1, 3) | (with homozygous ΔCFHR3/1) | |||||||
| DDD | Maternal grandfather (GN29GF) | — | — | — | — | — | — | — | |||
| Medjeral-Thomas et al. (2014) [6] | C3GN | Maternal male cousin (III-2) | Allograft C3 pos | Subendothelial, mesangial (native); subendothelial, IM (transplant recurrence) | — | — | — | CFHR5121–9 | Heterozygous internal duplication of exons 2–3 (SCRs 1–2) in CFHR5 | ||
| Index male (III-5) | Capillary wall C3; IgA/IgG/IgM neg | Segmental subendothelial, IM | Normal | Neg | Normal | (with absent CFHR1, 3) | (with homozygous ΔCFHR3/1) | ||||
| Chen et al. (2014) [7] | C3 glomerulopathy with some EM features of DDD | Sister (635) | GBM > mesangial C3c; Ig neg | Ribbon-like GBM > mesangial (limited quality) | Very low | Neg | Normal | Hybrid CFHR212–CFHR5 (with low levels of wild-type CFHR2, 5) | Heterozygous hybrid CFHR gene with deletion of CFHR2 exons 4–5 (SCRs 3–4) | ||
| Brother (638) | — | — | |||||||||
| Wong et al. (2014) [8]/Power (1990) | MPGN (and acquired partial lipodystrophy) | Mother (1.2) | C3 pos; IgM > IgG/IgA | Subendothelial, mesangial | Very low | Pos | — | — | Heterozygous CFH c.249G>T (p.R83S on SCR2) | ||
| First son (2.1) | — | Normal | Normal | (also C3 polymorphisms p.R102G/P314L) | (heterozygous CFH H3/H5 haplotypes) | ||||||
IF/IHC, immunofluorescence/immunohistochemistry; EM, electron microscopy; C3NeF, C3 nephritic factor; Ig, immunoglobulin.
Medjeral-Thomas et al. demonstrated segregation of a heterozygous mutation in CFHR5 that results in duplication of SCR domains 1 and 2 with C3GN in a family without Cypriot ancestry [6]. Although the abnormal protein, CFHR5121–9, is identical to that reported in Cypriot CFHR5 nephropathy, the respective genomic breakpoints within CFHR5 intron 3 are distinct. It is now recommended that assays to screen for the presence of CFHR5121–9 are performed in patients with C3 glomerulopathy irrespective of ancestry.
Malik et al. identified a novel heterozygous CFHR mutation in a large, Irish kindred in which C3GN had previously been linked to the RCA gene cluster [3]. Deletion of a genomic segment comprising exons 4–6 of CFHR3 and exon 1 of CFHR1 produced a hybrid protein in which SCR domains 1 and 2 of CFHR3 are linked to full-length CFHR1. The hybrid CFHR312–CFHR1 gene was located on an allele also bearing intact CFHR1 and CFHR3 genes, suggesting disease causation due to a gain of function.
Tortajada et al. reported a Spanish family with C3 glomerulopathy and a heterozygous mutation resulting in an abnormal CFHR1 protein with duplication of SCR domains 1–4 [5].
Chen et al. reported a UK sibship with C3 glomerulopathy associated with heterozygous deletion of exons 4–5 of CFHR2 [7]. This produced a hybrid protein consisting of SCR domains 1 and 2 of CFHR2 linked to full-length CFHR5 (CFHR212–CFHR51–9). Due to the similarity between the initial two SCR domains of CFHR2 and CFHR5, the hybrid CFHR212–CFHR51–9 protein is structurally similar to CFHR5121–9.
A number of earlier family studies in which C3 glomerulopathy was associated with mutations causing severe deficiency or dysfunction of CFH have been summarized previously [9]. Recently, Wong et al. demonstrated association of a heterozygous CFH non-synonymous coding variant (c.249G>T) with MPGN in a UK family [8]. This mutation resulted in a serine to arginine substitution (p.R83S) within SCR2. A recombinant mutant CFH protein fragment (SCR domains 1–4) displayed reduced C3b-binding and complement regulatory activity in vitro compared with a recombinant wild-type CFH protein.
Ozaltin et al. identified a homozygous coding variant (c.127C>T) in the diacylglycerol kinase ε (DGKε) gene in a consanguineous Turkish family (denoted in the manuscript as UT-062) with familial renal disease with features of MPGN and thrombotic microangiopathy [4]. DGKε is an intracellular lipid kinase that is expressed in podocytes. The coding variant produces a premature stop codon (p.Q43X) leading to a truncated protein. How this results in renal disease remains unclear. However, the disease association was strengthened by the identification of other homozygous DGKE variants leading to protein truncation in two other consanguineous families.
RECENT INSIGHTS INTO CFHR BIOLOGY
A key insight into the biology of CFHR1, CFHR2 and CFHR5 derived from the observation that a recombinant CFHR11–2 protein formed obligatory head-to-tail dimers via a unique dimerization motif located within the highly conserved N-terminal SCR domains 1 and 2 of CFHR1, -2 and -5 [10]. Recombinant CFHR5 was shown to be an exclusively dimeric protein and, when key amino acids within the putative dimerization domains were mutated, the recombinant protein was monomeric. The identification of the dimerization motif within CFHR1, -2 and -5 leads to the prediction that both homo- and heterodimers could exist between these proteins (Figure 3). Furthermore, a common structural polymorphism that results in combined deletion of the CFHR1 and CFHR3 genes (ΔCFHR3/1) would result in deficiency of not only the CFHR1 homodimer but also deficiency of heterodimers containing CHFR1 (CFHR1–CFHR2 and CFHR1–CFHR5 heterodimers).
FIGURE 3:

Schematic representation of potential homodimers and heterodimers within the CFHR1, CFHR2 and CFHR5 protein family. The dimerization motif is located within N-terminal SCR domains 1 and 2 of CFHR1, -2 and -5 (depicted in blue) enabling head-to-tail dimer formation. Potential homo- and heterodimers are shown. The combined deletion of the CFHR1 and CFHR3 genes (ΔCFHR3/1) would, in homozygosity, result in a complete loss of CHFR1-containing species. In this setting, the potential species comprise: CFHR2 and CFHR5 homodimers and a CFHR2–CFHR5 heterodimer (inset).
What is the relevance of these observations to patients with familial C3 glomerulopathy associated with abnormal CFHR proteins? Duplication of the dimerization motif and the presence or absence of the ΔCFHR3/1 allele could theoretically lead to diverse species of CFHR homo- and heterodimers (Table 2). Defining the composition of these proteins in vivo is a prerequisite to understanding the association of abnormal CFHR proteins with C3 glomerulopathy. In addition, it is important for our understanding of why the ΔCFHR3/1 allele modifies disease susceptibility in age-related macular degeneration [11], systemic lupus erythematosus [12] and IgA nephropathy [13].
Table 2.
Predicted homodimers and heterodimers associated with abnormal CFHR proteins in patients with C3 glomerulopathy
| Modified CFHR protein | Abnormal homodimers | Abnormal heterodimers |
|
|---|---|---|---|
| CFHR1, CFHR3 present | Homozygous ΔCFHR3/1 | ||
| CFHR112341–5 | (CFHR112341–5) × 2 | (CFHR112341–5)–CFHR1 (CFHR112341–5)–CFHR2 (CFHR112341–5)–CFHR5 |
CFHR112341–5)–CFHR2 (CFHR112341–5)–CFHR5 |
| CFHR5121–9 | (CFHR5121–9) × 2 | (CFHR5121–9)–CFHR1 (CFHR5121–9)–CFHR2 (CFHR5121–9)–CFHR5 |
(CFHR5121–9)–CFHR2 (CFHR5121–9)–CFHR5 |
| CFHR212–CFHR5 | (CFHR212–CFHR5) × 2 | (CFHR212–CFHR5)–CFHR1 (CFHR212–CFHR5)–CFHR2 (CFHR212–CFHR5)–CFHR5 |
(CFHR212–CFHR5)–CFHR2 (CFHR212–CFHR5)–CFHR5 |
| CFHR312–CFHR1 | (CFHR312–CFHR1) × 2 | (CFHR312–CFHR1)–CFHR1 (CFHR312–CFHR1)–CFHR2 (CFHR312–CFHR1)–CFHR5 |
(CFHR312–CFHR1)–CFHR2 (CFHR312–CFHR1)–CFHR5 |
At present, there is strong evidence to support the existence of all three homodimers [5, 10] but only one heterodimer (CFHR1–CFHR2). The latter was readily identified because CFHR1 but not CFHR2 interacts with heparin. When serum or plasma is subjected to heparin chromatography, CFHR2 is only retained (bound to heparin) in the presence of CFHR1 [5, 10]. The CFHR1–CFHR5 heterodimer has less clearly been identified [5, 10] while CFHR2–CFHR5 was not seen in one study [5] or was seen only in very low amounts in another [10]. In addition to dimerization, there is evidence that these proteins can form larger complexes [5]. Following heparin chromatography of CFHR1-containing plasma, protein complexes of various sizes were identified, consistent with formation of CFHR1 homo-oligomers and a variety of CFHR1–CFHR2 and CFHR1–CFHR5 hetero-oligomers [5].
What is the functional significance of dimerization and/or oligomerization? One obvious consequence would be an increase in avidity. Compared with monomeric recombinant CFHR5 (lacking the dimerization motif), dimeric recombinant CFHR5 displayed increased binding to GBM-bound mouse C3 in vivo in the Cfh-deficient mouse model of C3 glomerulopathy [10]. Monomeric proteins also interacted less effectively than dimeric proteins with human C3b in vitro [5, 10]. The significance of this increased avidity became clearer when it was proposed that following binding to C3b, unlike CFH, CFHR1, -2 and -5 do not inhibit C3b amplification but actually allow it to proceed [5, 10]. This led to the hypothesis that CFHR proteins, by competing with CFH for surface C3b, could influence the degree to which C3b amplification is inhibited (predominant CFH binding) or allowed to proceed (predominant CFHR binding). The latter process was termed CFH de-regulation since these CFHR proteins were devoid of any intrinsic complement regulatory activity.
CFH de-regulation has been shown in vitro. Normal CFHR proteins (CFHR1, -2 and -5) can compete with CFH for binding to shared surface ligands in vitro using heterologous erythrocytes (guinea-pig erythrocytes) as a complement-activating surface [5, 10]. By increasing the CFHR protein concentration, the interaction between CFH and surface ligands can be inhibited, resulting in lysis of the guinea pig erythrocytes. Notably, monomeric forms of these CFHR proteins were less efficient at CFH de-regulation than dimeric forms, presumably due to differences in avidity [5, 10]. Abnormal CFHR proteins are even more efficient de-regulators of CFH in this assay. Serum fractions, containing all CFHR1, -2 and -5 species from individuals with C3 glomerulopathy associated with either the CFHR5121–9 or CFHR312–CFHR1 abnormal protein, demonstrated enhanced red cell lysis (consistent with greater CFH de-regulation) [10]. Similarly, a CFHR1-containing protein preparation from a family member with the CFHR112341–5 protein (and accompanying homozygous ΔCFHR3/1) also mediated enhanced lysis of guinea-pig erythrocytes [5]. This patient preparation was then enriched for large multimeric complexes, which could be visualized using electron microscopy as ‘bundle-like structures' (whereas purified normal CFHR1 and CFH could not be seen). However, antagonism of CFH by normal or abnormal CFHR proteins has not been demonstrated in vivo or on GBM preparations. In addition, the physiological relevance of CFH de-regulation by normal CFHR proteins remains unknown. An important and interesting observation indicating that our knowledge in this area remains incomplete is that the hybrid CFHR212–CFHR5 protein [7], unlike the structurally very similar CFHR5121–9, protein [2], was associated with severe plasma C3 depletion. Recombinant CFHR212–CFHR5 protein enhanced the formation of the AP C3 convertase in the absence of CFH and inhibited its dissociation in the presence of CFH. This activity is similar to C3 nephritic factor and may be the reason that the phenotype was associated with very low plasma C3 levels.
TOWARDS A DISEASE MECHANISM
A number of processes by which C3 fragments accumulate along the GBM in C3 glomerulopathy can now be proposed (Figure 4). Under physiological conditions, C3b amplification is limited by the actions of CFH. It has been difficult to establish whether CFH prevents C3 accumulation along the GBM primarily by regulating the AP in the circulation, or by first attaching to the GBM in order to mediate its protective effects. Most likely, CFH performs both these actions in response to the various exogenous triggers of C3 activation encountered by the innate immune system (e.g. infection). In patients with abnormal CFH, the physiological mechanism of ‘C3 tickover’, whereby spontaneous C3 activation occurs via the AP in the circulation, proceeds without inhibition. This results in severe secondary consumption of plasma C3 and the AP activation protein, factor B, even in the absence of any exogenous trigger of C3 activation. Experimentally, accumulation of C3 fragments along the GBM has been demonstrated only in the setting of uncontrolled C3 activation due to complete CFH deficiency. Individuals with complete CFH deficiency and C3 glomerulopathy typically have very low plasma C3 levels in addition to abnormal glomerular C3 accumulation. In contrast, patients with C3 glomerulopathy associated with abnormal CFHR proteins but intact CFH often [2, 6], but not invariably [7], have normal or only moderately reduced plasma C3 levels. One explanation for the association between familial C3 glomerulopathy and abnormal CFHR proteins is that these interfere with the CFH regulation of C3 activation predominantly along the GBM. This would result in abnormal glomerular C3 deposition with less marked or no effect on plasma C3 regulation. Hence the CFHR212–CFHR5 [7] protein may interfere with CFH regulation of C3 both along the GBM (resulting in C3 glomerulopathy) and in the circulation (resulting in plasma C3 depletion).
FIGURE 4:

Schematic representation of pathogenesis of C3 glomerulopathy. Triggers of C3 activation (depicted at the centre) may be exogenous (e.g. infection) or endogenous (C3 tickover). C3 activation generates C3b molecules for attachment to surfaces including the GBM. In health (upper panel), C3b amplification is tightly controlled by factor H (CFH) in the circulation. Thus minimal C3b becomes available for attachment to surfaces, where amplification is further regulated by CFH on the GBM. In theory, any surface-attached C3b is then metabolised, leaving C3 fragments (iC3b, C3d) attached to the GBM, and releasing CFH back into the circulation. In practice, neither C3 nor CFH are detected along the GBM in normal glomeruli. In the setting of abnormal CFH (bottom left panel), uncontrolled C3 activation leads to excessive C3 fragments (iC3b, C3d) in the circulation, which later accumulate along the GBM. Some C3b amplification/metabolism may also occur on the GBM. Enhanced CFH de-regulation due to abnormal CFHR proteins (bottom right panel) has been proposed as one mechanism by which C3 accumulates along the GBM despite intact CFH. Competitive inhibition of CFH by abnormal CFHR proteins along the GBM would facilitate C3b surface amplification/metabolism. Increased C3b amplification in plasma due to an abnormal CFHR protein that stabilizes the C3 convertase has also been demonstrated (see the text).
THE CURRENT HISTOPATHOLOGICAL DEFINITION OF C3 GLOMERULOPATHY
Hou et al. conducted a review of 319 biopsies showing MPGN, in which different cut-off levels for glomerular immunoglobulin (Ig) deposition were evaluated as part of immunofluorescence criteria for diagnosis of C3 glomerulopathy [14]. The results (published simultaneously with the consensus report) showed that strict definition of C3 glomerulopathy based on ‘C3 only’ (i.e. total absence of glomerular Ig) was impractical, since it excluded half of the 42 DDD cases identified on EM. Instead, a more inclusive criterion of predominant glomerular C3 intensity of ≥2 levels of magnitude greater than any combination of IgG, IgM, IgA and C1q increased the sensitivity for DDD to 88%. Applying this criterion to 200 cases previously diagnosed as MPGN Type 1, and 77 cases of MPGN Type 3, in which secondary causes including hepatitis C infection, cryoglobulinaemia and paraproteinaemia had been excluded, 31% and 39%, respectively, could be classified as C3 glomerulopathy. The authors concluded that this approach identified those patients who were most likely to benefit from evaluation of the AP. It is now recommended that in practice ‘glomerulonephritis with dominant C3’ should be used as a morphological term for those cases with dominant staining for C3c. Dominant is defined as C3c intensity two orders of magnitude more than any other immune reactant on a scale of 0–3 (comprising 0, trace, 1, 2 and 3). This approach will identify the majority of cases of C3 glomerulopathy and trgger (depending on the clinical scenario) investigations to identify known complement abnormalities [1].
RECENT RETROSPECTIVE COHORT STUDIES
As C3 glomerulopathy is extremely rare, patient cohort studies provide valuable information regarding clinical and pathological features and their correlation with disease outcomes [15]. Medjeral-Thomas et al. conducted a retrospective cohort study in 80 adults and children in the UK and Ireland with C3 glomerulopathy [16]. Based on a strict definition of C3 glomerulopathy (in which cases were excluded if any amount of glomerular IgG/IgA or more than weak IgM was demonstrated on immunostaining), they estimated an annual incidence of one to two cases per million population. Electron microscopy was performed in all cases, enabling a number of important clinicopathologic distinctions to be drawn between DDD (21 patients) and C3GN (59 patients). Age at diagnosis was significantly lower for DDD (median age 12 versus 26 for C3GN), and low serum C3 levels at diagnosis significantly more common (79% versus 48% in C3GN among the 69 patients assessed). MPGN was the most common lesion on light microscopy in both groups (55% of all biopsies). Presence of >50% crescents was more common in DDD (19% of biopsies versus 5% in C3GN), whereas vascular disease and chronic changes including glomerulosclerosis and interstitial fibrosis were more common in C3GN. At a median follow-up of 28 months, 29% of patients had developed ESKD, with no difference in renal survival between DDD and C3GN. However using multivariate analysis, DDD emerged as a strong and independent predictor of ESKD, along with age ≥16 years at diagnosis and crescentic GN. Histological recurrence following renal transplantation in all six DDD recipients was associated with graft failure in three cases, while four of seven C3GN recipients had histological recurrence, with graft failure in three cases. Zand et al. reported a US retrospective cohort of 21 adult and paediatric patients transplanted for ESKD due to C3GN, of whom two-thirds had histological recurrence, with half of these developing subsequent graft failure [17]. Recurrence and graft failure rates have been similar in earlier DDD/C3GN cohort studies, although it is unclear whether graft survival is reduced in recipients with C3 glomerulopathy compared with other types of glomerulonephritis. Nevertheless, development of effective therapies for use in native C3 glomerulopathy may ultimately also be beneficial in the transplant setting.
RECENT TREATMENT STUDIES
As no treatment is proven to be beneficial for C3 glomerulopathy, research interest in therapeutic agents that modulate the complement pathways is high. Eculizumab is a humanized anti-complement C5 monoclonal antibody approved for use in patients with paroxysmal nocturnal haemoglobinuria and aHUS. Off-label use of eculizumab for treatment of C3 glomerulopathy was initially reported in three cases of DDD [18, 19] (one with transplant recurrence [21]) and an additional case of MPGN Type 1 [20]. In 2012, a prospective trial was reported by Bomback et al. in six patients with native or post-transplant recurrent DDD or C3GN [22]. Since the trial, at least seven additional case reports of eculizumab use in C3 glomerulopathy have been published, most involving patients with persistent disease despite immunosuppressive therapy (Table 3). In the trial, eculizumab therapy was associated with a significant fall in serum creatinine or proteinuria in only 3 out of 6 patients, whereas 9 out of the total 11 case reports showed clinical improvement, raising the possibility of publication bias. Nevertheless, further prospective evaluation of eculizumab in C3 glomerulopathy appears warranted, with judicious patient selection based on available prognostic data from retrospective cohort studies and the like. Information obtained from renal biopsy both before and after treatment, for example the degree of active inflammation, may ultimately also guide eculizumab use, although evidence for this approach from the existing studies is limited [19, 23, 29, 30]. It has not been established whether disease progression occurs in some cases despite therapeutic blockade of C5 activation, as a result of ongoing upstream activation of C3. This concept was highlighted by the family study involving CFHR212–CFHR5, in which eculizumab was associated in vitro with effective blockade of C5 activation but not C3 activation in patient serum [7]. A Phase 1 clinical trial in children and adults with DDD (NCT01791686) is currently evaluating recombinant soluble complement receptor 1 (CR1), which was previously shown to restore plasma C3 levels and reduce glomerular C3 in Cfh−/− mice [31], and inhibited C3 activation in vitro in the CFHR212–CFHR5 family study [7]. Elsewhere rituximab has been reported with success in individual cases of C3 glomerulopathy [17, 32] (as distinct from rituximab use in treatment of disease-associated paraproteinaemia). However treatment failure following sporadic use of rituximab, often in conjunction with immunosuppressive treatment (including in the renal transplant setting), has also been widely reported [17, 18, 21, 23, 26, 28, 29].
Table 3.
Published reports of eculizumab use in C3 glomerulopathy and MPGN
| Report (year) | Study design | Reported diagnosis (age at treatment/sex) | C3NeF | Treatment duration (months) | Clinical response |
||
|---|---|---|---|---|---|---|---|
| Daina et al. (2012) [18] | Case report | DDD (22F) | + | 11 | Yes | ||
| Vivarelli et al. (2012) [19] | Case report | DDD (17M) | + | 18 + 9 | Yes | ||
| Radhakrishnan et al. (2012) [20] | Case report | MPGN Type 1 (16F) | + | 1.5 | Yes | ||
| McCaughan et al. (2012) [21] | Case report | Allograft recurrent DDD (29F) | + | 2.5 | Yes | ||
| Bomback et al. (2012) [22] | Prospective, open-label, uncontrolled trial | DDD (22M) | − | 12 | Yes | 50% | 82% |
| DDD (42M) | + | 9 | No | ||||
| Allograft recurrent DDD (32M) | − | 12 | Yes | ||||
| C3GN (25M) | − | 12 | No | ||||
| Allograft recurrent C3GN (22M) | + | 12 | No | ||||
| Allograft recurrent C3GN (20M) | + | 12 | Yes | ||||
| Gurkan et al. (2013) [23] | Case report | Allograft recurrent C3GN (21M) | + | 12 | Yes | ||
| Besbas et al. (2013) [24] | Case report | C3 glomerulopathy (16F) | − | 10 | No | ||
| Kerns et al. (2013) [25] | Case report | C3 glomerulopathy (16M) | − | 3.5 | Yes | ||
| Rousset-Rouvière et al. (2014) [26] | Case report | DDD (10M) | + | 6.5 | Yes | ||
| Ozkaya (2014) et al. [27] | Case report | DDD (14F) | + | 7 | Yes | ||
| Berthe-Aucejo et al. (2014) [28] | Case report | DDD (17M) | + | 3.5 | No | ||
| Sánchez-Moreno et al. (2014) [29] | Case report | Allograft recurrent DDD (14F) | + | 30 | Yes | ||
CONCLUSION
Enhanced CFH de-regulation has been proposed as a distinct pathophysiological mechanism in several families in which C3 glomerulopathy segregates with mutations leading to abnormal CFHR proteins. These abnormal proteins display increased avidity for C3b and an enhanced ability to antagonize CFH in vitro. Hence, in patients with C3 glomerulopathy, abnormal CFHR proteins seem likely to prevent CFH-mediated inhibition of C3b amplification along the GBM. In patients with CFH deficiency/dysfunction, excessive production of C3 fragments in the circulation, which then accumulate within the kidney, provides an additional disease mechanism for C3 glomerulopathy. We now have an improved means of identifying patients with C3 glomerulopathy through the introduction of a renal biopsy classification: glomerulonephritis with dominant C3. As C5 inhibition is not effective in at least some patients, we need to look for new approaches, the most obvious being to inhibit C3 activation. How this is achieved will depend on how and where the defect in C3 regulation has arisen. In addition, we need continued clinicopathological phenotyping, a prerequisite to the design of informative clinical trials. There are many ongoing innovative approaches to these challenges so the future for patients with C3 glomerulopathy is bright.
CONFLICT OF INTEREST STATEMENT
The authors declare that the results presented in this paper have not been published previously in whole or part.
ACKNOWLEDGEMENTS
T.D.B. is a Kidney Research UK (KRUK) Clinical Research Fellow (TF/2011). M.C.P. is a Wellcome Trust Senior Fellow in Clinical Science (WT098476MA) and M.M.R. is funded by this fellowship.
REFERENCES
- 1.Pickering MC, D'Agati VD, Nester CM et al. C3 glomerulopathy: consensus report. Kidney Int 2013; 84: 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gale DP, Goicoechea de Jorge E, Cook HT et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010; 376: 794–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malik TH, Lavin PJ, Goicoechea de Jorge E et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol 2012; 23: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozaltin F, Li B, Rauhauser A et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol 2013; 24: 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tortajada A, Yebenes H, Abarrategui-Garrido C et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest 2013; 123: 2434–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medjeral-Thomas N, Malik TH, Patel MP et al. A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry. Kidney Int 2014; 85: 933–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Q, Wiesener M, Eberhardt HU et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest 2013; 124: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong EK, Anderson HE, Herbert AP et al. Characterization of a factor H mutation that perturbs the alternative pathway of complement in a family with membranoproliferative GN. J Am Soc Nephrol 2014; (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbour TD, Pickering MC, Cook HT. Dense deposit disease and C3 glomerulopathy. Semin Nephrol 2013; 33: 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goicoechea de Jorge E, Caesar JJ, Malik TH et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci USA 2013; 110: 4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes AE, Orr N, Esfandiary H et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet 2006; 38: 1173–1177 [DOI] [PubMed] [Google Scholar]
- 12.Zhao J, Wu H, Khosravi M et al. Association of genetic variants in complement factor H and factor H-related genes with systemic lupus erythematosus susceptibility. PLoS Genet 2011; 7: e1002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gharavi AG, Kiryluk K, Choi M et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 2011; 43: 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou J, Markowitz GS, Bomback AS et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int 2013; 85: 450–456 [DOI] [PubMed] [Google Scholar]
- 15.Bolignano D, Nagler EV, Van Biesen W et al. Providing guidance in the dark: rare renal diseases and the challenge to improve the quality of evidence. NDT 2014; 29: 1628–1632 [DOI] [PubMed] [Google Scholar]
- 16.Medjeral-Thomas NR, O'Shaughnessy MM, O'Regan JA et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol 2014; 9: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zand L, Lorenz EC, Cosio FG et al. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol 2014; 25: 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daina E, Noris M, Remuzzi G. Eculizumab in a patient with dense-deposit disease. N Engl J Med 2012; 366: 1161–1163 [DOI] [PubMed] [Google Scholar]
- 19.Vivarelli M, Pasini A, Emma F. Eculizumab for the treatment of dense-deposit disease. N Engl J Med 2012; 366: 1163–1165 [DOI] [PubMed] [Google Scholar]
- 20.Radhakrishnan S, Lunn A, Kirschfink M et al. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med 2012; 366: 1165–1166 [DOI] [PubMed] [Google Scholar]
- 21.McCaughan JA, O'Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant 2012; 12: 1046–1051 [DOI] [PubMed] [Google Scholar]
- 22.Bomback AS, Smith RJ, Barile GR et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 2012; 7: 748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurkan S, Fyfe B, Weiss L et al. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol 2013; 28: 1975–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besbas N, Gulhan B, Gucer S et al. A novel CFHR5 mutation associated with C3 glomerulonephritis in a Turkish girl. J Nephrol 2014; 27: 457–460 [DOI] [PubMed] [Google Scholar]
- 25.Kerns E, Rozansky D, Troxell ML. Evolution of immunoglobulin deposition in C3-dominant membranoproliferative glomerulopathy. Pediatr Nephrol 2013; 28: 2227–2231 [DOI] [PubMed] [Google Scholar]
- 26.Rousset-Rouvière C, Cailliez M, Garaix F et al. Rituximab fails where eculizumab restores renal function in C3nef-related DDD. Pediatr Nephrol 2014; 29: 1107–1111 [DOI] [PubMed] [Google Scholar]
- 27.Ozkaya O, Nalcacioglu H, Tekcan D et al. Eculizumab therapy in a patient with dense-deposit disease associated with partial lipodystropy. Pediatr Nephrol 2014; 29: 1283–1287 [DOI] [PubMed] [Google Scholar]
- 28.Berthe-Aucejo A, Sacquépée M, Fila M et al. Blockade of alternative complement pathway in dense deposit disease. Case Rep Nephrol 2014; 201568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sánchez-Moreno A, De la Cerda F, Cabrera R et al. Eculizumab in dense-deposit disease after renal transplantation. Pediatr Nephrol 2014; 29: 2055–2059 [DOI] [PubMed] [Google Scholar]
- 30.Herlitz LC, Bomback AS, Markowitz GS et al. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol 2012; 23: 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Nester CM, Holanda DG et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol 2013; 24: 1820–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giaime P, Daniel L, Burtey S. Remission of C3 glomerulopathy with rituximab as only immunosuppressive therapy. Clin Nephrol 2014; (epub ahead of print) [DOI] [PubMed] [Google Scholar]